
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical study of phenylbismuth anion as a blueprint for main-group single-molecule magnets†
Akseli
Mansikkamäki

NMR Research Unit, University of Oulu, P.O. Box 8000, Oulu, FI-90014, Finland. E-mail: akseli.mansikkamaki@oulu.fi
First published on 20th January 2023
Abstract
The hypothetical [BiPh]− anion obtained by a one-electron reduction from the respective bismuthinidene is proposed as a basis for constructing single-molecule magnets (SMMs) consisting purely of main-group elements. Based on high-level quantum-chemical calculations, the [BiPh]− anion is predicted to be a SMM with an effective barrier of 6418 cm−1 for the relaxation of magnetization. This barrier is much larger than any effective barrier observed so far in any experimentally characterized SMM. The reduction potential for the [BiPh]−/BiPh couple is calculated as −1.5 V, which implies that the [BiPh]− moiety is accessible from stable bismuthinidenes containing a BiPh moiety and sufficient steric protection for the reactive Bi atom. Thus, [BiPh]− provides a blueprint for the realization of purely main-group SMMs which can surpass in their properties the best known dysprosium-based SMMs.
Single-molecule magnets (SMMs) are molecular entities with a bistable magnetic ground state that displays slow relaxation of magnetization.1–6 The possibility to manipulate the magnetic state at a microscopic level can lead to possible applications in fields such as quantum information processing.7–9 The upper limit for the operational temperature of a SMM can be defined by the blocking temperature, below which the magnetization can be considered frozen within the timescale of the experiment. Although the blocking temperature does not have a universally accepted definition, it is a central concept in the design of new SMMs. An SMM that displays magnetic blocking at room temperature could be utilized, for example, in the design of room-temperature quantum computers.9
Currently the most successful approaches to SMMs with high blocking temperatures are based on dysprosium metallocene complexes and bimetallic dysprosium systems with a one-electron bond between the dysprosium ions. Both approaches have now lead to the observation of blocking temperatures above the boiling point of liquid nitrogen (77 K).10,11 The strong spin–orbit coupling (SOC) of lanthanides coupled with a weak crystal-field interaction enables the construction of strongly anisotropic magnetic ground states making them ideal candidates for design of SMMs.12–14 Important advancements have also been made with complexes of the 3d transition metals with low coordination numbers and non-aufbau ground configurations.15,16 However, SMMs with only 4d or 5d transition metals are much rarer.17–20 Despite the stronger SOC compared to the 3d transition metals, the much larger spatial extent of the 4d and 5d orbitals leads to strong crystal-field splitting and metal–ligand covalency. This then leads to stabilization of a low-spin state and quenching of orbital momentum. The tendency to form covalent bonds is even stronger for the p-block metals, and to this date no SMMs with only main-group elements have ever been characterized. Heavier main-group elements have been successfully used in the construction of SMMs, but the main magnetic moment still always originates from a d- or f-block element.3,21–23 The strong SOC of heavier main-group elements such as the 6p metals, however, should make it possible to construct SMMs where the main paramagnetic ion is a p-block element as long as some orbital near-degeneracy could be retained. This approach has so far not been considered in the design of SMMs.
Recently Neese, Cornella and co-workers characterized a mono-coordinate bismuthinidene with a σ26p2π electron configuration of the Bi(I) ion, where σ is the C–Bi bonding orbital, and 6pπ are the two non-bonded 6p orbitals that retain their p-orbital-like character in the molecule.24 The splitting between the 6pπ orbitals due to low-symmetry components of the crystal-field is weak enough that both orbitals are occupied by a single electron in a spin-triplet configuration. Strong SOC lifts the three-fold degeneracy of the spin triplet, leading to a massive zero-field splitting (ZFS) that stabilizes a non-magnetic ground state. The strong SOC and the triplet ground spin state indicate that bismuth could be used to construct SMMs from purely main-group elements if a degenerate ground state with large enough magnetic moment could be stabilized. The SOC constant of the Bi(I) ion was estimated at ∼8000 to 9000 cm−1.24 This can be compared to the value 1932 cm−1 calculated for the 4f orbitals of a free Dy3 + ion or to the value 2405 cm−1 calculated for the 5f orbitals of a Pu3 + ion, the heaviest metal ion used in SMMs.25
The main focus of the present work is a situation where an additional electron is introduced into the 6pπ orbitals of a Bi(I) ion in a bismuthinidene giving a Bi(0) atom with a σ26p3π configuration. The addition of one electron will lead to an odd-electron system that will have a doubly degenerate ground-state due to Kramers’ theorem. The results will be demonstrated on the simple bismuthinidene, phenylbismuth (BiPh), and the reduced anion [BiPh]−. Neither BiPh nor [BiPh]− as themselves represent viable synthetic targets as the unprotected mono-coordinated Bi would immediately dimerize or react further. They should instead be considered as the functional cores of larger molecules where the mono-coordinated Bi is sterically protected by large substituents of the phenyl group. Since the presence of the steric bulk should not affect the qualitative magnetic properties of the BiPh and [BiPh]− cores, the possible substituents are not considered in the calculations. In order to show that [BiPh]− can be utilized as a blueprint for a main-group SMM, it needs to be shown that (i) reduction of BiPh leads to the electron configuration σ26p3π at the Bi atom instead of electron delocalization to the phenyl group or the formation of a multiple C–Bi bond; (ii) the configuration σ26p3π leads to the stabilization of a pair of states with large axial magnetic moments; and (iii) the reduction potential of the [BiPh]−/BiPh couple is reasonable so that the reduction reaction can be carried out in practice.
The geometries of BiPh and [BiPh]− were optimized using density functional theory (DFT) with the hybrid PBE0 exchange–correlation functional26–29 as implemented in the Gaussian 16 code revision C.02.30 The electronic structure was calculated using multireference ab initio methods utilizing the Orca code version 5.0.2.31,32 First a state-averaged complete active space self-consistent field (SA-CASSCF) type calculations33 was carried out. The active space consisted of the two 6pπ orbitals, the σ-bonding and anti-bonding combinations and all six phenyl π orbitals. In case of BiPh this totals 10 electrons and 10 orbitals, and in the case of [BiPh]− 11 electrons and 10 orbitals. The phenyl π orbitals were included in the active space so that any charge-transfer configurations or C–Bi π bonding was properly taken into account. Electron correlation effects outside the active space were estimated using the N-electron valence state perturbation theory at second order (NEVPT2),34–36 and SOC was introduced using the quasi-degenerate perturbation theory (QDPT) approach.37 See the ESI† for further details.
Both BiPh and [BiPh]− have a planar C2v-symmetric geometry. The calculated orbitals are shown in Fig. 1 along with the leading electron configuration in the ground state. The orbital structures of both systems are very similar. The two 6pπ orbitals retain their atomic-like p-orbital character and are nearly degenerate. The electron configurations can be described as σ26p2π and σ26p3π for BiPh and [BiPh]−, respectively. No multiple C–Bi bonding is observable in the orbitals, and the lowest-energy spin states with significant metal-to-ligand or ligand-to-metal charge transfer character lie more than 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 above the respective ground spin states. Thus, the Bi oxidation states can be rather unambiguously assigned as Bi(I) in BiPh and Bi(0) in [BiPh]−.
000 cm−1 above the respective ground spin states. Thus, the Bi oxidation states can be rather unambiguously assigned as Bi(I) in BiPh and Bi(0) in [BiPh]−.
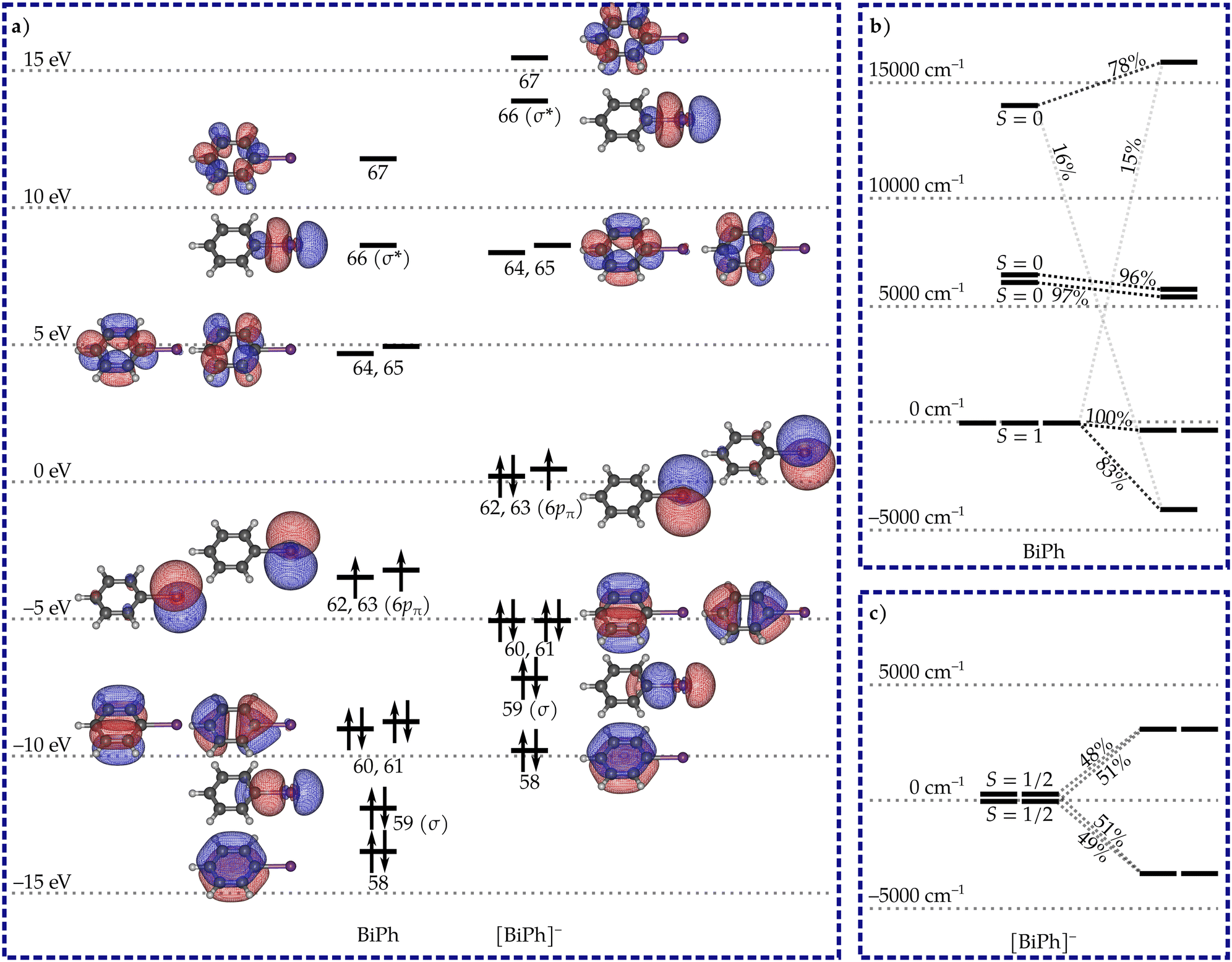 | ||
| Fig. 1 (a) The most important valence orbitals of BiPh and [BiPh]− along with the most important electron configuration in the respective ground states. The orbitals are state-averaged CASSCF natural orbitals and the orbital energies are expectation values of the generalized Fock operator. (b) The splitting of pure spin states under the effect of spin–orbit coupling in BiPh showing the non-degenrate ground state with zero magnetic moment. The percentages indicate the relative contribution from a given pure spin state to the respective spin–orbit coupled state. (c) The splitting of pure spin states in [BiPh]− into Kramers doublets showing the stabilization of a ground state with non-zero anisotropic magnetic moment. | ||
The lowest-energy spin-state of BiPh is a spin-triplet that is strongly split under SOC to give a non-degenerate ground state, and an excited quasi-doublet with two states 3391 cm−1 above the ground state (Fig. 1b). The non-degenerate ground state consists 83% of the MS = 0 component of the ground spin-triplet and is strongly mixed with an excited spin-singlet state. The results are similar to those observed by Neese, Cornella and co-workers in a mono-coordinate bismuthinidene, although the splitting they observed was over twice as large.24 The three lowest states can be associated with a pseudopin triplet to give ZFS parameters D = 3391 cm−1 and E = −0.06 cm−1. The large positive axial ZFS parameter D means that the system will not behave as an SMM.
The σ26p3π configuration of the Bi(0) atom in [BiPh]− leads to a doublet spin-ground state which is nearly degenerate with the first excited spin-doublet. The splitting of the two doublets before the inclusion of SOC is 166 cm−1. The near-degeneracy leads to unquenched orbital angular momentum, and strong splitting under SOC. The two spin-doublets mix and split into two Kramers doublets (KDs) separated by 6418 cm−1 (Fig. 1c). Each of the states in the two KDs consist of more than 97% of the states in the two nearly degenerate spin-doublets with no significant mixing with other excited spin states. The principal components of the g tensors calculated for the two KDs are listed in Table 1. The tensor of the ground KD is strongly axial with small but non-vanishing transverse components. The principal magnetic axis lies along the molecular C2 axis (Fig. 2). The excited KD is much less axial and its principal magnetic axis lies at a 90° angle relative to the principal axis of the ground KD.
| E/cm−1 | g x | g y | g z |
|---|---|---|---|
| 0 | 0.05637 | 0.05681 | 3.94501 |
| 6418 | 0.03999 | 0.14832 | 0.27211 |
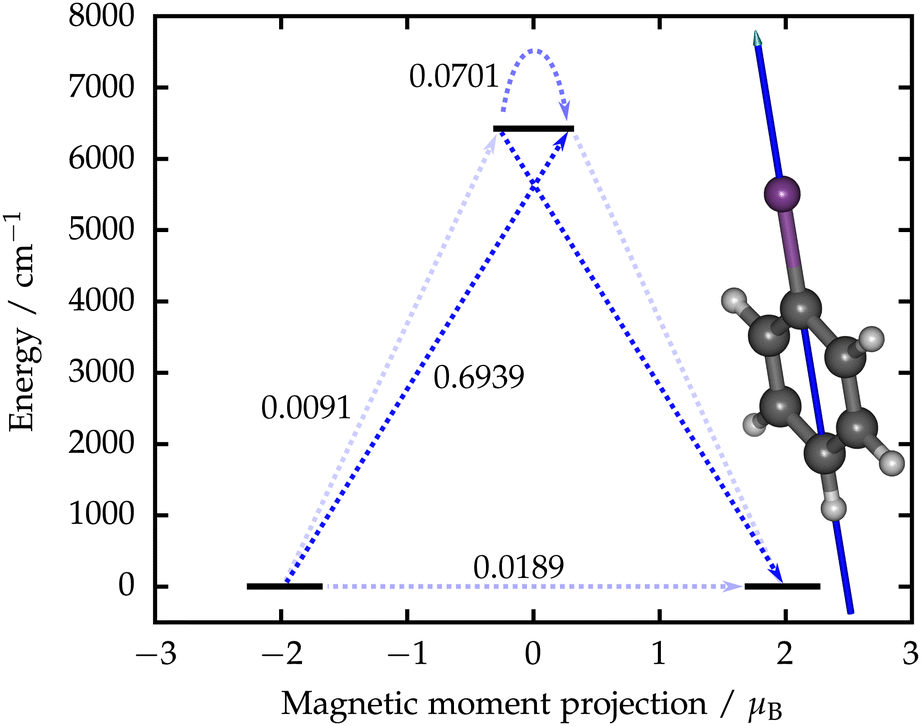 | ||
| Fig. 2 Qualitative barrier for the relaxation of magnetization calculated for [BiPh]− along with the principal magnetic axis of the ground Kramers doublet. The arrows and the corresponding numbers indicate magnitudes of transition magnetic moment matrix elements between the states in units of Bohr magneton. The excited state consists of two overlapping states. The strongest arrows indicate the most probable route for the relaxation of magnetization. | ||
The qualitative barrier for the relaxation of magnetization in [BiPh]− was constructed using the well-established method38 (Fig. 2). The calculated transition magnetic moments show that the most probable route for the relaxation of magnetization is via the excited KD giving rise to an effective energy barrier of 6418 cm−1. This is almost four times as high as the highest barrier obtained from fits to experimental data.11 The non-vanishing transverse components of the g tensor in the ground KD mean that quantum-tunneling of magnetization (QTM) is not fully blocked12 and will make a contribution to the relaxation dynamics at low temperatures. In case the blocking temperature was determined solely by the barrier height, it is likely that [BiPh]− would have a higher blocking temperature than any SMM characterized to date. However, this still requires study by experimental measurements, as various spin-phonon mechanisms and QTM can lead to “underbarrier” relaxation.39–41
The reduction potential for the [BiPh]−/BiPh couple in acetonitrile was calculated using an implicit solvation model as −1.5 V versus the SHE (see the ESI† for further details). The calculated value suggests that the [BiPh]− moiety is within the reach of redox chemistry. To further probe the relative stability of [BiPh]−, the proton affinities of BiPh and [BiPh]− in acetonitrile were calculated as 1058 kJ mol−1 and 1239 kJ mol−1, respectively (see the ESI† for further details). While [BiPh]− is a stronger base than BiPh, the proton affinities of the two specie are close enough that it is reasonable to assume that if a molecule containing the BiPh moiety can be sterically stabilized and isolated, the same also applies to [BiPh]−.
In summary, it was shown that SMMs constructed from purely main-group elements can be realized in practice in a molecule that contains the [BiPh]− anion with a Bi(0) atom as its core. The strong SOC of the 6p element will lead to a very large effective barrier for the reversal of magnetization that surpasses all barriers in SMMs characterized so far. The results demonstrate that main-group chemistry can be used to construct new generations of SMMs, and that, in principle, these can supersede even dysprosium-based SMMs.
Financial support was provided by the Academy of Finland (project 332294) and the University of Oulu (Kvantum Institute). Computational resources were provided by CSC-IT Center for Science in Finland and the Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533).
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Benelli and D. Gatteschi, Introduction to Molecular Magnetism. From Transition Metals to Lanthanides, Wiley-VCH, Weinheim, Germany, 2015 Search PubMed.
- M. Feng and M.-L. Tong, Chem. – Eur. J., 2018, 24, 7574–7594 CrossRef CAS PubMed.
- F.-S. Guo, A. K. Bar and R. A. Layfield, Chem. Rev., 2019, 119, 8479–8505 CrossRef CAS PubMed.
- Z. Zhu, M. Guo, X.-L. Li and J. Tang, Coord. Chem. Rev., 2019, 378, 350–364 CrossRef CAS.
- Z. Zhu and J. Tang, Chem. Soc. Rev., 2022, 51, 9469–9481 RSC.
- Z. Zhu and J. Tang, Natl. Sci. Rev., 2022, 9(12), nwac194 CrossRef PubMed.
- C. Godfrin, A. Ferhat, R. Ballou, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, Phys. Rev. Lett., 2017, 119, 187702 CrossRef CAS PubMed.
- A. Gaita-Ariño, F. Luis, S. Hill and E. Coronado, Nat. Chem., 2019, 11, 301–309 CrossRef PubMed.
- M. Atzori and R. Sessoli, J. Am. Chem. Soc., 2019, 141, 11339–11352 CrossRef CAS PubMed.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed.
- C. A. Gould, K. R. McClain, D. Reta, J. G. C. Kragskow, D. A. Marchiori, E. Lachman, E.-S. Choi, J. G. Analytis, R. D. Britt, N. F. Chilton, B. G. Harvey and J. R. Long, Science, 2022, 375, 198–202 CrossRef CAS PubMed.
- L. Ungur and L. F. Chibotaru, Phys. Chem. Chem. Phys., 2011, 13, 20086–20090 RSC.
- L. Ungur and L. F. Chibotaru, Inorg. Chem., 2016, 55, 10043–10056 CrossRef CAS PubMed.
- K. L. M. Harriman, D. Errulat and M. Murugesu, Trends Chem., 2019, 1, 425–439 CrossRef CAS.
- P. C. Bunting, M. Atanasov, E. Damgaard-Møller, M. Perfetti, I. Crassee, M. Orlita, J. Overgaard, J. van Slageren, F. Neese and J. R. Long, Science, 2018, 362, eaat7319 CrossRef CAS PubMed.
- D. Errulat, K. L. M. Harriman, D. A. Gálico, J. S. Ovens, A. Mansikkamäki and M. Murugesu, Inorg. Chem. Front., 2021, 8, 5076–5085 RSC.
- J. Martínez-Lillo, T. F. Mastropietro, E. Lhotel, C. Paulsen, J. Cano, G. De Munno, J. Faus, F. Lloret, M. Julve, S. Nellutla and J. Krzystek, J. Am. Chem. Soc., 2013, 135, 13737–13748 CrossRef PubMed.
- A. Sanchis-Perucho, M. Orts-Arroyo, J. Camús-Hernández, C. Rojas-Dotti, E. Escrivà, F. Lloret and J. Martínez-Lillo, CrystEngComm, 2021, 23, 8579–8587 RSC.
- K. S. Pedersen, M. Sigrist, M. A. Sørensen, A.-L. Barra, T. Weyhermüller, S. Piligkos, C. A. Thuesen, M. G. Vinum, H. Mutka, H. Weihe, R. Clérac and J. Bendix, Angew. Chem., Int. Ed., 2014, 53, 1351–1354 CrossRef CAS PubMed.
- J.-L. Liu, K. S. Pedersen, S. M. Greer, I. Oyarzabal, A. Mondal, S. Hill, F. Wilhelm, A. Rogalev, A. Tressaud, E. Durand, J. R. Long and R. Clérac, Angew. Chem., Int. Ed., 2020, 59, 10306–10310 CrossRef CAS PubMed.
- F.-S. Guo, M. He, G.-Z. Huang, S. R. Giblin, D. Billington, F. W. Heinemann, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Inorg. Chem., 2022, 61, 6017–6025 CrossRef CAS PubMed.
- P. Evans, D. Reta, G. F. S. Whitehead, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2019, 141, 19935–19940 CrossRef CAS PubMed.
- P. Zhang, F. Benner, N. F. Chilton and S. Demir, Chem, 2021, 8, 717–730 Search PubMed.
- Y. Pang, N. Nöthling, M. Leutzsch, L. Kang, E. Bill, M. Gastel, E. Reijerse, R. Goddard, L. Wagner, D. SantaLucia, S. DeBeer, F. Neese and J. Cornella, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-d3jl7.
- J. Jung, M. Atanasov and F. Neese, Inorg. Chem., 2017, 56, 8802–8816 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- M. J. Frisch, et al., Gaussian16 Revision C.02, 2016, Gaussian Inc., Wallingford, CT, USA Search PubMed.
- F. Neese, WIREs Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- B. O. Roos, in Advances in Chemical Physics: Ab Initio Methods in Quantum Chemistry II, ed. K. P. Lawley, Wiley, New York, NY, USA, 1987, vol. 69, pp. 399–455 Search PubMed.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, Chem. Phys. Lett., 2001, 350, 297–305 CrossRef CAS.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, J. Chem. Phys., 2002, 117, 9138–9153 CrossRef CAS.
- F. Neese, T. Petrenko, D. Ganyushin and G. Olbrich, Coord. Chem. Rev., 2007, 251, 288–327 CrossRef CAS.
- L. Ungur, M. Thewissen, J.-P. Costes, W. Wernsdorfer and L. F. Chibotaru, Inorg. Chem., 2013, 52, 6328–6337 CrossRef CAS PubMed.
- A. Lunghi, F. Totti, R. Sessoli and S. Sanvito, Nat. Commun., 2017, 8, 14620 CrossRef PubMed.
- L. Gu and R. Wu, Phys. Rev. Lett., 2020, 125, 117203 CrossRef CAS PubMed.
- L. Gu and R. Wu, Phys. Rev. B, 2021, 103, 014401 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, additional computational data, optimized Cartesian coordinates and sample input files. See DOI: https://doi.org/10.1039/d3cc00042g |
| This journal is © The Royal Society of Chemistry 2023 |