
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-electrocatalytic CO2 reduction mediated by a dibenzophosphole oxide and a chromium complex†‡
Connor A.
Koellner
,
Amelia G.
Reid
and
Charles W.
Machan
 *
*
Department of Chemistry, University of Virginia, McCormick Rd, PO Box 400319, Charlottesville, Virginia 22904-4319, USA. E-mail: machan@virginia.edu
First published on 25th April 2023
Abstract
We report a co-electrocatalytic system for the selective reduction of CO2 to CO, comprised of a previously reported molecular Cr complex and 5-phenylbenzo[b]phosphindole-5-oxide (PhBPO) as a redox mediator. Under protic conditions, the co-electrocatalytic system attains a turnover frequency (TOF) of 15 s−1 and quantitative selectivity for CO. It is proposed that PhBPO interacts with the Cr-based catalyst, coordinating in an axial position trans to an intermediate hydroxycarbonyl species, M–CO2H, mediating electron transfer to the catalyst and lowering the barrier for C–OH bond cleavage.
The continued advancement of global civilization drives an increasingly immense demand for energy, resources, and materials, with corresponding adverse environmental consequences. In the interest of mitigating the environmental impact of this energy demand, research has focused on the use of electrocatalysts for important chemical processes.1–3 Electrocatalysts interconvert electrical and chemical energy, allowing the use of petrochemical energy to be supplanted by renewable sources. A key chemical transformation to achieving carbon neutrality is the catalytic reduction of carbon dioxide (CO2) to carbon monoxide (CO), which could address both the issue of environmental effects and the need for electrochemical pathways to industrially important molecular precursors.4–7
The electrochemical reduction of CO2 to CO requires the transfer of two electrons, typically mediated to the substrate via a catalytic system.8–10 When electrons are transferred sequentially to a single site, their associated reduction potentials can be separated by hundreds of millivolts as the required driving force increases with each added charge. Biological systems address these energetic challenges with redox cofactors that mediate electron transfer to active sites, in order to perform multielectron catalysis with high efficiency and selectivity.11 However, examples of analogous co-electrocatalytic CO2 reduction in synthetic homogenous catalytic systems with redox mediators (RMs) are limited.12–15 The area is promising as a general strategy, with examples known for co-electrocatalytic alcohol oxidation, nitrogen reduction, dioxygen reduction, and hydrogenation.16–21
Previously, our research group reported a co-electrocatalytic system based on a molecular Cr complex Cr(tbudhbpy)Cl(H2O) (1) with dibenzothiophene-5,5-dioxide (DBTD) as a RM that is capable of the selective reduction of CO2 to CO under protic and aprotic conditions.13 It was proposed that the coordination of the redox mediator to the Cr metal center promotes catalytic turnover under aprotic conditions because of three factors: through-space electronic conjugation (TSEC, sharing of a single electron between two vertically aligned π systems), dispersion effects, and the formation of an axial Cr–O bond trans to the site of CO2 substrate binding.13 TSEC broadly describes the transport of energy and charge density between stacked π-systems, which occurs in the previous scenario between the π-system of DBTD and the bipyridyl ligand backbone of 1.22–25
Under protic conditions we proposed that a pancake-bonding (PB) interaction occurred (sharing of two electrons in two π systems) that could be tuned by expanding the aromatic character of the DBTD core.14,26 Since sulfones are generally poor ligands, with few reports on their coordination chemistry,27,28 we became interested in how the coordinating strength of the redox mediator might alter the co-electrocatalytic response. Modifying the RM species to a different ligand type could favor the formation of the co-catalytic assembly and increase the observed activity. Therefore, we selected 5-phenylbenzo[b]phosphindole-5-oxide (PhBPO) as the RM to pair with Cr(tbudhbpy)Cl(H2O) 1 (Fig. 1 and Fig. S1, ESI‡). PhBPO shares some structural similarities with DBTD and we anticipated that a R3P![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O moiety would serve as a better ligand than R2S(O)2.13,29
O moiety would serve as a better ligand than R2S(O)2.13,29
 | ||
| Fig. 1 Overview of a co-electrocatalytic system with inner-sphere electron transfer based on PB between PhBPO and Cr(tbudhbpy) (1) under aprotic and protic conditions; S = N,N-DMF. | ||
Cyclic voltammetry (CV) experiments were carried out with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in N,N-dimethylformamide (N,N-DMF) as the solvent. Under argon (Ar) saturation conditions, PhBPO displays a reversible one-electron redox feature with an E1/2 = −2.42 V versus ferrocenium/ferrocene (Fc+/Fc) (Fig. S2 and S3, ESI‡). Control CVs under saturated CO2 conditions show a slight loss of reversibility concomitant with an increase in current relative to Ar saturation, suggesting a slow chemical reaction follows reduction (Fig. S5, ESI‡), unlike what was observed previously for DBTD.13 Similar loss of reversibility is observed when phenol (PhOH) is added under both saturated CO2 and Ar conditions, however, a less dramatic increase in current is observed (Fig. S5, ESI‡). A control electrolysis experiment was performed with the PhBPO mediator and PhOH under CO2 saturation conditions (Fig. S24 and S25, ESI‡), resulting in production of CO with ca. 9% FE, but not achieving a single turnover with respect to [PhBPO], indicating that this compound is not intrinsically a catalyst for CO2 reduction under protic conditions (Table 1).
| Conditions | Potential (V vs. Fc+/0) | FECO (%) | Turnovers ([CO]/[1]) | TOFCPE (s−1) | η (V) |
|---|---|---|---|---|---|
| —[Cr] = Cr(tbudhbpy)Cl(H2O) 1 at a concentration of 0.5 mM. [PhBPO] and [PhOH] were 1.25 mM and 0.6 M respectively when included. Turnovers correspond to moles of CO produced divided by the moles of 1 present and do not represent a measurement of the system to inactivity. NQ—not quantifiable.a Previously reported results.13 | |||||
| 1 + PhBPO + PhOH | −2.58 | 101 ± 4% | 5.1 | 15 | 0.63 |
| 1 + PhOHa | −2.10 | 96 ± 8% | 15 | 4.35 | 0.11 |
| PhBPO + PhOH | −2.58 | 8.8 ± 1% | 0.14 | — | — |
| PhBPO | −2.58 | NQ | NQ | NQ | — |
| PhOH (Rinse Test) | −2.58 | NQ | NQ | NQ | — |
The addition of PhBPO (2.5 mM) to a solution of 1 (1 mM) under Ar saturation conditions suggests that no interaction occurs at the PhBPO0/− reduction under inert conditions (Fig. 2A—green), as evidenced by an unchanged redox wave. Conversely, under CO2 saturation conditions this mixture generates a large irreversible increase in current at the PhBPO0/− redox couple, suggestive of a catalytic process (Fig. 2A—blue).30 Compound 1 does not present a reduction feature near −2.50 V vs. Fc+/Fc, which indicates that the one-electron reduction of PhBPO results in the formation of a new adduct that modifies the electronic structure of 1, enabling co-electrocatalytic CO2 reduction.
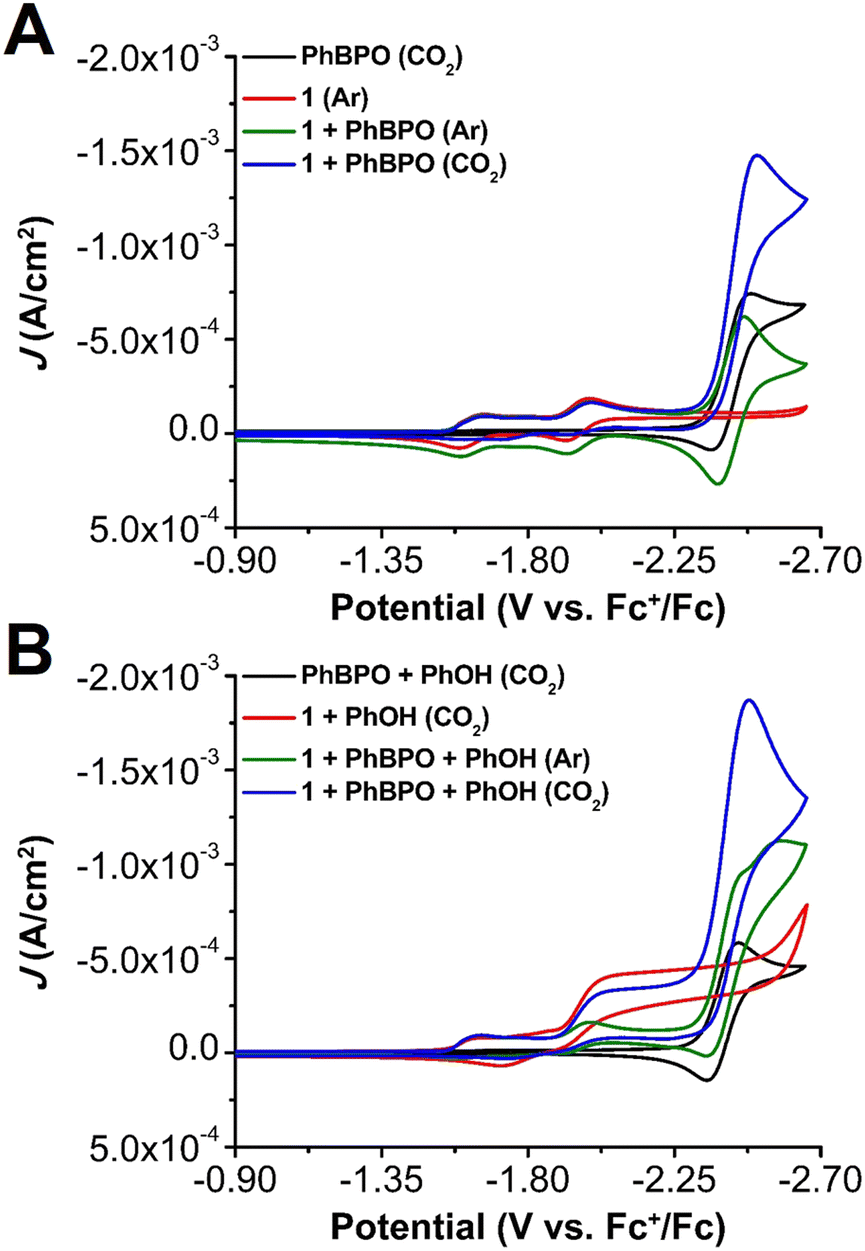 | ||
| Fig. 2 CVs comparing the reactivity of Cr(tbudhbpy)Cl(H2O) 1 (1.0 mM) and PhBPO (2.5 mM) in saturated Ar and CO2 solutions under (A) aprotic conditions and (B) protic conditions with added PhOH (0.1 M). | ||
When 0.1 M PhOH is added to 1 and PhBPO under CO2 saturation, a minimal difference with identical conditions in the absence of PhBPO is observed until approximately −2.25 V vs. Fc+/Fc where a large increase in current occurs (Fig. 2). This second catalytic regime appears at the PhBPO0/− reduction potential (Fig. S6 and S7, ESI‡).19,29 Variable concentration studies demonstrate that the catalytic current is proportional to the concentration of 1, PhBPO, a fixed ratio of 1 and PhBPO, PhOH, and CO2 (Fig. S10–S19, ESI‡). A slight shift to positive potentials is observed with increasing [PhBPO], which is suggestive of a favorable binding equilibrium for PhBPO under catalytic conditions (Fig. S10, ESI‡).30 Again, this behavior is comparable to the previously reported DBTD-mediated system.13,14,26
A bulk electrolysis experiment at −2.58 V vs. Fc+/Fc with 1, PhBPO, and PhOH present revealed a Faradaic efficiency for CO (FECO) of 101 ± 4% (Table 1 and Fig. S20 and S21, ESI‡). The observed Faradaic efficiency is comparable to the unmediated 1 + PhOH catalytic system, however, a higher TOF (15 s−1) is observed for the PhBPO-mediated system (Table 1). Interestingly, the activity enhancement under co-electrocatalytic conditions using PhBPO as the RM is not as significant as that observed for the DBTD-mediated system13,14,26 even though a stronger interaction with the Cr center is anticipated, vide infra. PhBPO remains stable during electrolysis experiments as evidenced by the conserved 31P-NMR spectral feature of the RM at about 32 ppm in pre- and post-electrolysis samples (Fig. S28 and S29, ESI‡). Furthermore, the ratio of the integrated spectral feature for PhBPO in Fig. S28 and S29 (ESI‡) is consistent in both pre- and post-electrolysis solutions when compared to the integral of the 80% aqueous phosphoric acid reference peak.
In order to assess the nature of the inner-sphere interaction between PhBPO and the Cr center in comparison to DBTD, we examined the reduction of the mediator, adduct formation and transition state for C–OH bond cleavage by previously optimized DFT methods.29 The calculated reduction potential for PhBPO0/− of −2.48 V vs. Fc+/0 showed excellent agreement with the experimentally determined value of E1/2 = −2.42 V.29 Examination of the spin density plot and Kohn-Sham (KS) representation of the SOMO for the PhBPO− radical suggested that the added electron density was distributed in the dibenzophosphole moiety in a similar manner to that observed previously in the dibenzothiophene fragment of DBTD− (Fig. S33 and S34, ESI‡).13,14
We have established in prior work that the co-electrocatalytic cycle is initiated by the displacement of an axial DMF ligand in a [Cr(tbudhbpy)(CO2H)(DMF)]− complex (S = 1) by the reduced RM (S = 1/2) to generate a new adduct [Cr(tbudhbpy)(CO2H)(RM)]2− (S = 3/2) when using sulfone-based mediators (RM) like DBTD.13,14 In the case of PhBPO, the same S = 3/2 spin manifold is preferred in the [Cr(tbudhbpy)(CO2H)(PhBPO)]2− adduct. The alternative S = 5/2 manifold is 4.7 kcal mol−1 higher in energy while the S = 1/2 is both 1.1 kcal mol−1 higher in energy and exhibits significant spin contamination. The binding of PhBPO− to the solvento complex [Cr(tbudhbpy)(CO2H)(DMF)]− is substantially more exoergic at −4.8 kcal mol−1 than the binding of DBTD−, which was previously determined to be approximately isoergic (−0.1 kcal mol−1).14 In this RM-Cr adduct, the cleavage of the C–OH bond assisted by a proton donor is proposed to be the rate-determining step of the co-electrocatalytic cycle. The barrier for the PhBPO adduct [Cr(tbudhbpy)(CO2H)(PhBPO)]2−·PhOH sits at ΔG = +10.4 kcal mol−1, which is 1.2 kcal mol−1 lower in energy than that determined for Cr(tbudhbpy)(CO2H)(DBTD)]2−·PhOH at the same level of theory.14 C–OH bond cleavage to produce [Cr(tbudhbpy)(CO)(RM)]− with water as a co-product has comparable exothermic parameters between the two RMs: −20.3 kcal mol−1 for the DBTD adduct and −21.8 kcal mol−1 for the PhBPO adduct. Furthermore, the loss of the CO product and neutral RM from [Cr(tbudhbpy)(CO)(RM)]− to produce the four-coordinate monoanion [Cr(tbudhbpy)(CO)(RM)]− (S = 3/2) is slightly exothermic in both cases: −2.2 kcal mol−1 for the DBTD adduct and −3.5 kcal mol−1 for PhBPO, both in the S = 3/2 state.
The binding interaction between PhBPO− and the Cr complex is the result of (1) the formation of a bond between R3PO and Cr; (2) dispersive interactions; and (3) PB between the aromatic fragments.13,14 From the computational and experimental results presented here, it is clear that the coordinating ability of PhBPO relative to DBTD in the reduced state is stronger, but the relative impact on the TS barrier does not manifest in improved kinetics during CPE. We attribute this to the increased binding strength of the PhBPO RM relative to the strength of PB, vide infra. Importantly, the vertical atom–atom alignment was found to be poor for the PhBPO adduct prior to the RDS (Fig. S35, ESI‡), with significant rotation about the O–Cr bond observed (Fig. 3), suggesting that the PB contribution to the interaction with PhBPO has decreased relative to DBTD. Short distances and vertically aligned atoms have previously been established as requirements to maximize PB between aromatic frameworks, since these allow the best orbital overlap.31
 | ||
| Fig. 3 Geometry (A) and spin density plot (B) of [Cr(tbudhbpy)(CO2H)(PhBPO)]2− in the S = 3/2 manifold highlighting the poor spatial overlap of the aromatic regions and spin distribution consistent with pancake bonding. Select H atoms and tBu groups omitted for clarity. | ||
For both the DBTD and PhBPO adducts, a rotation about this bond was performed (Fig. S37 and S38, ESI‡), to determine the nature of the energy landscape around the optimal RM and catalyst backbone orientation. Interestingly, the potential energy surface of possible configurations for PhBPO is flat relative to that of DBTD. We propose that the increased binding strength of PhBPO to Cr diminishes the relative importance of PB involving the bpy fragment during the formation of the co-catalytic assembly in comparison to DBTD. Consistent with this, PhBPO shows significantly less co-planarity with the bpy fragment of the Cr complex than DBTD. Since DBTD is a poor ligand, PB is relatively more important to the thermodynamic favorability of adduct formation, which directs the vertical fragments into close alignment. Since PhBPO doesn’t require the same amount of PB to form the co-catalytic assembly, near isoenergetic conformations exist which do not bring the aromatic components of RM and Cr complex into ideal vertical-vertical alignment. Thus, catalyst speciation is prevented from completely coming under thermodynamic control to the configuration which would cause the greatest co-electrocatalytic current increase under reaction conditions. Indeed, the decrease in ΔG‡ when PhBPO− forms the co-catalytic assembly relative to DBTD− is consistent with the proposal of stronger interactions with the Cr center. The lower ΔG‡ when PhBPO serves as the RM is comparable to the trend observed when more electron-donating moieties are incorporated into the catalyst ligand framework,14 implying that PhBPO, being reduced at potentials which are 170 mV more negative than DBTD, as well as being more electron-rich in nature, increases the ability of the Cr center to donate electron density to the CO2H fragment during C–OH bond cleavage.
In conclusion, we report a new example of an inner-sphere electron transfer RM in a co-electrocatalytic system for CO2 reduction. The 1 + PhBPO co-catalytic system in the presence of PhOH as a proton source exhibits an enhancement of catalytic activity compared to just the Cr catalyst under protic conditions while retaining selectivity for CO. Furthermore, the PhBPO-containing system behaves similarly to the previously reported DBTD co-electrocatalytic systems,13,14 albeit at a slower rate with a higher overpotential. These observations suggest that although the strength of the axial bonding interaction between the RM and Cr complex can be used to access lower energy pathways, axial coordination strength must be balanced against the contribution of PB to bring the aromatic components into the best alignment for co-catalytic activity. Importantly, these results suggest that the use of electron-poor phosphine oxide derivatives with greater aromatic character will result in better kinetic enhancements by improving the interaction strength between RM and catalyst through better vertical orbital overlap of the aromatic π-systems, which we are exploring in ongoing studies.
Conflicts of interest
There are no conflicts to declare.References
- B. Das, A. Thapper, S. Ott and S. B. Colbran, Sustainable Energy Fuels, 2019, 3, 2159–2175 RSC.
- D. L. Dubois, Inorg. Chem., 2014, 53, 3935–3960 CrossRef CAS PubMed.
- V. Masson-Delmotte, P. Zhai, H.-O. Pörtner, D. Roberts, J. Skea, P. R. Shukla, A. Pirani, W. Moufouma-Okia, C. Péan, R. Pidcock, S. Connors, J. B. R. Matthews, Y. Chen, X. Zhou, M. I. Gomis, E. Lonnoy, T. Maycock, M. Tignor and T. Waterfield, Global Warming of 1.5 °C. An IPCC Special Report on the impacts of global warming of 1.5 °C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty, Intergovernmental Panel on Climate Change, Cambridge University Press, UK, 2018 Search PubMed.
- E. Rode, A. S. Agarwal, N. Sridhar and D. Hill, Handbook of Climate Change Mitigation and Adaptation, Springer, New York, New York, 2014 Search PubMed.
- E. Alper and O. Y. Orhan, Petroleum, 2017, 3, 109–126 CrossRef.
- B. H. Davis and G. Jacobs, in Thermochemical Conversion of Biomass to Liquid Fuels and Chemicals, ed. M. Crocker, The Royal Society of Chemistry, Cambridge, UK, 2010, pp. 95–124 Search PubMed.
- J. Kibsgaard., Z. W. Seh, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355(6321) DOI:10.1126/science.aad4998.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- C. W. Machan, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, and G. Parkin and L. Que Jr, Elsevier, Oxford, 2021, pp. 1101–1124 DOI:10.1016/B978-0-08-102688-5.00077-5.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- F. Melin and P. Hellwig, Chem. Rev., 2020, 120, 10244–10297 CrossRef CAS PubMed.
- S. Dey, F. Masero, E. Brack, M. Fontecave and V. Mougel, Nature, 2022, 607, 499–506 CrossRef CAS PubMed.
- S. L. Hooe, J. J. Moreno, A. G. Reid, E. N. Cook and C. W. Machan, Angew. Chem., Int. Ed., 2022, 61, e202109645 CAS.
- A. G. Reid, J. J. Moreno, S. L. Hooe, K. R. Baugh, I. H. Thomas, D. A. Dickie and C. W. Machan, Chem. Sci., 2022, 13, 9595–9606 RSC.
- P. T. Smith, S. Weng and C. J. Chang, Inorg. Chem., 2020, 59, 9270–9278 CrossRef CAS PubMed.
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef CAS PubMed.
- C. M. Galvin and R. M. Waymouth, J. Am. Chem. Soc., 2020, 142, 19368–19378 CrossRef CAS PubMed.
- P. Garrido-Barros, J. Derosa, M. J. Chalkley and J. C. Peters, Nature, 2022, 609, 71–76 CrossRef CAS PubMed.
- S. L. Hooe, E. N. Cook, A. G. Reid and C. W. Machan, Chem. Sci., 2021, 12, 9733–9741 RSC.
- E. A. McLoughlin, K. C. Armstrong and R. M. Waymouth, ACS Catal., 2020, 10, 11654–11662 CrossRef CAS.
- C. W. Anson and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 18472–18475 CrossRef CAS PubMed.
- M. Bai, J. Liang, L. Xie, S. Sanvito, B. Mao and S. Hou, J. Chem. Phys., 2012, 136, 104701 CrossRef PubMed.
- Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2006, 45, 6430–6437 CrossRef CAS PubMed.
- Y. Morisaki and Y. Chujo, Polym. Chem., 2011, 2, 1249–1257 RSC.
- P. Shen, J. Li, Z. Zhao and B. Z. Tang, CCS Chem., 2019, 1, 181–196 Search PubMed.
- A. G. Reid, M. E. Moberg, C. A. Koellner, J. J. Moreno, S. L. Hooe, K. R. Baugh, D. A. Dickie and C. W. Machan, Organometallics, 2023 DOI:10.1021/acs.organomet.2c00600.
- F. A. Cotton and T. R. Felthouse, Inorg. Chem., 1981, 20, 2703–2708 CrossRef CAS.
- E. V. Dikarev, R. Y. Becker, E. Block, Z. Shan, R. C. Haltiwanger and M. A. Petrukhina, Inorg. Chem., 2003, 42, 7098–7105 CrossRef CAS PubMed.
- J. J. Moreno, S. L. Hooe and C. W. Machan, Inorg. Chem., 2021, 60, 3635–3650 CrossRef CAS PubMed.
- C. Costentin and J.-M. Saveant, Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry, Wiley, Hoboken, 2019 Search PubMed.
- M. Kertesz, Chem. – Eur. J., 2019, 25, 400–416 CrossRef CAS PubMed.
Footnotes |
| † We thank Dr Juan J. Moreno for initial contributions to the project. |
| ‡ Electronic supplementary information (ESI) available: UV-vis, NMR, and CV data. CCDC: 2219200. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc00166k |
| This journal is © The Royal Society of Chemistry 2023 |