
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransient imine as a directing group for the Pd-catalyzed anomeric C(sp3)–H arylation of 3-aminosugars†
Juba
Ghouilem
a,
Sokna
Bazzi
a,
Nicolas
Grimblat
 b,
Pascal
Retailleau
c,
Vincent
Gandon
bd and
Samir
Messaoudi
*a
b,
Pascal
Retailleau
c,
Vincent
Gandon
bd and
Samir
Messaoudi
*a
aUniversite Paris-Saclay, CNRS, BioCIS, 92290, Châtenay-Malabry, France. E-mail: samir.messaoudi@universite-paris-saclay.fr; Tel: +(33) 0146835887
bLaboratoire de Chimie Moléculaire (LCM), CNRS UMR 9168, Ecole Polytechnique, Institut Polytechnique de Paris, route de Saclay, Palaiseau 91120, France
cInstitut de Chimie des Substances Naturelles, CNRS UPR 2301, Universite Paris-Saclay, avenue de la terrasse, Gif-sur-Yvette 91198, France
dUniversité Paris-Saclay, CNRS, ICMMO, 91405, Orsay cedex, France
First published on 31st January 2023
Abstract
The first example of Pd(II)-catalyzed anomeric arylation of 3-aminosugars is reported by using an L,X-type transient directing group (TDG) approach combined with an external 2-pyridone ligand. The released free amine was in situ transformed into an azide function, which was then exploited in a CuAAC to increase the molecular complexity and prepare a variety of complex substituted C3-triazolo C-glycosides in good yields.
The aryl C-glycoside motif, which is found in many natural products and drug candidates, has attracted tremendous attention in medicinal chemistry and drug discovery.1 These glycomimetics often feature enhanced resilience to enzymatic and hydrolytic cleavage under biological conditions compared to their O/N-glycoside congeners.2 In particular, aryl C-glycosides bearing a nitrogen atom at the C3 position of the sugar moiety (e.g. 3-amino or 3-triazolo C-glycosides) are one of the most important families of C-glycosides found in natural bioactive compounds.3 These glycosides have emerged as a privileged class with diverse potential applications (Fig. 1).
 | ||
| Fig. 1 Aryl C-glycoside-based bioactive molecules. | ||
Classical methods for the synthesis of aryl C-glycosides have largely relied on the use of metal-catalyzed cross-coupling strategies involving the use of stoichiometric organometallic coupling partners.3f–h The persistent need from drug discovery programs to elaborate complex aryl C-glycosides has led to pioneering developments in the field of C–H functionalization.4 This approach, which avoids the preactivation of the coupling partners, has led to new paradigms for the synthesis of aryl C-glycosides. In this context, several groups have reported elegant methods by applying metal-catalyzed activation of C(sp2)–H bonds. However, methods involving a direct activation of the less reactive C(sp3)–H bonds of the sugar partners are rare.5 Thus far, precise structural design and selective modification of glycosides remain challenging tasks. Recently, Ackermann's group5e and our group5d reported independently two studies in which the anomeric, as well as C-3 positions of the sugar moieties, were functionalized selectively through a Pd-catalyzed C(sp3)–H activation reaction (Scheme 1A and B). Nevertheless, these strategies require synthetic steps to prepare the activated sugar substrate and remove the directing group. To address this issue, we sought to develop a transient imine directing group approach6 for the Pd-catalyzed anomeric C(sp3)−H arylation of 3-aminosugars to access aryl C-glycosides in an expedient way (Scheme 1C).
 | ||
| Scheme 1 Strategies to access aryl C-glycosides. | ||
We hypothesized that the stereoselective introduction of the aryl moiety could be accomplished through a C(sp3) arylation of the in situ generated axial imine intermediate (A, Fig. 2) as an L,X-type transient directing group, to direct the palladium(II) catalyst selectively to the α-anomeric C(sp3)–H bond through a bidentate coordinating mechanism (intermediates B and C). Before testing the coupling experimentally, a computational study of the key CMD step was performed at the M06//6-311++G(d,p)/SDD level of theory in order to evaluate the free energy demand. It was found that when the axial imine pyridol directing group was used (R = Ac), the CMD transition state (CMD-TS) that gives rise to the α-configuration necessitates a barrier of 19.71 kcal mol−1 (Fig. 2), which is comparable to our previous reported study with the use of a classical directing group strategy.5d
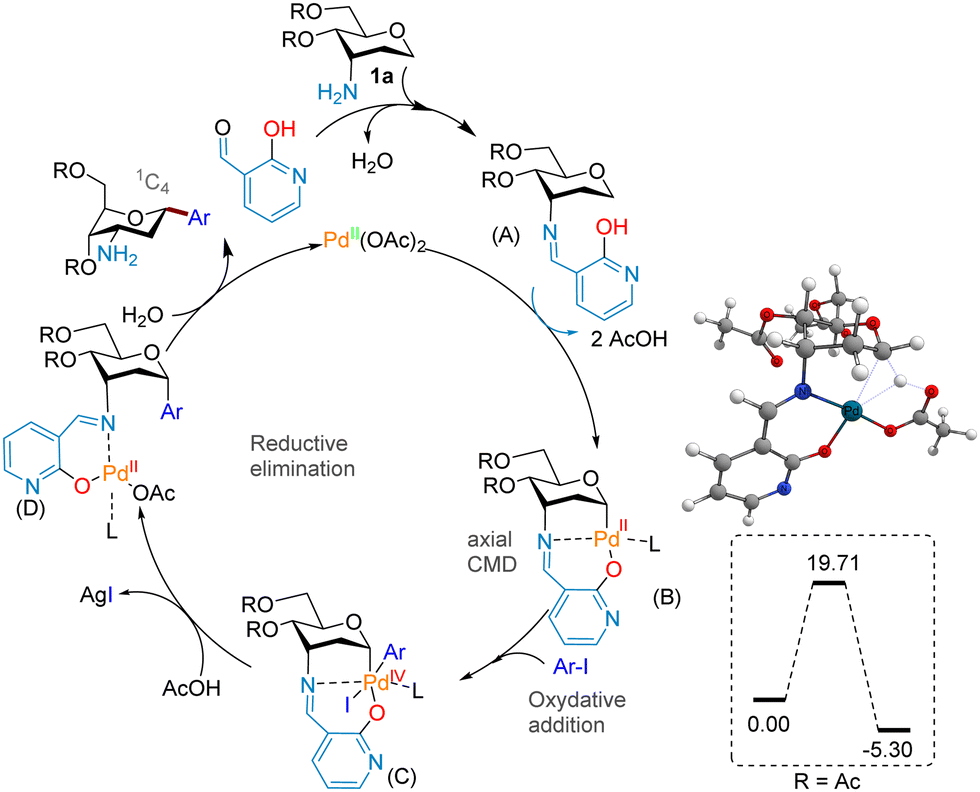 | ||
| Fig. 2 Proposed mechanism and DFT calculations. | ||
We anticipated at this stage that the final C-aryl glycosides bearing a free amine will be extremely polar and tricky to purify, so to ease their isolation and analysis, we envisioned transforming the released free amine products without purification into azides. The later could then undergo a CuAAC process to increase the molecular complexity and prepare a variety of complex substituted C3-triazolo C-glycosides. This approach is conceptually attractive in terms of diversifying the C-aryl glycoside pharmacophore, aiming to identify novel scaffolds of biological interest.
To initiate this study, we selected O-protected 3-amino sugars 1a–c and 4-iodotoluene 2a as models. To facilitate isolation and purification of the desired C3-azido C-glycosides and reduce the generation of waste, we decided to examine both transformations in a one-pot manner as depicted in Table 1: (i) the Pd-catalyzed C(sp3) activation directed by the in situ generated transient imine group and (ii) the Cu-catalyzed azidation of the released free amine. Representative results are summarized in Table 1.
|
|
|||||
|---|---|---|---|---|---|
| Entry | TDG | Ligand | [Pd] | [Ox] | Yieldb (%) |
a Reactions were conducted with substrate 1a–c (0.4 mmol), 2a (1.2 mmol), Pd. cat. (10 mol%), TDG (50 mol%), L (50 mol%), Ag-source (2 equiv.), and H2O (10 equiv.) in a mixture of HFIP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOH (19:1) [0.1 M] at 90 °C for 16 h.
b Yield of isolated product 3a.
c 30 min heating under microwave irradiation. :AcOH (19:1) [0.1 M] at 90 °C for 16 h.
b Yield of isolated product 3a.
c 30 min heating under microwave irradiation.
|
|||||
| 1 | TDG1 | L1 | Pd(OAc)2 | AgTFA | 27 |
| 2 | TDG2 | L1 | Pd(OAc)2 | AgTFA | 0 |
| 3 | TDG3 | L1 | Pd(OAc)2 | AgTFA | 23 |
| 4 | TDG4 | L1 | Pd(OAc)2 | AgTFA | 32 |
| 5 | TDG5 | L1 | Pd(OAc)2 | AgTFA | 0 |
| 6 | TDG4 | L2 | Pd(OAc)2 | AgTFA | 34 |
| 7 | TDG4 | L3 | Pd(OAc)2 | AgTFA | 41 |
| 8 | TDG4 | L4 | Pd(OAc)2 | AgTFA | 43 |
| 9 | TDG4 | L5 | Pd(OAc)2 | AgTFA | 42 |
| 10 | TDG4 | L6 | Pd(OAc)2 | AgTFA | 37 |
| 11 | TDG4 | L7 | Pd(OAc) 2 | AgTFA | 51 |
| 12 | TDG4 | L8 | Pd(OAc)2 | AgTFA | 40 |
| 13 | TDG4 | — | Pd(OAc)2 | AgTFA | 27 |
| 14 | TDG4 | L7 | Pd(OAc)2 | Cu(TFA)2 | 28 |
| 15 | TDG4 | L7 | Pd(OAc)2 | AgOAc | 0 |
| 16 | TDG4 | L7 | Pd(OAc)2 | Ag2O | 0 |
| 17 | TDG4 | L7 | Pd(OAc)2 | AgOMs | 0 |
| 18 | TDG4 | L7 | PdCl2 | AgTFA | 20 |
| 19 | TDG4 | L7 | Pd(OAc)2/Pd(TFA)2 | AgTFA | 42 |
| 20 | TDG4 | L7 | Pd(OAc)2 | AgTFA | 51c |

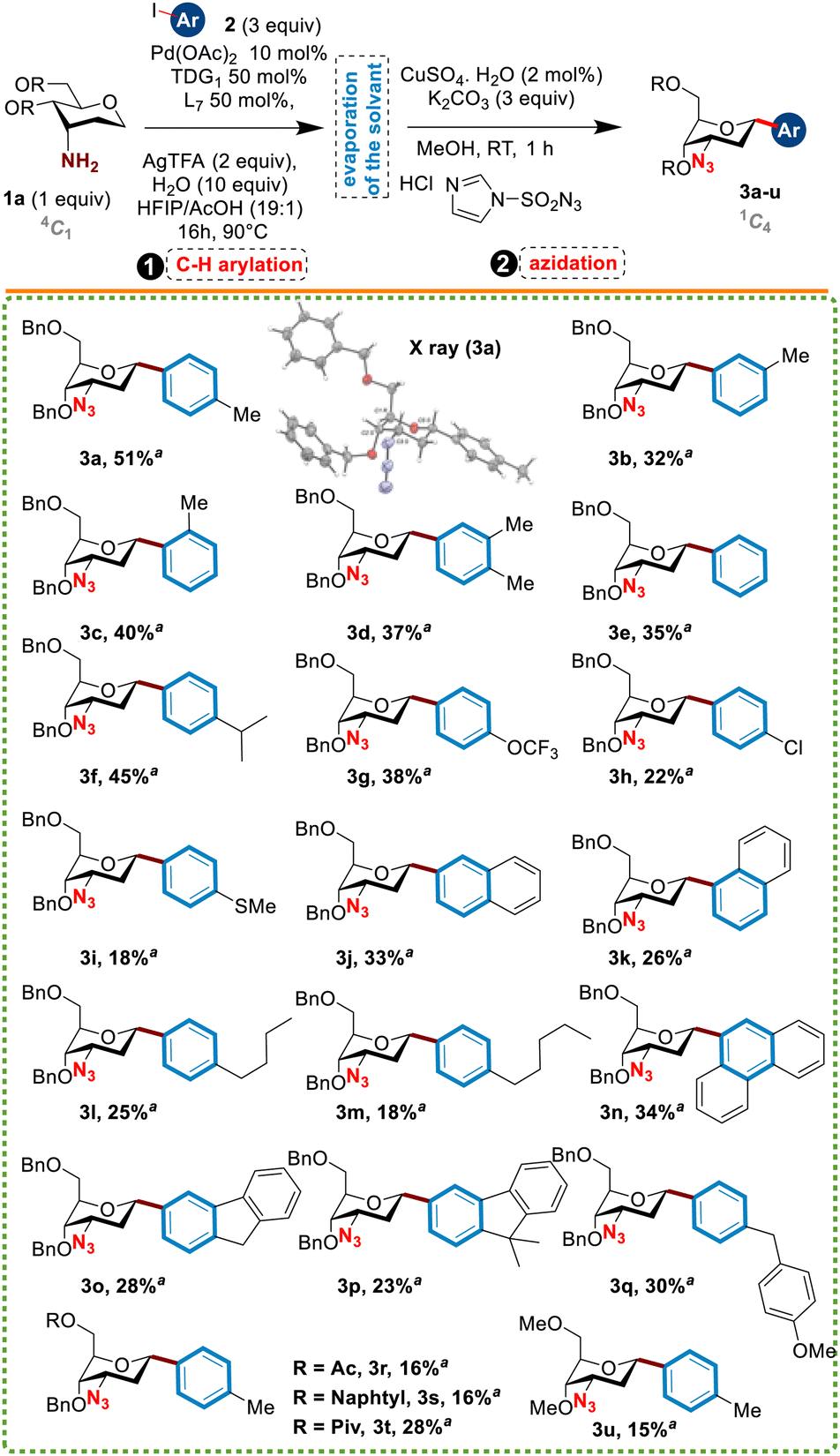
In preliminary experiments, the C(sp3)-arylation was examined first by using Pd(OAc)2 as a catalyst (10 mol%), TDG1 (50 mol%), L1 (50 mol%) as a ligand, AgTFA (2 equiv.) and H2O (10 equiv.) in a mixture of HFIP/AcOH (19:1 ratio, 0.1 M) at 110 °C for 16 h. The reaction mixture was monitored by LCMS and once the disappearance of the starting material was observed and the formation of the 3-amino C-tolyl glycoside intermediate was detected, the diazo-transfer reagent imidazole-1-sulfonyl azide7 (3 equiv.), CuSO4·H2O (10 mol%) and K2CO3 (3 equiv.), was added to the mixture which was stirred for two additional hours at RT. Under these conditions, no desired product was detected by LCMS analysis when both starting substrates 1b and 1c in which the hydroxyl functions were protected by acetyl or benzylidene groups, were used (Table 1). However, the use of O-benzylated sugar 1a led to the formation of the desired C3-azido C-aryl glycoside in a modest 27% yield for the two steps (entry 1). X-ray analysis clearly showed the α-configuration of the aryl group and the 1C4 conformation of the sugar nucleus, demonstrating the high diastereoselectivity of this reaction. This first set of results clearly demonstrated that the choice of the protecting group of the sugar moiety plays a pivotal role in this reaction.
It is worth noting that all our attempts to isolate the 3-amino C-tolyl glycoside intermediate [3a-NH2] were not an easy task and revealed that this aryl C-glycoside displays unexpected photophysical properties. Precisely, no absorbance at ∼240 nm and at ∼360 nm wavelength was detected despite the presence of three aromatic nuclei (ESI†). This issue renders the monitoring of the reaction by LCMS or TLC much more complicated, thus prompting us to continue our study by using the initial approach involving the in situ transformation of the amino group to an azide.
Knowing that the azidation step works quantitatively, we screened several parameters (Pd-catalyst, TDG, ligand, base, solvent, temperature, and reaction time, Table 1) to increase the yield of the first steps. The screening of various transient directing groups revealed that TDG3 and TDG4 display the same reactivity as TDG1 (23% and 32% yields, respectively) while quinoline benzaldehyde TDG2 or TDG5 were completely unreactive. A characteristic aspect of the developed method lies in the pyridone ligands used for this coupling. 5-Nitro-pyridone L2 was firstly examined, resulting in a similar yield as with 5-CF3 pyridone L1 (34% vs. 32% yields). Substitution of the C3 position of the pyridine with electron-withdrawing groups further improved the yield up to 51% (L3–L7), except for L6, which produced 3a in a yield similar to L1. Not surprisingly, only a 25% yield of 3a was obtained when the reaction was performed without ligand. Various other parameters were examined (Pd-catalyst, source of Ag, other oxidants, reaction temperature) but without success to improve the yield. Finally, we found that the arylation/azidation occurred smoothly with 51% yield when using Pd(OAc)2 as a catalyst (10 mol%), TDG4 (50 mol%), L7 as the ligand (50 mol%), AgTFA (2 equiv.), and H2O (10 equiv.) as an additive in a mixture of HFIP:AcOH (19:1) at 90 °C for 16 h (entry 11). Interestingly, the reaction time was reduced to only 30 min when the coupling was performed under microwave irradiation (entry 20). Of note, the use of a Pd-catalyst and TDG was necessary to achieve this transformation since no reaction took place when the coupling was conducted in their absence.
Motivated by these results, we next explored the scope of this new anomeric arylation procedure of 1a with aryl iodides. At first, we were pleased to see that various aryl iodides bearing diverse functions such as –OMe, –Me, –SMe, –Cl, –OCF3, and –alkyl chain (butyl or pentyl) reacted smoothly with 1a to afford the desired aryl C-glycosides 3a–w in moderate to good yields and exclusive α-anomeric stereoselectivity (Table 2). Surprisingly, the presence of an ortho substituent at the aromatic ring of the coupling partner did not affect the reaction process, as compound 3c was obtained in a satisfactory yield. Moreover, bulky polycyclic aromatic partners such as fluorene and phenanthrene were also tolerated under these conditions (compounds 3n–p). Finally, the synthetic utility of this methodology was demonstrated by the synthesis of the C-glycoside 3q as an analog of the dapagliflozin drug (FORXIGAs) used to treat type 2 diabetes (Fig. 1). Although the yields remained modest due to the unreacted anomeric C–H bond of sugars of type 1a, this constitutes the first example of intermolecular C(sp3)–H functionalization using a transient directing group.
|
a Reactions were performed in a flame dried re-sealable tube using sugar 1a (0.4 mmol), ArI (1.2 mmol), Pd(OAc)2 (10 mol%), TDG (50 mol%), L (50 mol%), AgTFA (2 equiv.), and H2O (10 equiv.) in a mixture of HFIP:AcOH (19:1) [0.1 M] at 90 °C for 16 h. a Yield of isolated product.
|
|---|
|
It is worth noting that the role of protecting groups turned out to be crucial for this reaction, particularly the primary alcohol at the C6 position of the sugar. Low yields were obtained when acetyl and naphthyl groups were used instead of benzyl one (compounds 3r and 3s); however, the pivalyl group seems to be better (compound 3t, 28% yield). Furthermore, switching from benzyl- to methoxyl-protecting groups led to the desired compound 3u in only a 15% yield.
Another important application of this method is the orthogonal preparation of a series of complex 3-triazolo-aryl C-glycosides through the simple copper-catalyzed azide-alkyne cycloaddition (CuAAC reaction).8 Different motifs were introduced at the azide function such as the trifluorinated benzene 4a, which can be considered as a direct analog of GB1107 (Fig. 1).
In addition, other triazolo aryl C-glycosides bearing biologically valuable functions such as a fluorescein glycoconjugate 4b, as well as the uridine-glycoconjugate 4c were synthesized in 59% and 65% yields, respectively (Scheme 2).
 | ||
| Scheme 2 Reactions were performed in a flame dried re-sealable tube using 3a (0.4 mmol), alkyne (0.4 mmol), CuSO4·H2O (0.04 mmol, 10 mol%), ascorbic acid (40 mol%) in t-BuOH:H2O (2:1) 0.1 M at RT. a Yield of isolated product. | ||
In conclusion, we report the first example of intermolecular anomeric C(sp3)–H functionalization using a transient directing group. This approach obviates the need for installation and removal of the directing group and furnishes in one pot manner a diverse collection of synthetically valuable 3-azido aryl C-glycosides with excellent α-selectivity. Moreover, the azide function allowed us to increase the structural complexity of this series of glycosides through a CuAAC reaction. Notably, the facile nature of this reaction has successfully been applied to the synthesis of various substituted triazolo aryl C-glycosides.
The authors acknowledge the support of this project by CNRS, Universite Paris-Saclay and the ANR SelFSuCHi (ANR-18-CE07-0012).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Štambaský, M. Hocek and P. Kočovský, Chem. Rev., 2009, 109, 6729 CrossRef PubMed; (b) E. De Clercq, J. Med. Chem., 2016, 59, 2301 CrossRef CAS PubMed; (c) Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281 CrossRef CAS PubMed; (d) K. Kitamura, Y. Ando, T. Matsumoto and K. Suzuki, Chem. Rev., 2018, 118, 1495 CrossRef CAS PubMed; (e) É. Bokor, S. Kun, D. Goyard, M. Tóth, J.-P. Praly, S. Vidal and L. Somsák, Chem. Rev., 2017, 117(3), 1687 CrossRef PubMed; (f) C.-F. Liu, Molecules, 2022, 27, 7439 CrossRef CAS PubMed.
- (a) R. Duval and C. Duplais, Nat. Prod. Rep., 2017, 34, 161 RSC; (b) C. Jaramillo and S. Knapp, Synthesis, 1994, 1 CrossRef CAS; (c) K. Kitamur, U. Ando, T. Matsumoto and K. Suzuki, Angew. Chem., Int. Ed., 2014, 53, 1258 CrossRef PubMed.
- (a) T. Cañeque, F. Gomes, T. Thi Mai, G. Maestri, M. Malacria and R. Rodriguez, Nat. Chem., 2015, 7, 744 CrossRef PubMed; (b) Y. Chang, L. Xing, Q. Sun, S. Liang, T. Liu, X. Zhang, T. Zhu, B. A. Pfeifer, Q. Che and G. Zhang, J. Nat. Prod., 2020, 83, 2749 CrossRef CAS PubMed; (c) G. D. A. Martin, L. T. Tan, P. R. Jensen, R. E. Dimayuga, C. R. Fairchild, C. Raventos-Suarez and W. Fenical, J. Nat. Prod., 2007, 70, 1406 CrossRef CAS PubMed; (d) H. Huang, T. Yang, X. Ren, J. Liu, Y. Song, A. Sun, J. Ma, B. Wang, Y. Zhang and C. Huang, J. Nat. Prod., 2012, 75, 202 CrossRef CAS PubMed; Y. Song, G. Liu, J. Li, H. Huang, X. Zhang, H. Zhang and J. Ju, Mar. Drugs, 2015, 13, 1304 Search PubMed; (e) K. Toshima, H. Ouchi, Y. Okazaki, T. Kano, M. Moriguchi, A. Asai and S. Matsumura, Angew. Chem., Int. Ed. Engl., 1997, 36, 2748 CrossRef CAS; (f) K. Toshima, Y. Maeda, H. Ouchi, A. Asaib and S. Matsumura, Bioorg. Med. Chem. Lett., 2000, 10, 2163 CrossRef CAS PubMed; (g) C. Liu, W. Wang, B. J. A. Elisworth and A. regueiro-ren, WO/2021/242982, 2021; (h) C. Liu, P. Rao Jalagam, J. Feng, W. Wang, T. Raja, M. Reddy Sura, R. K. V. L. P. Manepalli, B. Reddy Aliphedi, S. Medavarapu, S. K. Nair, V. Muthalagu, R. Natesan, A. Gupta, B. Beno, M. Panda, K. Ghosh, J. K. Shukla, H. Sale, P. Haldar, N. Kalidindi, D. Shah, D. Patel, A. Mathur, B. A. Ellsworth, D. Cheng and A. Regueiro-Ren, J. Med. Chem., 2022, 65, 11084 CrossRef CAS PubMed.
- J. Ghouilem, M. de Robichon, F. Le Bideau, A. Ferry and S. Messaoudi, Chem. – Eur. J., 2021, 27, 491 CrossRef CAS PubMed.
- (a) N. Probst, G. Grelier, N. Ghermani, V. Gandon, M. Alami and S. Messaoudi, Org. Lett., 2017, 19, 5038 CrossRef CAS PubMed; (b) N. Probst, G. Grelier, S. Dahaoui, M. Alami, V. Gandon and S. Messaoudi, ACS Catal., 2018, 8, 7781 CrossRef CAS; (c) J. Ghouilem, R. Franco, P. Retailleau, M. Alami, V. Gandon and S. Messaoudi, Chem. Commun., 2020, 56, 7175 RSC; (d) J. Ghouilem, C. Tran, N. Grimblat, P. Retailleau, M. Alami, V. Gandon and S. Messaoudi, ACS Catal., 2021, 11, 1818 CrossRef CAS; (e) J. Wu, A. Kopp and L. Ackermann, Angew. Chem., Int. Ed., 2022, 61, e202114993 CAS.
- For recent reviews, see: (a) J. I. Higham and J. A. Bull, Org. Biomol. Chem., 2020, 18, 7291 RSC; (b) M. I. Lapuh, S. Mazeh and T. Besset, ACS Catal., 2020, 10, 12898 CrossRef CAS; (c) C. Jacob, B. U. W. Maes and G. Evano, Chem. – Eur. J., 2021, 27, 13899 CrossRef CAS PubMed; (d) P. Gandeepan and L. Ackermann, Chemistry, 2018, 4, 199 CrossRef CAS.
- (a) S. P. Green, K. M. Wheelhouse, A. D. Payne, J. P. Hallett, P. W. Miller and J. A. Bull, Org. Process Res. Dev., 2020, 24, 67 CrossRef CAS PubMed; (b) A. Vasella, C. Witzig, J.-L. Chiara and M. Martin-Lomas, Helv. Chim. Acta, 1991, 74, 2073 CrossRef CAS; (c) P. B. Alper, S.-C. Hung and C.-H. Wong, Tetrahedron Lett., 1996, 37, 6029 CrossRef CAS; (d) P. T. Nyffeler, C.-H. Liang, K. M. Koeller and C.-H. Wong, J. Am. Chem. Soc., 2002, 124, 10773 CrossRef CAS PubMed.
- (a) N. Nebra and J. García-Álvarez, Molecules, 2020, 25, 2015 CrossRef CAS PubMed; (b) E. Haldon, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC; (c) D. H. Hilko, L. F. Bornaghi and S.-A. Poulsen, J. Org. Chem., 2018, 83, 11944 CrossRef CAS PubMed; (d) G. G. Crisp and B. L. Flynn, J. Org. Chem., 1993, 58, 6614 CrossRef CAS; (e) K. Peterson, R. Kumar, O. Stenström, P. Verma, P. R. Verma, M. Hakansson, B. Kahl-Knutsson, F. Zetterberg, H. Leffler, M. Akke, D. T. Logan and U. J. Nilsson, J. Med. Chem., 2018, 61(3), 1164 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures for the starting materials and 1H and 13C spectra for all new compounds. CCDC 2225278 and 2225279. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc00046j |
| This journal is © The Royal Society of Chemistry 2023 |