
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective dearomatization reactions of heteroarenes by anion-binding organocatalysis
Martin
Aleksiev
and
Olga
García Mancheño
 *
*
Organic Chemistry Institute, University of Münster, Corrensstraße 36/40, 48149 Münster, Germany. E-mail: olga.garcia@uni-muenster.de
First published on 15th February 2023
Abstract
Catalytic asymmetric dearomatization of heteroaromatic compounds has received considerable attention in the last few years, since it allows for a fast expansion of the chemical space by converting relatively simple, flat molecules into complex, three dimensional structures with added value. Among different approaches, remarkable progress has been recently achieved by the development of organocatalytic dearomatization methods. In particular, the anion-binding catalysis technology has emerged as a potent alternative to metal catalysis, which together with the design of novel, tunable anion-receptor motifs, has provided new entries for the enantioselective dearomatization of heteroarenes through a chiral contact ion pair formation by activation of the electrophilic reaction partner. In this feature, we provide an overview of the different methodologies and advances in anion-binding catalyzed dearomatization reactions of different heteroarenes.
 Martin Aleksiev | Martin Aleksiev was born in Pleven (Bulgaria) in 1996. He received his BSc in chemistry from University of Alcala in 2020, and his MSc degree in Organic Chemistry from University Autonomous of Madrid in 2021. In 2022, he joined the research group of Professor Olga García Mancheño at the Organic Chemistry Institute of the University of Munster. His current studies focus on Anion Binding Catalysis. |
 Olga García Mancheño | Olga García Mancheño studied chemistry at the University Autonomous of Madrid, where she obtained her PhD in 2005 in the group of Prof. J. C. Carretero. After a postdoctoral stay in the group of Prof. C. Bolm at RWTH Aachen, she carried out her habilitation at the University of Münster. She was appointed as professor for organic chemistry at the University of Regensburg in 2013, and in 2017 at the University of Münster. Her main research interests focus on the design of novel catalytic systems for their application in photocatalysis and asymmetric anion-binding catalysis. |
1. Introduction
Aromatic compounds are broadly distributed in nature and excellent substrates of choice for synthesis,1 since there are plentiful commercially and readily available derivatives of generally low cost. The dearomatization of these compounds is very useful for expanding the chemical space by converting relatively simple, flat molecules into more decorated, complex, three dimensional structures with added value for several research fields such as organic synthesis, agrochemical and pharmaceutical chemistry.2–6 Despite the synthetic advantages of this straightforward strategy, the high stability of the aromatic compounds makes the control of the regioselectivity and stereoselectivity of this process very challenging.7 However, in the last decade, a progress in asymmetric catalytic dearomati-zation reactions has shown the great potential of this area.8–10The enantioselective dearomatization of heteroarenes is of particular interest due to the fact that heterocycles are key elements of natural products and pharmaceuticals.11–13 Catalytic asymmetric dearomatization reactions of heteroarenes could be carried out by different methodologies, in which the arene can be enrolled as nucleophile or electrophile depending of its intrinsic nature (Fig. 1a).8,14 In this regard, transition metal asymmetric catalysis has been extensively used in this type of processes due to the extended possibilities provided by organometallic chemistry and the availability of versatile chiral ligands.8,15–17 Nevertheless, most of these methods are still limited regarding to the dearomatization of electron poor arenes, such as pyridines or (iso)quinolines, as they are generally more efficient for electron rich systems. In contrast, the development of several organocatalytic methods has allowed an efficient activation of electrophilic heteroarenes and the introduction of chirality by dearomatization through ionic intermediates.8,18,19 Due to the fact that most organic reactions proceed via charged species or the presence of some degree of charge polarization, finding efficient ways to control the stereochemistry with this kind of species lies at the heart of organic synthesis. Among the different catalytic approaches involving charged intermediates or reagents, anion-binding has recently emerged as a powerful toolkit in asymmetric catalysis.19–26 Anion-binding catalysis19 is based on the activation of an ionic electrophile by the binding of a catalyst to its counter anion through non-covalent interactions such as hydrogen-bonding, forming a chiral contact ion-pair complex, which can induce chirality into the final product after a nucleophilic attack (Fig. 1b). It is worthy to mentioned that, in the last decades, a large number of small molecules have been described to operate through an anion-binding activation mechanism.19 Most of these catalysts rely on hydrogen bonding,27,28 but present additional structural elements to help for the substrate activation and stabilization of the transition state by different contacts such as additional hydrogen bonds or π–π interactions.19–31 Some of the most prominent catalysts that operate through this mechanism and have been employed for the development of enantioselective dearomatization of heteroarenes are chiral thioureas, silenodienols and oligo-triazoles, among others.
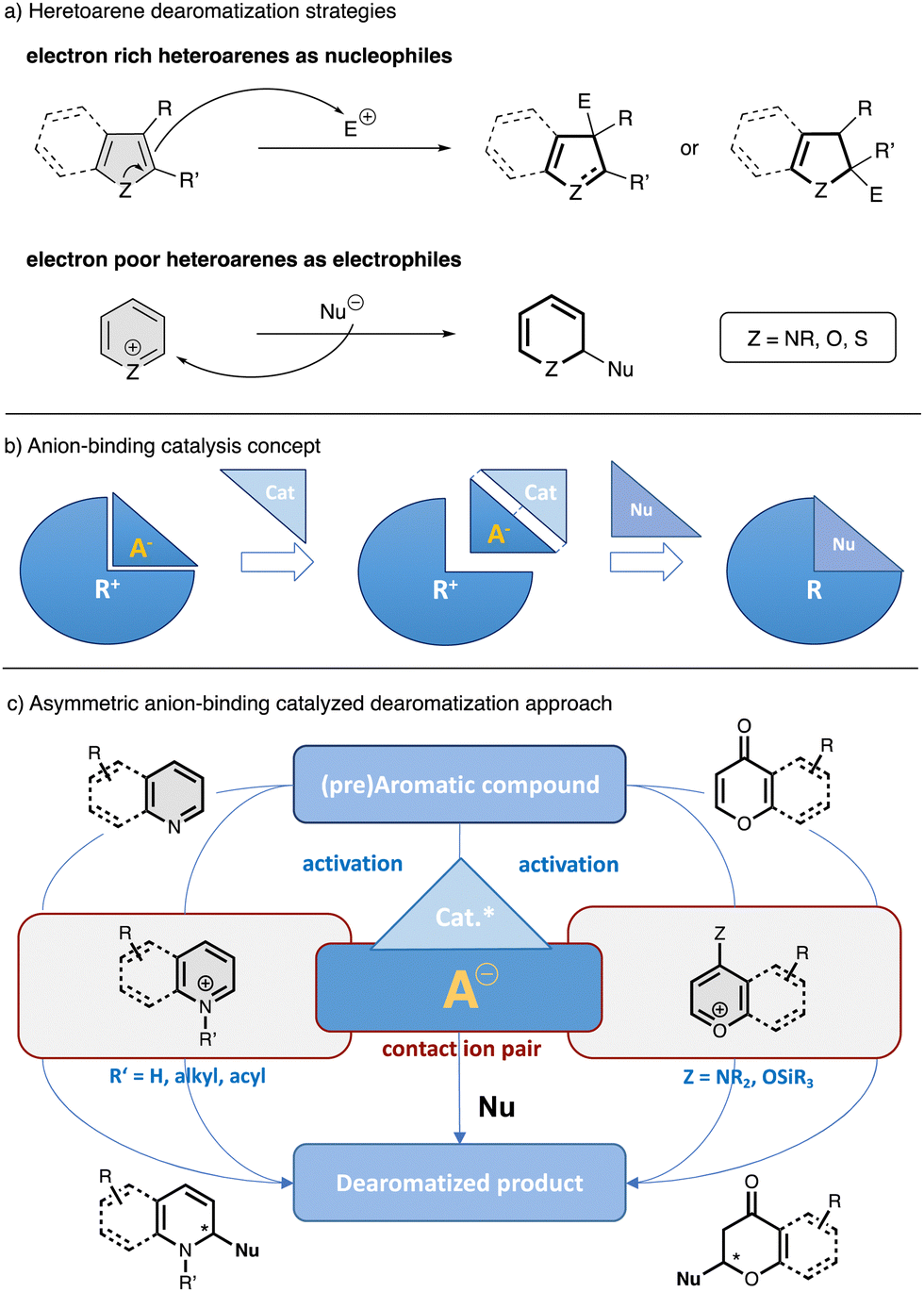 | ||
| Fig. 1 General scheme for the dearomatization of heteroarenes (a) anion-binding concept (b) and asymmetric anion-binding dearomatization approach via activation-nucleophilic addition (c). | ||
In the field of anion-binding catalyzed dearomatization reactions, typically the in situ generation of the reactive ionic electrophilic heteroarene is required (Fig. 1c).19 Thus, a previous activation of the heteroarene or pre-aromatic precursor by nitrogen atom quaternization though protonation, alkylation or acylation (N-heteroarenes), or by enamine formation or enolization (O-heteroarenes), allows the following enantioselective nucleophilic addition in the presence of a chiral catalyst. Additionally, there is also possible the dearomatization of electron rich hetereoarenes, such as indoles, by facilitating their stereocontrolled attack to anion-binding activated electrophilic species.
Several reviews on the broad use of anion-binding in catalysis,19–26 as well as asymmetric dearomatization reactions of arenes8–10 have been published. However, this feature just focusses on the different methodologies and advances in enantioselective anion-binding catalyzed strategies. We aim at giving an overview of the dearomatization of different types of heteroarenes embracing this activation mode, in which our group has contributed significantly in the past few years. In this feature, the examples reported are arranged according to the type of aromatic substrate, and it has been further divided by the nature of the nucleophile and/or catalyst employed in each reaction.
2. Dearomatization of isoquinolines
2.1. Silyl ketene acetals as nucleophiles
The first example of enantioselective anion-binding catalyzed dearomatization of an N-heteroarene was published in 2005 by Jacobsen and co-workers (Scheme 1).32 It consists in an N-acyl Mannich reaction of isoquinolines 2 employing 10 mol% of the chiral thiourea catalyst 1 and tert-butyldimethylsilyl (TBS) protected ketene acetal 3a as a nucleophile. In this case, strong dependence in the solvent and the acylating group was found. The best results were obtained with non-polar solvents as diethyl ether and 2,2,2-trichloroethyl chloroformate (TrocCl) as acylating agent. As in Reissert-type reactions,33,34 the activation of the substrate by forming the N-acyl iminium cation was first performed, followed by the formation of the chiral contact ion pair in presence of the chiral organocatalyst. Lastly, the nucleophilic addition takes place towards the corresponding 1,2-dihydroquinolines 4, which were obtained with good yields and high enantioselectivities up to 92% ee.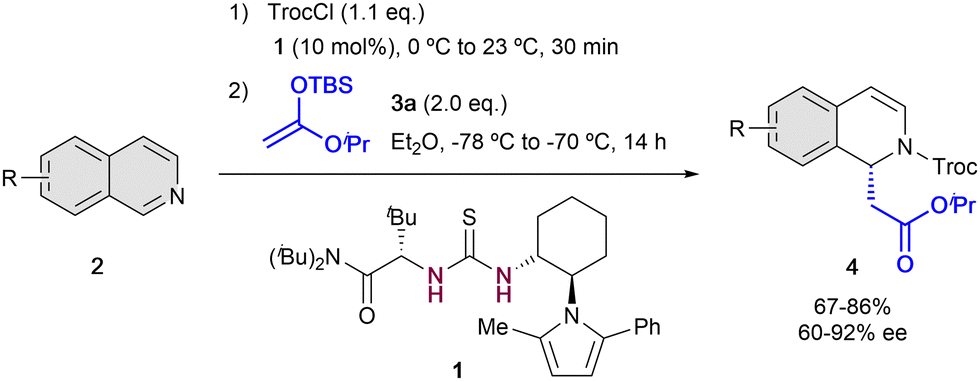 | ||
| Scheme 1 First anion binding dearomatization reaction reported by Jacobsen et al. | ||
After this pioneering work, in the following years other several catalytic systems have been introduced as alternative anion-binding organocatalysts. In 2013, the Mattson group developed the first example of a C2-symmetric 1,1′-bi-2-naphtol (BINOL) based silanediol HB-donor 5 as anion-binding catalyst (Scheme 2).35 They were able to achieve moderated yields and enantioselectivities up to 50% ee in the presence of 20 mol% of (R)-5a. The best results were obtained employing TrocCl, and triisopropylsilyl (TIPS) dimethyl ketene acetal 3b in toluene at −78 °C. Motivated by the initial success, in 2015 they further improved the levels of enantiocontrol to up to 78% ee by employing the re-designed tetraphenyl-substituted silanediol catalyst (R)-5b at −55 °C.36
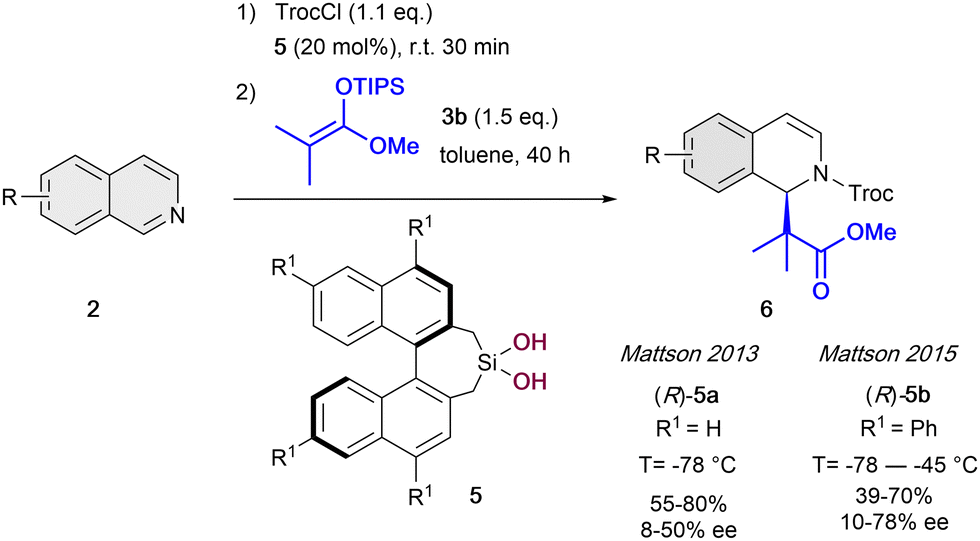 | ||
| Scheme 2 Silanediol-catalyzed dearomatization of isoquinolines by Mattson et al. | ||
Regarding to the hardly explored silanediol catalysts, it can be remarked the later development of biphenyl-2,2-bisfenchol-based (BIFOL) silanediol by Goldfuss et al. in 2019.37 This alkoxysilanediol has an unexpectedly stable BIFOL backbone against hydrolysis, and is able to achieve the dearomatization of isoquinolines by N-acyl Mannich reaction in high yields, though only in low enantioselectivities (up to 12% ee).
On the other hand, in 2014, the García Mancheño group developed a new family of helical triazole-based HB-donors 7 as anion-binding catalysts.38 1,2,3-Triazoles are heterocycles with interesting properties such as large dipole moment (μ = 4.38 D)39 and highly polarizable C–H bonds,40 what makes them promising HB-donor moieties for anion-binding catalysis.41 The chirality in the triazole catalysts 7 is introduced by a chiral trans-1,2-diamine unit that acts as a central backbone to pre-orientate the formation of the chiral helical structure upon binding to the substrate counter-anion. Nevertheless, this central chirality is not enough to induce high level chirality transfer, given that four triazole rings are required to have a cooperative effect towards the effective formation of a chiral helical triazole-anion complex.38 In 2016, our group employed the triazole organocatalysts 7 for the N-acyl Mannich reaction of isoquinolines 2 with silyl ketene acetals 3 (Scheme 3).42 The reaction was carried out in presence of 10 mol% of (R,R)-7a, TrocCl as acylating agent, and methyl tert-butyl ether (MTBE) as solvent at −78 °C, achieving high yields and enantioselectivities up to 72% ee.
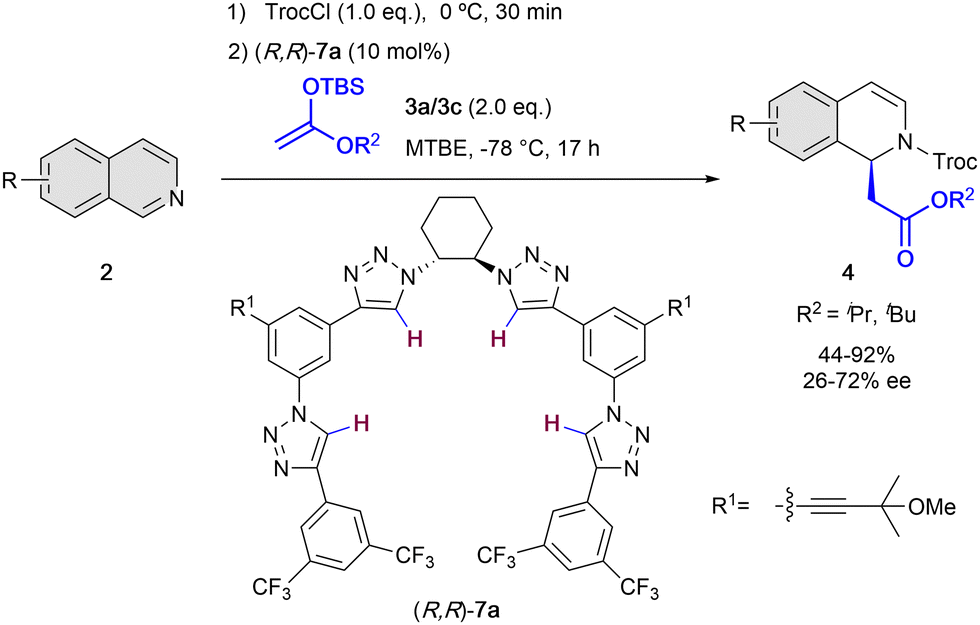 | ||
| Scheme 3 Chiral oligotriazole-catalyzed dearomatization of isoquinolines by García Mancheño et al. | ||
2.2. Extension to other nucleophiles with thiourea catalysts
In addition to the pioneering work of Jacobsen (Scheme 1),32 in the last years thiourea organocatalysts were also used together with different nucleophiles for the reaction with the same type of in situ formed N-acyl isoquinolinium cation by acylation of isoquinolines with TrocCl (Scheme 4).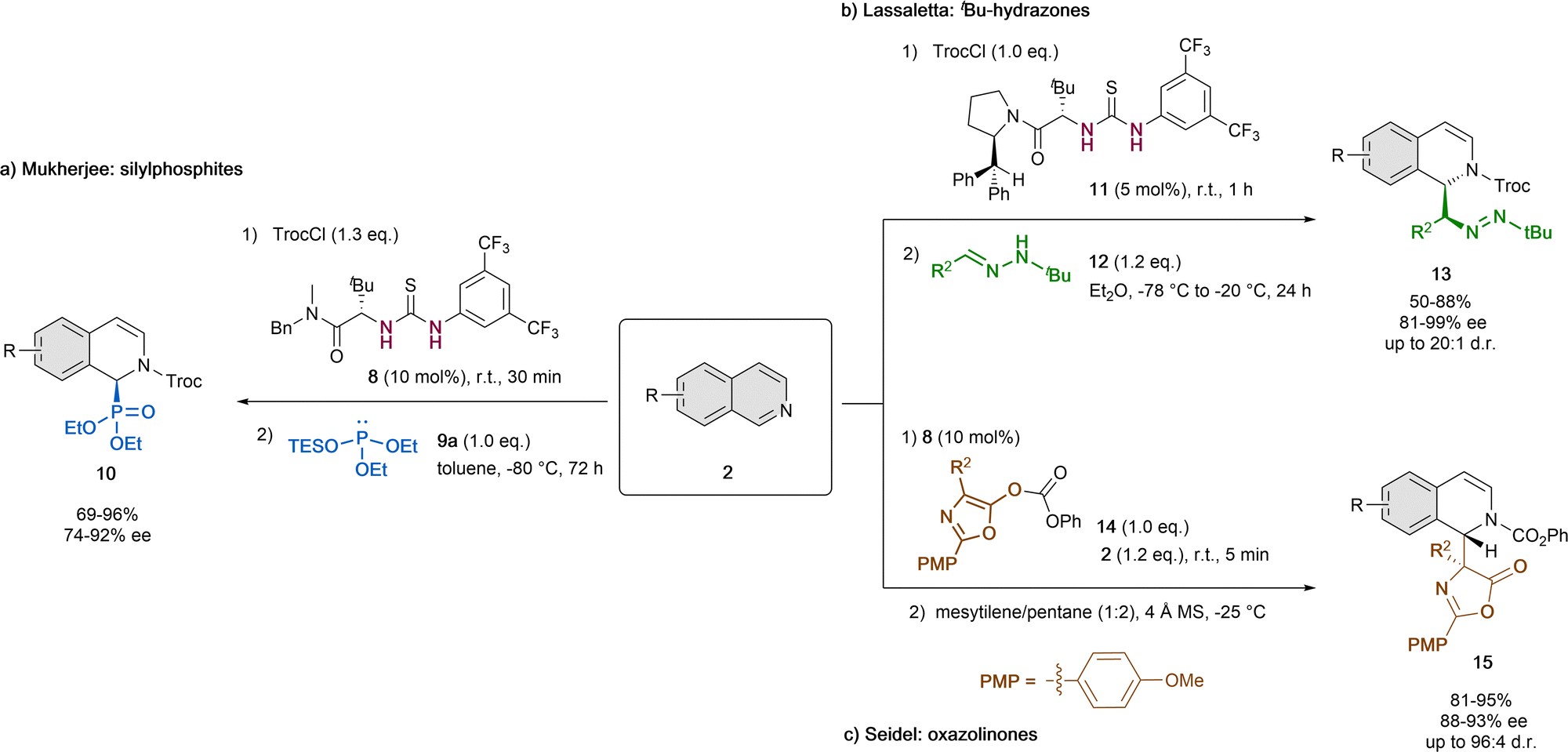 | ||
| Scheme 4 Thiourea-catalyzed asymmetric dearomatization of isoquinolines by (a) Choudhury and Mukherjee, (b) Lassaletta et al. and (c) Seidel et al. | ||
In 2016, Choudhury and Mukherjee reported the asymmetric dearomatization of isoquinolines 2 employing 10 mol% of the tert-leucine-based thiourea derivative 8 and diethyl triethylsilyl (TES) phosphite 9a as a nucleophile in toluene at −80 °C (Scheme 4a).43 As a result, the corresponding cyclic α-aminophosphonates 10 were generated with high yields and enantioselectivities up to 92% ee. This method provides an easy way to obtain chiral α-aminophosphonates, that are widely used in medicinal and pharmaceutical science.44
Alternatively, non-silylated nucleophiles were also applied. In 2021, Lassaletta and co-workers described the ability of tert-leucine-derived thiourea 11 to catalyze the dearomatization reaction of isoquinolines 2 with N-tert-butyl hydrazones 12 as polarity-reversed nucleophiles in Et2O (Scheme 4b).45 The resulting dihydro-isoquinolines 13 present two contiguous stereogenic centers and were obtained in high yields, diastereoselectivities up to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r. and enantioselectivities up to 99% ee.
:1 d.r. and enantioselectivities up to 99% ee.
Moreover, azalactones (3-oxazolin-5-ones) could also be enrolled in this type of catalytic dearomatization reactions via a Steglich rearrangement, which was reported by Seidel et al. in 2011 (Scheme 4c).46 To this end, they used 10 mol% of the thiourea organocatalyst 8, 4 Å molecular sieves and a mixture of mesitylene and pentane (1:2) as solvent at −25 °C. Upon prior formation of the N-acylisoquinolinium ion by acylation from the azalactone 14, the attack of the enolate species evolves to the final disubstituted amino acid products 15 in high yields, enantioselectivities up to 93% ee and diastereoselectivities up to 96:4 d.r.
3. Dearomatization of quinolines
3.1. Strategies based on hydrogen-bonding catalysis
Compared to isoquinolines, quinolines proved to be more challenging substrates, most probably because of the lower reactivity and inefficient interaction to the mostly employed bidentate organocatalysts. In contrast, dual or multidentate systems are able to achieve better stereocontrol in the dearomatization process of quinolines through a better fixation of the N-acylquinolinium salt.The presumably first anion-binding catalytic dearomatization of quinolines 17 relied on a Petasis-type reaction reported by Takemoto et al. in 2007 (Scheme 5).47 The reaction was carried out by employing 10 mol% of the dual catalyst 16 in the presence of phenyl chloroformate as acylating agent, vinyl boronic acid (18) as a nucleophile, a mixture of H2O and NaHCO3 for the regeneration of the catalyst and CH2Cl2 as solvent. While the 1,2-amino alcohol group of the catalyst activates the boronic acid, the thiourea unit can bind the chloride counter-anion and form a contact ion pair with the N-phenoxycarbonyl quinolinium salt. Thus, the corresponding dihydroquinolines 19 were achieved in moderate yields and enantioselectivities up to 97% ee.
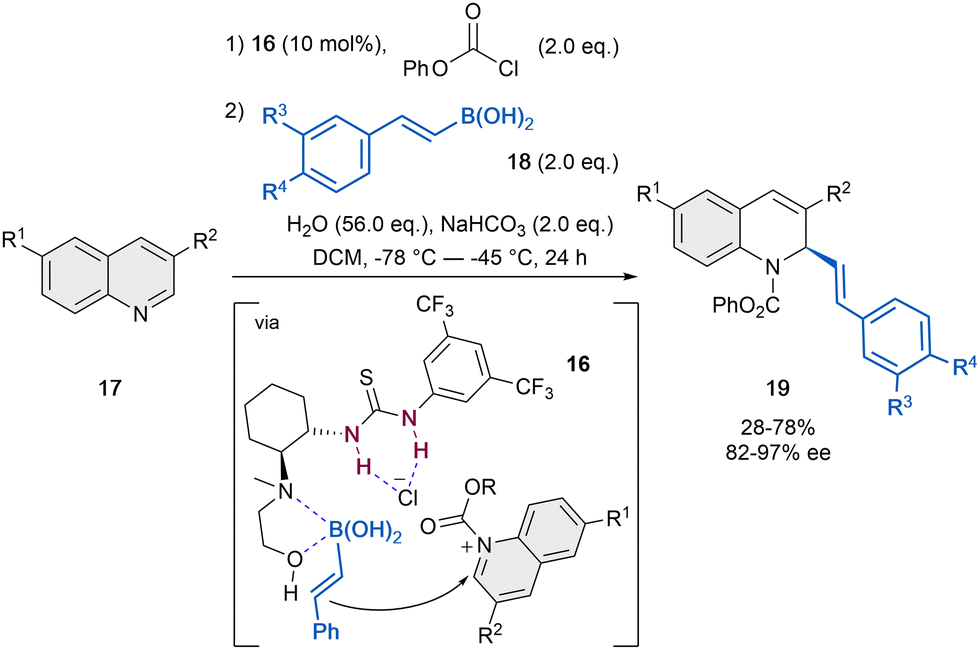 | ||
| Scheme 5 Asymmetric Petasis-type reaction of quinolones reported by Takemoto et al.47 | ||
In the following years, the García Mancheño group has made a significant contribution to the development of different catalytic systems for the dearomatization of quinolines, by in situ standard activation through acylation with TrocCl (Scheme 6).38,48–52 In 2014, we reported Reisert-type reaction of quinolines 17 employing 5 mol% of the regioisomer tetrakistriazole (R,R)-7b, in presence of iso-propyl ketene acetal 3a in MTBE as solvent (Scheme 6a).38 Electron rich and electron poor substituents were well tolerated and the desired products 20 were obtained with high yields and enantioselectivities up to 96% ee. Only substituents in C-8 were not tolerated, and substituents in C-3 hindered the correct evolution of the reaction, leading to significantly lower enantioselectivities.
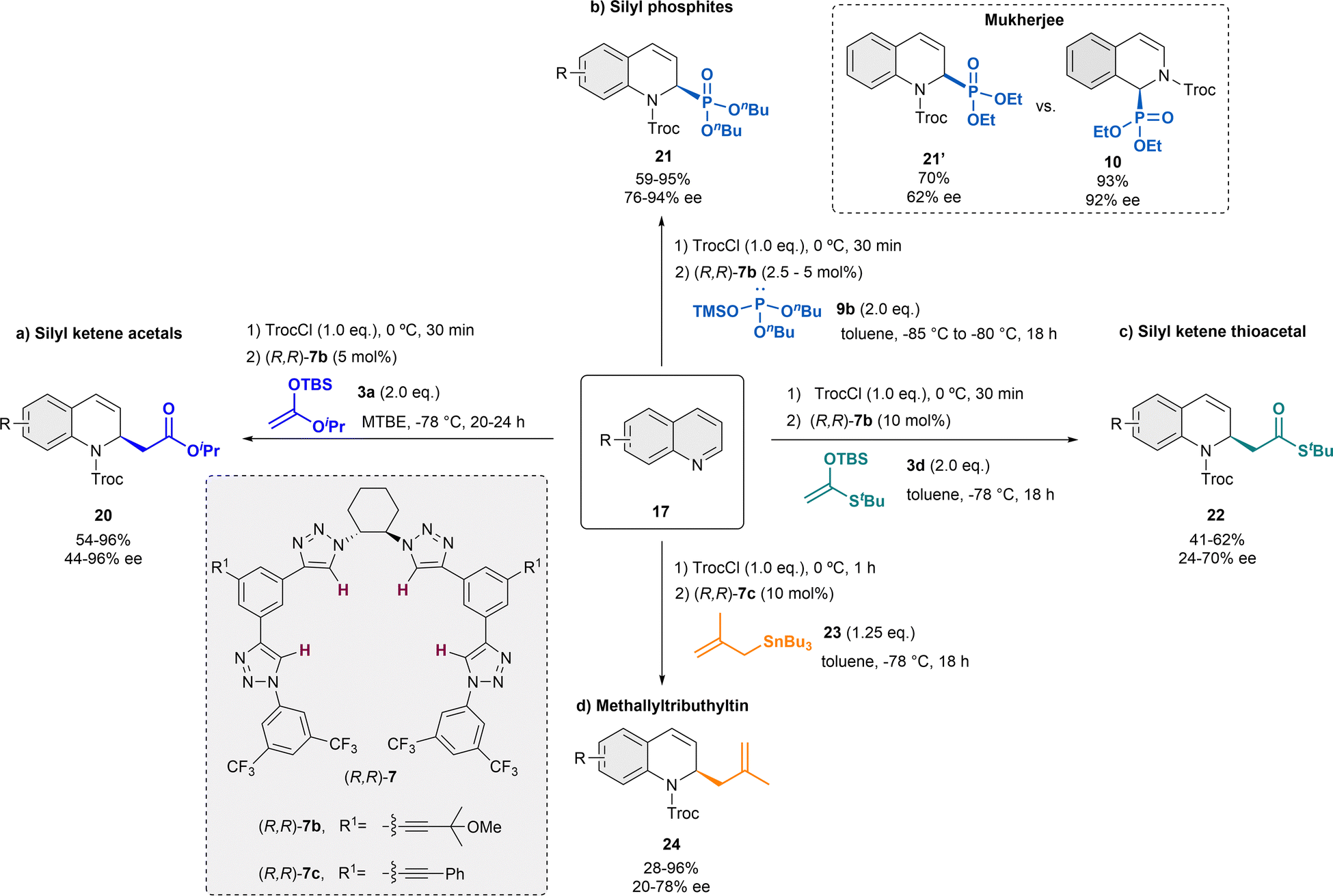 | ||
| Scheme 6 Asymmetric dearomatization of quinolines employing several nucleophiles in the presence of tetrakis-triazole catalysts 7. | ||
The application of this catalytic system could be efficiently extended to different nucleophiles. Inspired by the thiourea-catalyzed studies of Mukherjee with phosphites, which were not performing that well with quinolines (e.g. 62% ee, quinoline vs. 92% ee, isoquinoline),43 the related asymmetric triazole-catalyzed dearomatization towards the synthesis of the corresponding dihydroquinoline α-aminophosphonic acid derivatives 21 was developed (Scheme 6b).48 In order to achieve the desired products, silyl protected alkylphosphites such as 9b [(nBuO)2P-OTMS] were used in presence of 2.5 or 5 mol% of (R,R)-7b in toluene. Both electron donating and withdrawing groups were well tolerated, obtaining the products 21 high yields and enantioselectivities up to 94% ee.
After the successful studies with silyl ketene acetals and phosphites presenting a similar nucleophilicity according to the Mayr's nucleophilicity scale53,54 (N ∼ 10), our group further screened several nucleophiles for this approach.49 The selected nucleophiles presented N-values between 1.7 and 10. It was observed that nucleophiles with N-value below 6 do not react under the standard conditions and strong nucleophiles, such as Grignard reagents with expected N > 20, aggressively reacted with the substrate leading to decomposition products. However, it was noticed that the yield and enantioselectivity could not be perfectly correlated to the N-value. The best chiral inductions were obtained with the related silyl ketene thioacetal 3d (Scheme 6c).49 Hence, after the corresponding optimization process, the desired dihydroquinolines 22 were obtained with moderate yields and enantioselectivities up to 72% ee, in presence of 10 mol% of (R,R)-7b in toluene.
Furthermore, methallyltributhyltin 23 (N = 7.5) also provided promising results during the nucleophile screening.49 Therefore, an asymmetric α-allylation of quinolines was developed embracing a similar strategy (Scheme 6d).50,55 This approach opens a fast access to chiral N-heterocycles bearing a versatile allyl group that could be transformed into further valuable groups. For this reaction, 10 mol% of catalyst (R,R)-7c bearing additional aromatic substituents in the backbone for a better interaction with the allyltin reagent was used in dry toluene. Hence, the α-allylated dihydroquinolines 24 were obtained in high yields and enantioselectivities up to 78% ee.
Combined experimental and computational mechanistic studies were carried out to gain important information on the key interactions between the catalyst and heteroarene substrate (Fig. 2).51
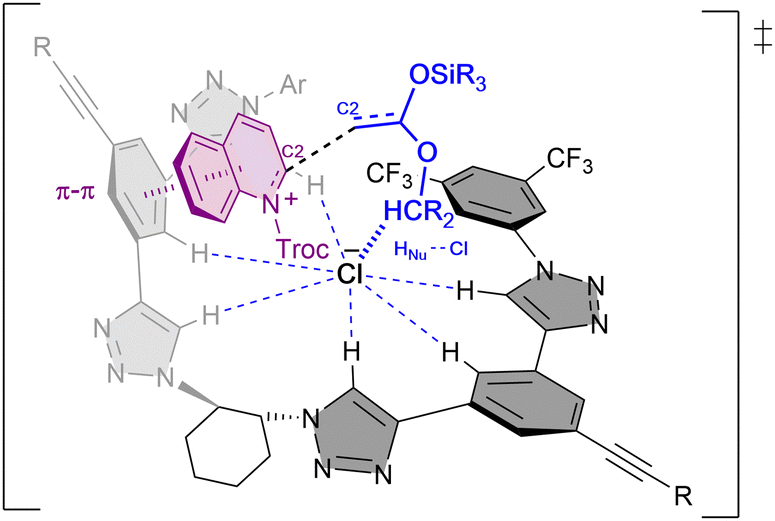 | ||
| Fig. 2 Plausible transition state and key interactions between (R,R)-7b and the quinolinium substrate in the chiral contact ion-pair complex based on experimental and computational studies. | ||
These studies showed that the main interaction towards the catalyst-anion complex formation is produced between the chloride and the hydrogen atoms of the triazole rings of the catalyst (R,R)-7b (e.g. Ka(Cl−) = 536 M−1 in acetone-d6, 2 mM; Bu4NCl as Cl− source). Moreover, the chloride is also interacting through hydrogen bonding with the substrate and the nucleophile, such as a silyl ketene acetal, guiding the addition process. Lastly, a π–π interaction between the substrate and one of the arms of the catalyst leads to a parallel orientation, allowing only the attack from the less hindered Re-face. All together it explains the obtention of the (R)-enantiomer as major product.
Based on these findings, we re-designed the triazole-based catalyst to (R,R)-7d, aiming at being able to enrol challenging formaldehyde N,N-dialkylhydrazones (DAHs)56 as polarity reverse nucleophiles (N ∼7–8) for the addition to the quinolinium salts (Scheme 7).52 Hence, in order to enhance the anion-binding affinity and allow new F–H bonds between the catalyst and the hydrogen atoms of the hydrazone, a CF3 group instead of the alkyne substituent was introduced in the side-arms of the tetrakistriazole catalyst. Moreover, the N,N-dialkyl substitution should guarantee for an additional hydrogen bond interaction with the counter-anion57 and a better fixation of both substrate and nucleophile in the catalyst cavity.
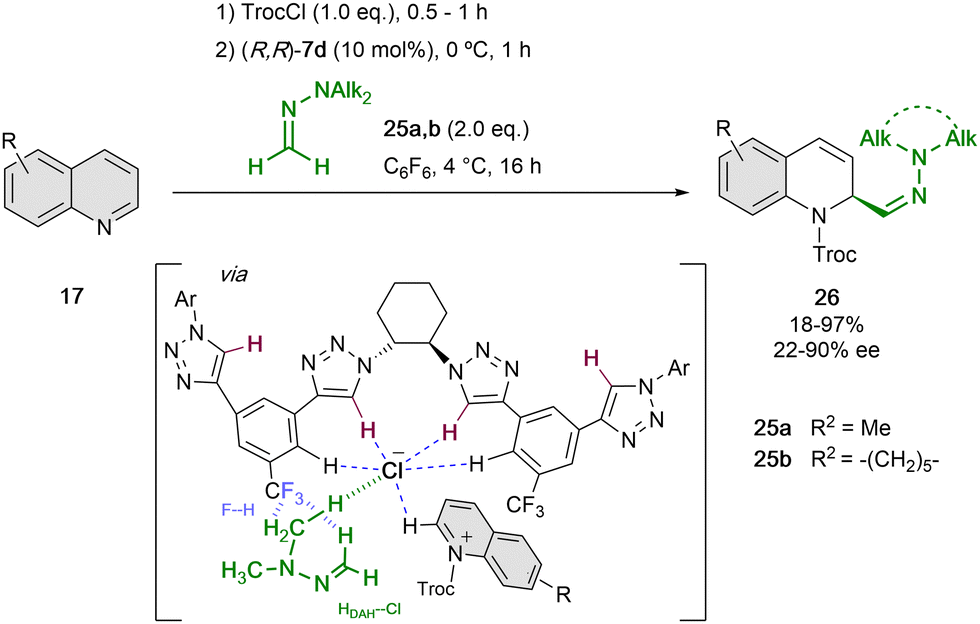 | ||
| Scheme 7 Enantioselective dearomatization of quinolines employing N,N-dialkylhydrazones reported by García Mancheño et al. | ||
Computational studies showed that only one of the arms of the catalyst is interacting with the counterion, and the approach of the nucleophile is directed by the previously mentioned F–H bonds. For this reaction, 10 mol% of (R,R)-7d was used in C6F6, with the purpose to disfavour the possible cation–π interactions between the substrate and solvent.58 Different N,N-dialkylhydrazones 25 could be employed. However, the N-substitution affected significantly to the enantiocontrol, which is in concordance with the hypothesis of the participation of the N-alkyl group in the chiral contact ion pair.57 Different substitution of the quinoline was also well tolerated and the desired dihydroquinolines 26 were obtained in high yields and enantioselectivities up to 90% ee.
3.2. Strategies based on halogen-bonding catalysis
In the last years, halogen bonding (XB)59,60 has become a promising tool in organocatalysis,61–63 resembling and, in some cases, showing a stronger activation that the one exhibited by the more classical hydrogen bonding analogous interaction. The presence of an EWG attached to a halogen atom produces an anisotropically distributed electron density along the EWG–X bond, thus generating an electropositive region called σ-hole.64 This region, provides Lewis acidity properties to the halogen atoms, which allowed them to form non-covalent interactions with Lewis basic substrates.59,60,65,66 The strength of this interactions increases with the polarizability of the halogen. Consequently, most of the employed XB-organocatalysts are based on the iodine atom.67Plausibly, the first reported reaction employing XB activation was published by Bolm and co-workers in 2008.68 In this work, a quinoline reduction was achieved in presence of a perfluoroalkene catalyst and a Hantzsch ester as a reducing agent. However, the first asymmetric version based primarily on halogen bonding activation was achieved by Huber et al. in 2020.69 They used a dicationic XB-donor catalyst based on two iodoimidazolium units with rigidly attached chiral sidearms for the asymmetric Mukaiyama aldol type reaction between an 1,2-dicarbonilic compound and a silyl-protected enolate. Unfortunately, only up to 33% ee could be attained, most probably due to the challenging nature of the XB-interaction that requires a high directionality (∼180°) and the larger distance between the substrate and the chiral backbone produced by the large size of the iodine atoms.
Inspired by our previous results with chiral-triazole based anion-binders, in 2021 we reported the chiral tetrakis-iodo-triazole (R,R)-27 as a novel XB-donor organocatalyst (Scheme 8).70 To test its enantioinduction capability, the Reissert-type reaction of quinolines 17 with silyl ketene acetals 3 was chosen as a benchmark reaction. The anion binding properties of (R,R)-27 were examined by NMR- and CD-titrations using the corresponding tetrabutylammonium salt, showing significant stronger anion affinities compared to the analogous HB-donor (R,R)-7b (Ka(Cl−) = 1125 vs. 99 M−1 in 1:1 acetone-d6:CDCl3, 2.5 mM; Bu4NCl as Cl− source). The best result was observed using 10 mol% of (R,R)-27 in CHCl3 at −60 °C. The desired (S)-product was the obtained in 30% ee; at the same level of the current state-of-the-art stereocontrol in XB-activation catalysis. Interestingly, the opposite enantiomer respect to the obtained with the parent proto-triazole catalyst (R,R)-7b was observed, which was in concordance to the examined pseudo-inverted CD-spectra and DFT calculations. These studies indicate that the bigger size of the iodine atoms in the upper triazoles produces a distortion of the helical cavity, leading to a bidentate binding mode instead of the tetradentate exhibited in the case of 7.
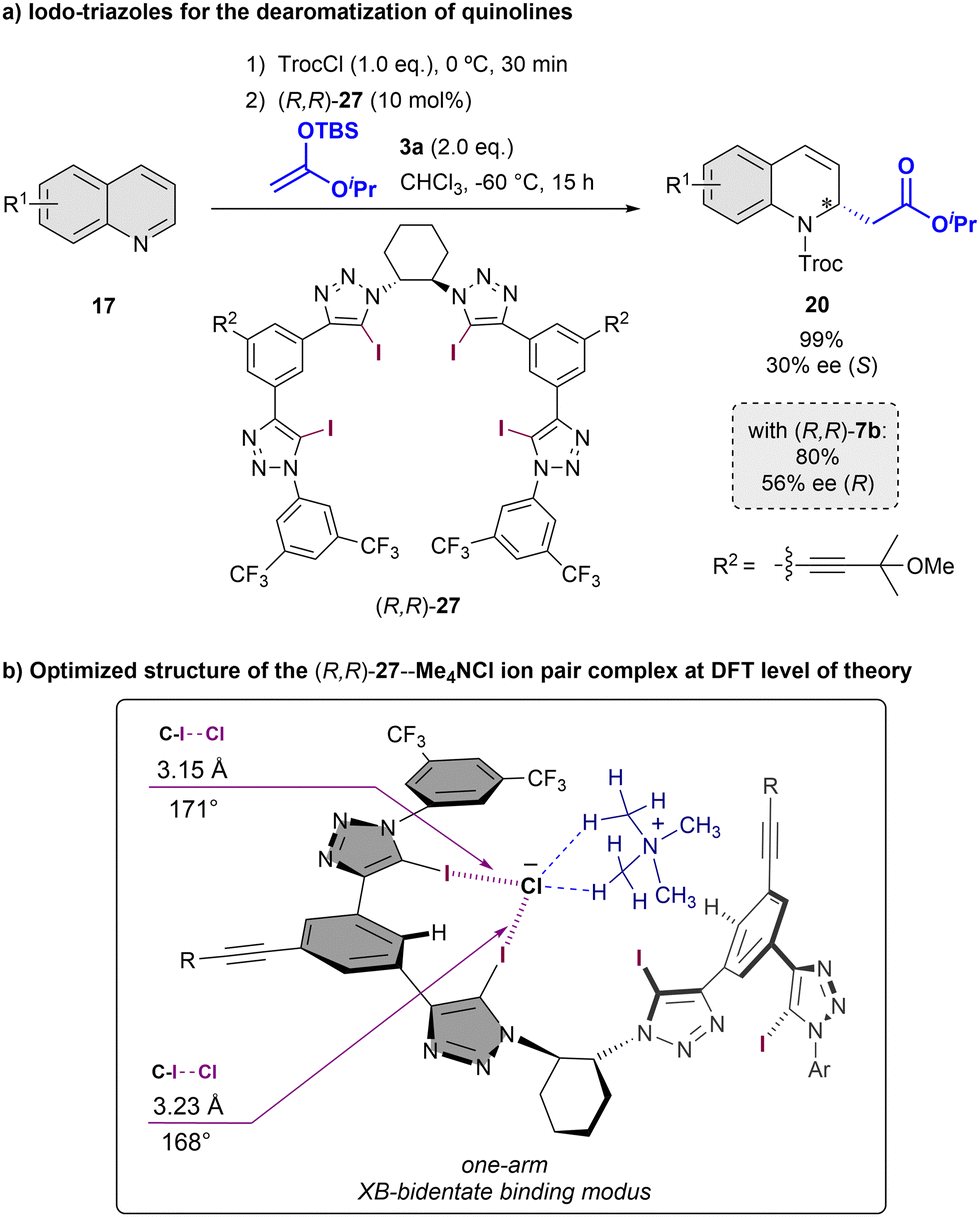 | ||
| Scheme 8 Asymmetric Reissert type reaction of quinolines employing a chiral triazole-based XB-donor catalyst (a) and computed binding mode with Me3NCl as model substrate (b). | ||
4. Dearomatization of pyridines
Pyridines, the simplest six-membered-ring N-heteroarenes, are very interesting molecules because present a broad bioactive spectrum.71–73 The dearomatization of pyridines is more challenging compared to the quinolines or isoquinolines. This could be attributed to two main issues. In the one hand, the complete loss of aromaticity in the case of pyridines, whereas quinolines or isoquinolines are able to partially retained it in the other aromatic ring. On the other hand, pyridines show additional difficulties in terms of regioselectivity due to the two possible positions, C2 and C4, prone to nucleophilic attack.74Challenged by their intrinsic reactivity and regioselectivity issues, in 2015 we developed the first enantioselective dearomatization of pyridines 28 mediated by anion binding catalysis (Scheme 9a).75 We were delighted to observed that (R,R)-7b was superior to other classical catalysts such as thiourea or squaramides regarding to both stereo- and regioselectivity. The reaction was carried out with 5 mol% of (R,R)-7b and iso-propyl ketene acetal 3a in Et2O at −78 °C, providing selectively the corresponding C2-substituted (R)-dihydropyridines 29 in high yields and up to 98% enantioselectivity. Moreover, the opposite (S)-enantiomer could also be obtained in a high enantiomeric ratio (−92% ee) when using the enantiomeric catalyst (S,S)-7b. It is important to remark that the C4 addition was preferred only with pyridines substituted at the C3 with medium or bulky groups or in absence of catalyst.
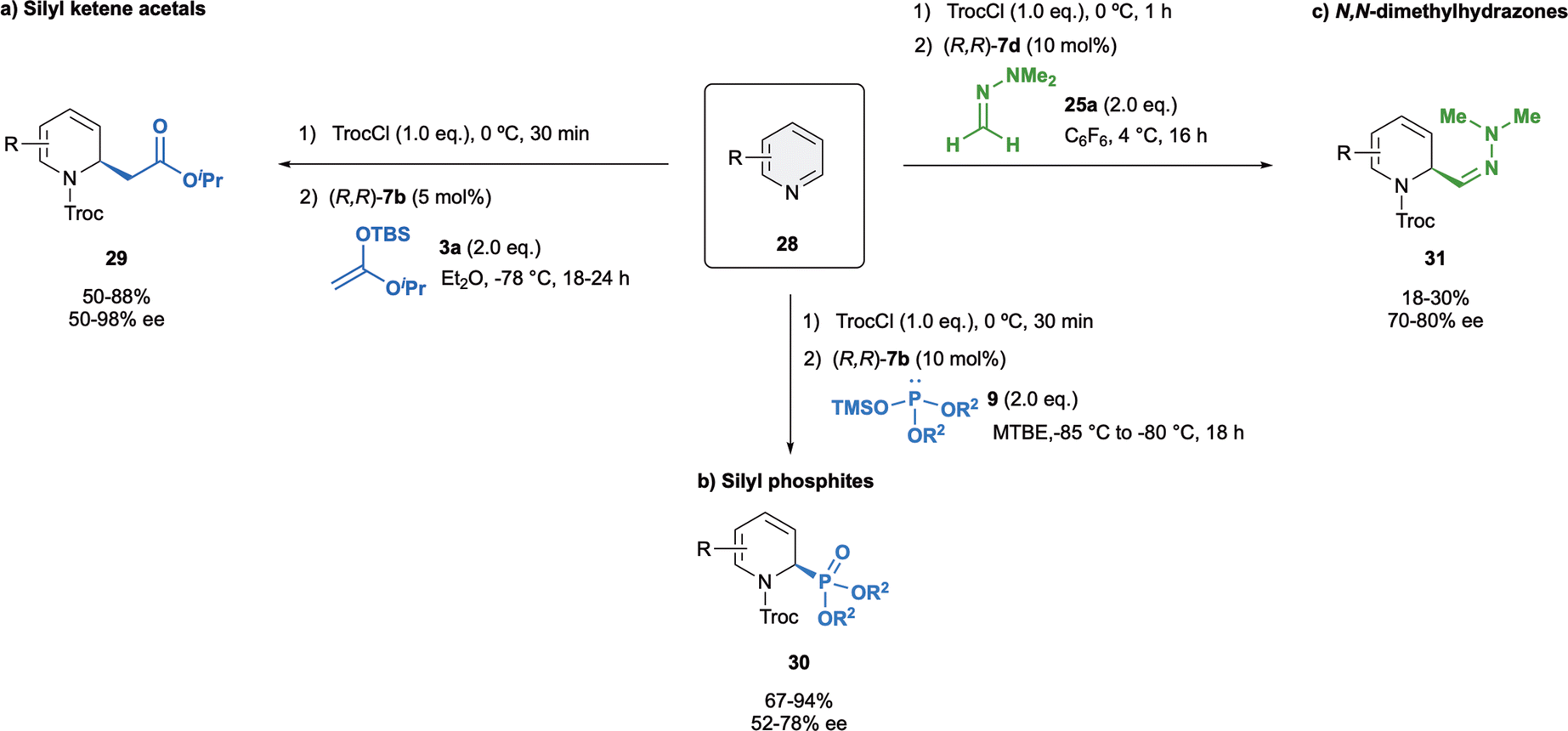 | ||
| Scheme 9 Dearomatization of pyridines with several nucleophiles reported by García Mancheño et al. | ||
Our group also exemplary demonstrated the asymmetric dearomatization of pyridines with different silyl phosphites 9 with (R,R)-7b in MTBE as a solvent (Scheme 9b),48 as well as N,N-dimethylhydrazone 25a with catalyst (R,R)-7d in C6F6 (Scheme 9c).52 The desired products 30 and 31 were obtained in good to moderate yields, and enantioselectivities up to 78% ee and up to 80% ee, respectively.
Thiourea-based catalysts have been employed for the dearomatization of pyridines too. In 2016, Caruana, Fochi and Bernardi reported the dearomatization of N-alkylpyridinium salts 33 instead of the more reactive acyl analogues by nucleophilic addition of indoles 32 (Scheme 10).76 The reaction was performed in presence of 10 mol% of 34 and stoichiometric amounts of 1,8-bis(dimethylamino)naphthalene that acts as a proton sponge (PS) in toluene as a solvent. The resulting 1,4-dihydropyridine derivatives 35 were obtained in high yields and up to 93% ee. The formation of the adduct 36 in the course of the reaction was observed by NMR. In addition to the expected interaction with the bromide counter-anion, it shows a covalent bond between the C6 atom of the pyridinium salt and the tertiary amine of 34. The simultaneous anion-binding activation and C2-blocking by covalent-bond formation with the catalyst explain the observed selective nucleophilic addition to the C4 position, resembling the normal reactivity of pyridinium salts.
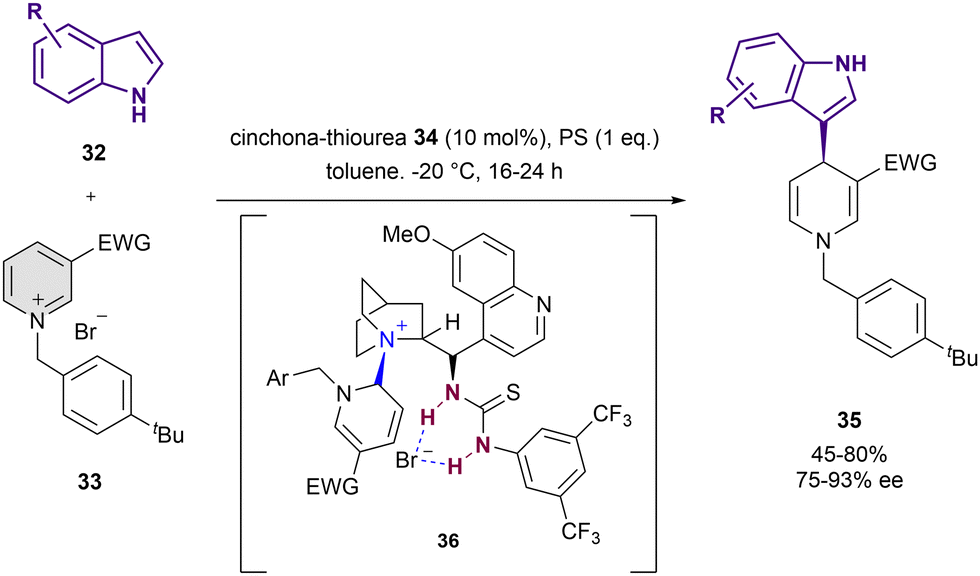 | ||
| Scheme 10 Asymmetric dearomatization of N-benzylpyridinium salts with indoles by Bernardi et al. | ||
5. Dearomatization of other N-heteroarenes
Besides isoquinolines, quinolines and pyridines, the anion-binding dearomatization approach has been also extended to more complicated structures which present more than one heteroatom such as diazarenes.7,77 These derivatives present a larger number of reactive sites, making a challenge to achieve not only stereo-, but also regiocontrol in the formation of the corresponding chiral di- or tetrahydrodiazaheterocycles.In 2016, our group reported the first anion binding dearomatization reaction of various types of diazarenes with silyl ketene acetal 3a in presence of 10 mol% of (R,R)-7b in MTBE (Scheme 11a).78 The studied substrates 37 contained the two nitrogen atoms either in the same or in different rings, leading to the desired products 38 in high yields and enantioselectivities up to 92% ee. Moreover, more challenging six- and five-membered monocyclic diazarenes were also explored. In the case of the six membered heterocycle 39, the desired C2 product 40 was obtained with high regioselectivity, whereas the five membered heterocycles mostly led to decomposition products.
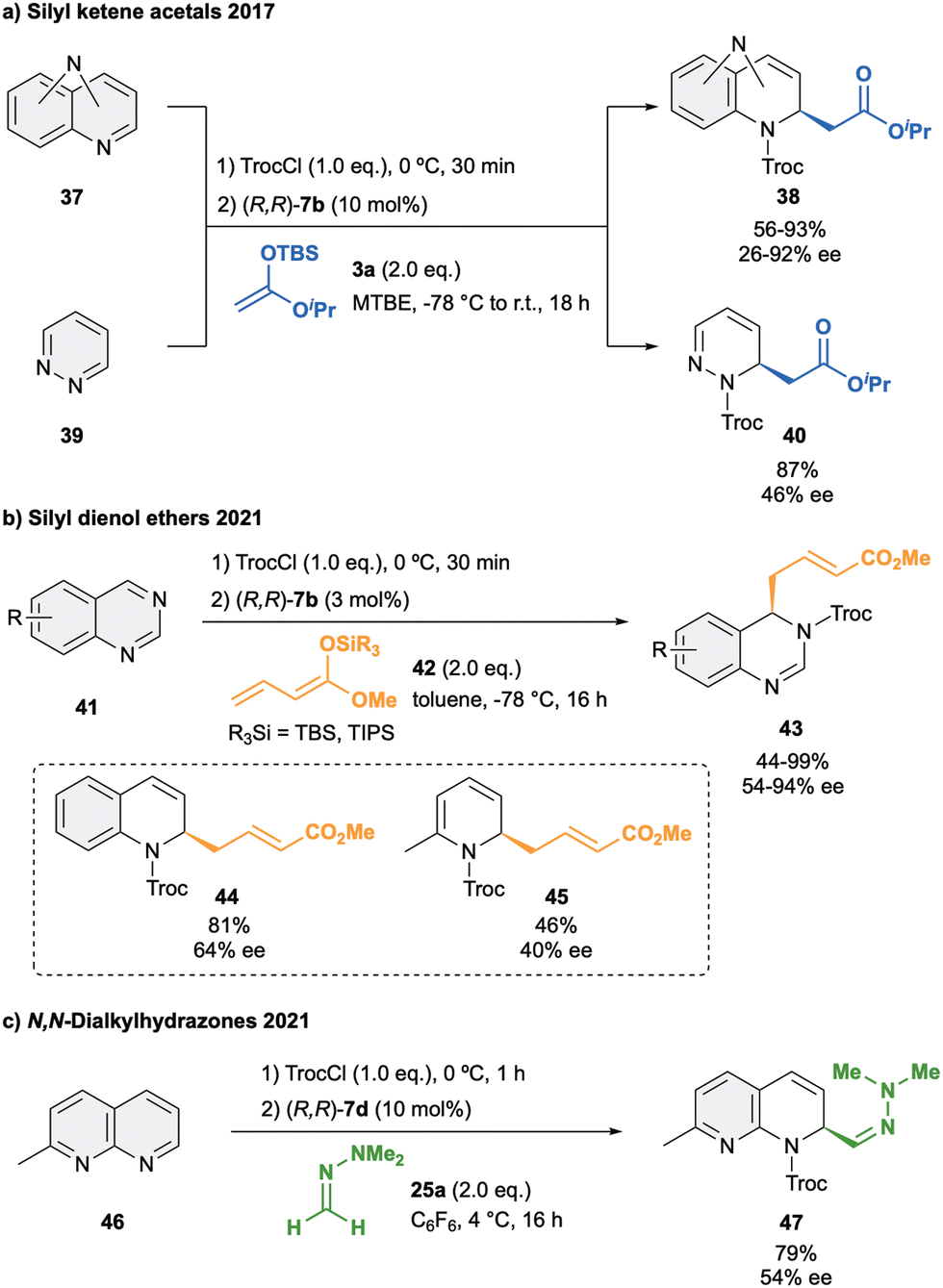 | ||
| Scheme 11 Enantioselective dearomatization of diazarenes with (a) silyl kentene acetals, (b) silyl dienol ethers and (c) dialkylhydrazones reported by García Mancheño et al. | ||
In 2021, we developed the vinylogous Mukaiyama-type dearomatization reaction of N-Troc activated quinazolines 41 by addition of silyl protected dienol ethers 42 in toluene (Scheme 11b).79 The use of 3 mol% of (R,R)-7b allowed for high selectivity, leading to the exclusive formation of the C4 addition products 43 in moderate to high yields and enantioselectivities up to 94% ee. A similar contact ion pair complex formation involving the fixation of the cationic substrate by π–π stacking with one of the arms of the catalyst and coulomb interactions with the chloride anion was proposed (Fig. 3). However, in this case, the additional interactions between the counterion and the dienolate (HNu–Cl) orientate its vinylogous nucleophilic site towards the addition at the C4 position of the quinazoline from the less hindered Re-face. Interestingly, a less efficient orientation and stereocontrol was observed for other N-heteroarenes such as quinolines (44, up to 64% ee) or pyridines (45, up to 40% ee).
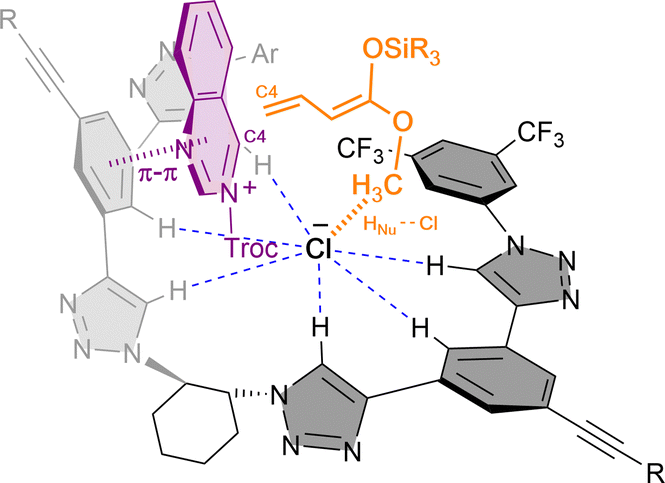 | ||
| Fig. 3 Catalyst-substrate-nucleophile complex via multiple non-covalent interactions proposed for the vinylogous Mukaiyama-type reaction reported by García Mancheño et al. | ||
Lastly, an isolated example on the addition of N,N-dimethylhydrazone 25a to 2-methyl-1,8-naphthyridine (46) in presence of 10 mol% of (R,R)-7d was reported (Scheme 11c).52 The corresponding hydrazone 47 was then obtained in 79% of yield and a moderate 54% ee, showing again the importance of appropriate matching of the substrate, nucleophile and structural features of the used catalyst.
6. O-Heteroarenes: pyrylium-type derivatives
O-Heteroarenes, similarly to N-heteroarenes, are important synthetic building blocks with a rich chemistry towards the preparation of interesting naturally occurring and bioactive oxygen heterocyclic compounds.80–82 In this regard, pyrylium derivatives are well-suited substrates to test the anion-binding catalytic dearomatization approach. Nevertheless, some complications regarding their reactivity exists, because most of nucleophilic reactions with pyrylium salts lead to ring opening or decomposition reactions.836.1. Cycloaddition strategies
In 2011, the Jacobsen group published the first anion-binding dearomatization reaction of O-heteroarenes by an intramolecular [5+2]-cycloaddition of 3-pyranones 48 bearing a carboxyl leaving group (LG) in the C2-position and a tethered alkene in C6 (R2) (Scheme 12, left).84 This method enabled the preparation of 8-oxabicyclo[3.2.1]octane derivatives 51 with α,β-unsaturated units. A dual cooperative thiourea organocatalytic system formed by the chiral primary amino-thiourea 49 (15 mol%), and an achiral thiourea 50 (15 mol%) was employed with acetic acid as a cocatalyst in toluene. First, the condensation between 49 and the ketone group of the substrate takes place to generate the corresponding reactive oxidopyrylium species upon enamine formation, followed by the benzoate-LG abstraction by 50 towards the assembling of the chiral contact ion pair between the 49-pyrylium and 50-carboxylate complexes. The desired cycloadducts 51 were obtained in moderate yields and enantioselectivities up to 95% ee.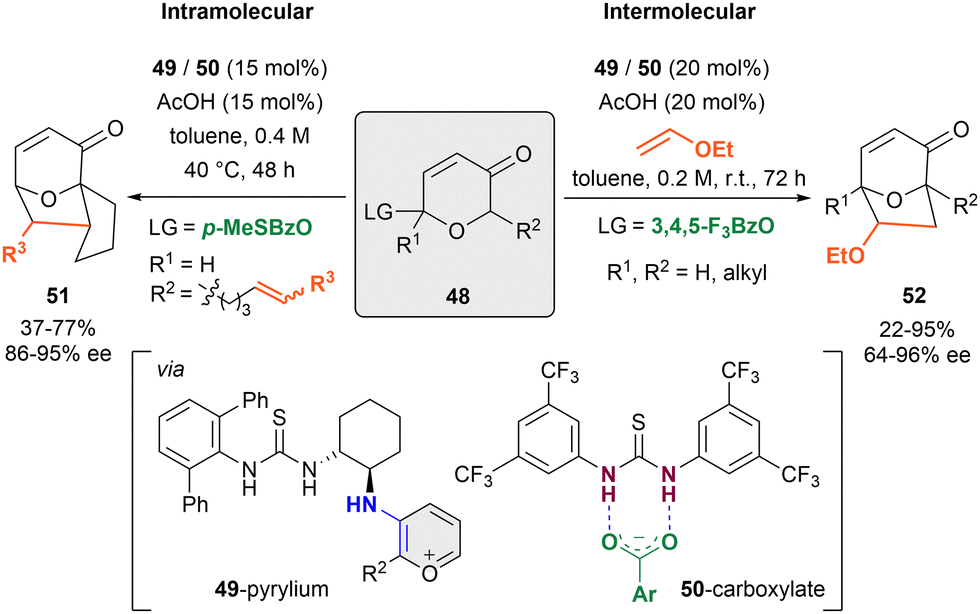 | ||
| Scheme 12 [5+2] cycloaddition of pyrylium derivates reported by Jacobsen et al. | ||
Later on, the same group was able to achieve higher yields and enantioselectivities through a related intermolecular [5+2]-cycloaddition between substituted pyrylium derivatives and electron rich terminal alkenes such as vinylethers (Scheme 12, right).85 However, a slightly higher catalytic loading (20 mol% vs. 15 mol%) and the use of 3,4,5-trifluorobenzoate (3,4,5-F3BzO) as leaving group were required for an optimal stereochemical outcome in the products 52 (up to 96% ee).
6.2. Nucleophilic additions
Alternative to the dearomatization of pyrylium derivatives by cycloaddition approaches, nucleophilic dearomatizations reactions have been also employed for this type of heteroarenes.86,87 In 2016, Mattson et al. described the first anion-binding catalytic addition of carbonyl nucleophiles such as silyl ketene acetals 3 to chromanones 53 (Scheme 13, left).86 These substrates are previously activated in situ with TIPSOTf to form the reactive siloxybenzopyrylium salt substrates. The development of a new 3,3′-naphtalen-2-yl silanediol (R)-5c was necessary to achieve the desired products 54 in high yields and enantioselectivities up to 56% ee. Compared to the previously used silanediols (see Scheme 2), (R)-5c presents a bulkier environment around the active binding site, which allows the formation of a unique chiral pocket.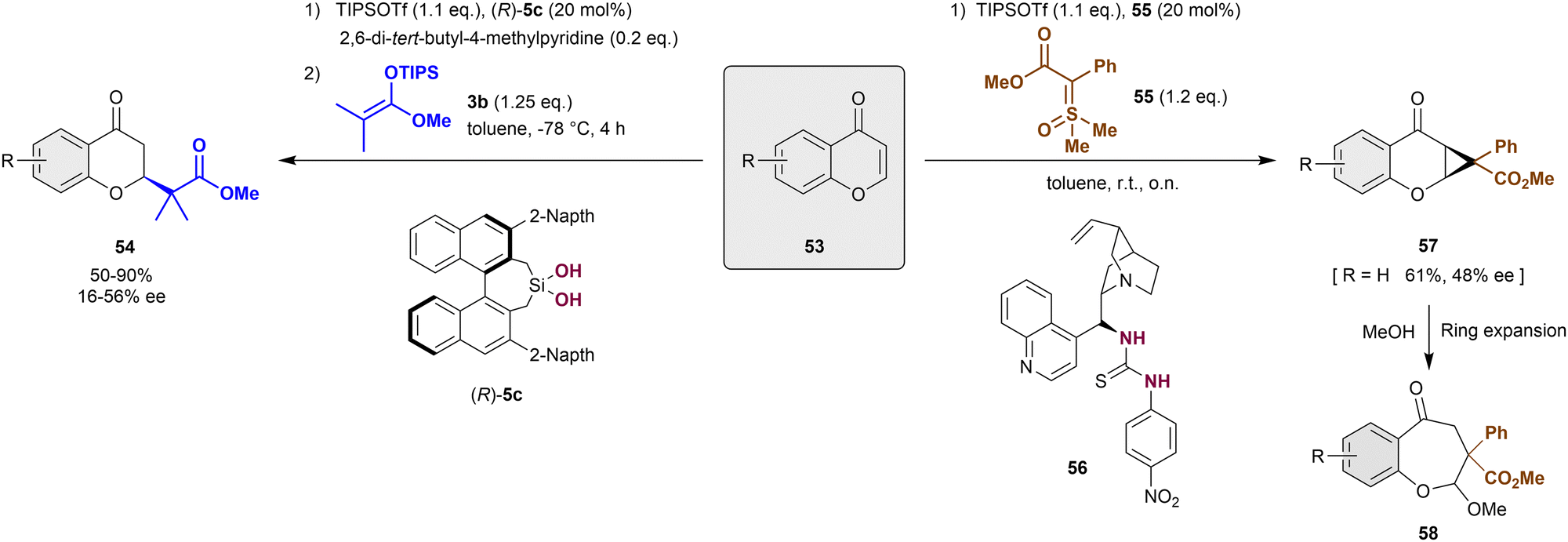 | ||
| Scheme 13 Enantioselective dearomatization of in situ formed benzylpyrylium derivatives reported by Mattson. | ||
More recently, the Mattson group reported the racemic cyclopropanation-nucleophilic ring expansion sequence towards the synthesis of benzo[b]oxepines 58 (Scheme 13, right).87 To achieve this, sulfoxonium ylides 55 were employed for the cyclopropanation of benzylpyrylium triflates, which were prepared in situ from chromanones 53 through the classical approach depicted above. Although the method was mainly developed as racemic version, the use of the chiral thiourea-based anion-binding catalyst 56 allowed a facially selective cycloaddition reaction. Thus, the corresponding model product 57 (R = H) was achieved in 61% yield and 48% ee, which was improved to 74% ee after recrystallization.
In 2019, and inspired by the previous work of Mattson,86 we developed the one-pot dearomatization reaction of pyrylium-type derivatives with differently substituted silyl ketene acetals 3 in presence the tetrakistriazole (R,R)-7b (Scheme 14).88 This catalytic system showed a high reactivity, allowing for extremely low loadings (typically 1 mol%, and down to 0.05 mol%)89 compared to the previous reported studies, while maintaining a high enantiocontrol. The active benzylpyrylium salts were formed in situ by treatment of 4-chromanones 53 with 2,6-lutidine and tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf) in toluene. Differently substituted OTBS-benzylpyrylium salts formed in situ from 4-chromones were well tolerated providing the corresponding products 59 with high yields and enantioselectivities up to 96% ee. Remarkably, this approach was successfully extended to the more challenging 4-pyrones 60 in presence of 5 mol% of (R,R)-7b, leading to no ring opening but the desired products 61 in moderate yields and enantioselectivities up to 92% ee.
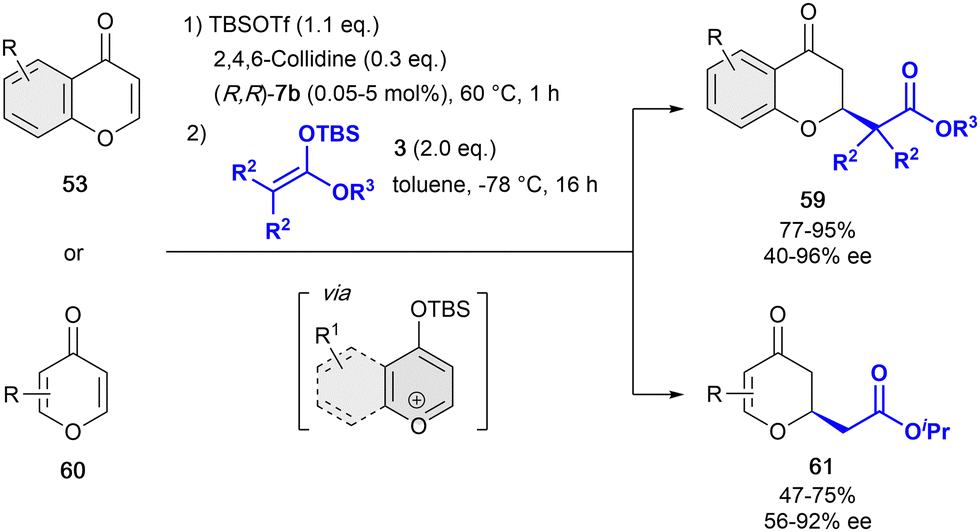 | ||
| Scheme 14 Dearomatization of pyrylium derivatives by García Mancheño et al. | ||
7. Heteroarenes as nucleophiles in anion-binding dearomatization reactions
Although most of the dearomatization strategies based on anion-binding catalysis are based on the activation of the heteroarene as electrophile, electron rich heteroarenes can also be dearomatized through this type of catalysis upon reacting with the appropriate electrophilic species.19,90,91 Embracing this idea, in 2014, Porco Jr., Jacobsen et al. reported the nucleophilic addition of 3-substituted indoles 62 to silyl protected γ-pyrone derivatives 63 (Scheme 15).90 They employed an arylpyrrolidino-derived thiourea organocatalyst 64 in combination with benzoic acid as a cocatalyst. The desilylation of the pro-electrophile 63 promoted by the Brønsted acid92 takes first place, followed by the bromide-anion abstraction by 64 towards the formation of the chiral contact ion pair (64-Br-substrate). Finally, after the nucleophilic addition of the indole 62 to the in situ generated exo-conjugated oxonium electrophile, the desired chiral indoline products 65 bearing a quaternary stereocenter in the C3-position were obtained in moderate to high yields and enantioselectivities up to 96% ee.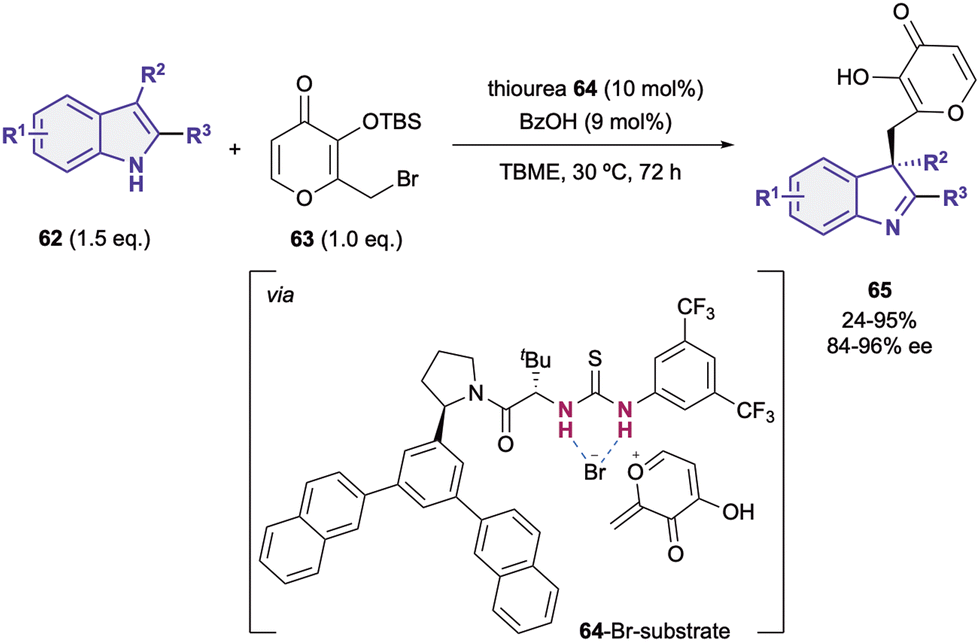 | ||
| Scheme 15 Indole addition to pyrones by Jacobsen and co-workers. | ||
As last example, Liu and Tian employed the bifunctional thiourea-pyridine organocatalyst 66 for the acyl transfer in both a Steglich rearrangement of oxazolyl carbonates 67 and the Black rearrangement of benzofurane substrates 68 (Scheme 16).91 This bifunctional catalyst behaves in a similar way as the one described by Seidel in 2011,46 in which the dialkylaminopyridine moiety activates the acyl group and the thiourea behaves as a chiral anion receptor. In both cases, 5 mol% of 66 was enough to obtain moderate to high yields and enantioselectivities up to 97% ee for the Steglich reaction (69), and 87% ee for the Black rearrangement (70).
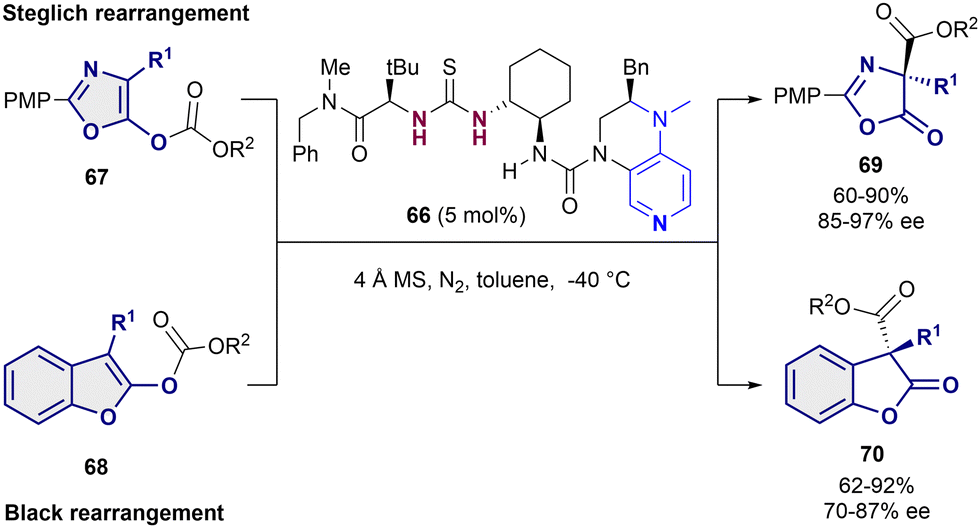 | ||
| Scheme 16 Enantioselective Steglich and Black rearrangements reported by Liu and Tian. | ||
8. Conclusions & outlook
Anion binding organocatalytic approaches have proved to be highly efficient to achieve the dearomatization of heteroarenes. After the pioneering studies in this field with chiral thioureas by the group of Jacobsen, several other catalytic systems such as silanediols, oligo-triazoles and bifunctional catalysts have been introduced for the dearomatization of electron poor N-hetereoarenes such as (iso)quinolines, pyridines and diazarenes using silyl ketene acetals as nucleophiles. Additionally, other nucleophiles such as silyl phosphites, allyltin reagents, dienolates, or N,N-dialkylhydrazones were also successfully applied. Furthermore, this approach was efficiently extended to highly challenging pyrylium-type O-heterocycles, which was also possible through a pre-activation of non-aromatic chromanone or pyrone derivatives by enamine formation or enolization followed by anion-binding catalysis. Alternative to the most employed activation and dearomatization of electrophilic heteroarenes, a few examples with electron rich aromatic compounds have been also described. In these cases, the heteroarene plays the role of the nucleophile, whereas the anion-binding catalyst activates the electrophilic reaction partner. Finally, inspired by classical HB-catalysis, the first application of XB-activation was also employed for this approach, opening new possibilities for the future design of catalysts relying in σ-hole interactions. However, despite some important breakthroughs in the field covered here, the design of more effective anion-binding catalysts for enantioselective dearomatization reactions combined with a low catalyst loading is still a real challenge due to the fact that ion pairing is less directional than covalent interactions.The results discussed in this feature provides a solid platform and shows broad possibilities for the future exploration of the chemical landscape within this research field, where we anticipate that dual and bifunctional synergetic catalytic systems will have an important impact in the design of more efficient catalytic dearomatization processes beyond the to date addressed heteroarenes and the discovery of new catalytic transformations.
Author contributions
Both authors have discussed the organization and presentation of the contents, and participated on the writing and editing of the Feature Article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The DFG (Deutsche Forschungsgemeinschaft) within the IRTG-2678 and the European Research Council (ERC-CG 724695) are gratefully acknowledged for generous support. M. A. also thanks the DFG within the IRTG-2678 for a PhD contract.Notes and references
- J. Mortier, Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, Wiley & Sons Inc, Hoboken, New Jersey, 2016 Search PubMed.
- F. L. Ortíz, M. J. Iglesias, I. Fernández, C. M. A. Sánchez and G. R. Gómez, Nucleophilic dearomatizing (DNAr) reactions of aromatic C, H-systems. A mature paradigm in organic synthesis, Chem. Rev., 2007, 107, 1580–1691 CrossRef PubMed.
- F. Lovering, J. Bikker and C. Humblet, Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- S. P. Roche and J. A. Porco Jr., Dearomatization strategies in the synthesis of complex natural products, Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS PubMed.
- W. C. Wertjes, E. H. Southgate and D. Sarlah, Recent advances in chemical dearomatization of nonactivated arenes, Chem. Soc. Rev., 2018, 47, 7996–8017 RSC.
- C. J. Huck and D. Sarlah, Shaping molecular landscapes: recent advances, opportunities, and challenges in dearomatization, Chem, 2020, 6, 1589–1603 CAS.
- R. Waack, The stability of the aromatic sextet, J. Chem. Educ., 1962, 39, 469–472 CrossRef CAS.
- Asymmetric Dearomatization Reactions, ed. S.-L. You, Wiley-VCH, Weinheim, 2016 Search PubMed.
- C.-X. Zhuo, W. Zhang and S.-L. You, Catalytic asymmetric dearomatization reactions, Angew. Chem., Int. Ed., 2012, 51, 12662–12686 CrossRef PubMed.
- C. Zheng and S.-L. You, Advances in Catalytic Asymmetric Dearomatization, ACS Central Sci., 2021, 7, 432–444 CrossRef CAS PubMed.
- Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, Wiley-VCH, Weinheim, 2011 Search PubMed.
- Heterocyclic Chemistry in Drug Discovery, ed. J. J. Li, Wiley & Sons Inc, Hoboken, New Jersey, 2013 Search PubMed.
- Applications of Heterocycles in the Design of Drugs and Agricultural Products, ed. N. Meanwell and M. Lolli, Elsevier, 2021 Search PubMed.
- Heterocyclic Chemistry, ed. J. A. Joule and K. Mills, 5th edn, 2010 Search PubMed.
- B. M. Trost, Asymmetric catalysis: An enabling science, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5348–5355 CrossRef CAS PubMed.
- A. Fanourakis, P. J. Docherty, P. Chuentragool and R. J. Phipps, Recent Developments in Enantioselective Transition Metal Catalysis Featuring Attractive Noncovalent Interactions between Ligand and Substrate, ACS Catal., 2020, 10, 10672–10714 CrossRef CAS PubMed.
- Special Issue: Recent Advances in Transition-Metal Catalysis, Chem. Rec. ed. H. Yorimitsu, M. Kotora and N. T. Patil, 2021, vol. 21, pp. 3335–4173 Search PubMed.
- See also: Z.-L. Xia, Q.-F. Xu-Xu, C. Zheng and S.-L. You, Chiral phosphoric acid-catalyzed asymmetric dearomatization reactions, Chem. Soc. Rev., 2020, 49, 286–300 RSC.
- Anion-Binding Catalysis, ed. O. García Mancheño, Wiley-VCH, Weinheim, 2022 Search PubMed.
- S. Beckendorf, S. Asmus and O. García Mancheño, H-Donor Anion Acceptor Organocatalysis - The Ionic Electrophile Activation Approach, ChemCatChem, 2012, 4, 926–936 CrossRef CAS.
- K. Brak and E. N. Jacobsen, Asymmetric ion-pairing catalysis, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed.
- M. Mahlau and B. List, Asymmetric Counteranion-Directed Catalysis: Concept, Definition, and Applications, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed.
- M. D. Visco, J. Attard, Y. Guan and A. E. Mattson, Anion-binding catalyst designs for enantioselective synthesis, Tetrahedron Lett., 2017, 58, 2623–2628 CrossRef CAS.
- L. Schifferer, M. Stinglhamer, K. Kaur and O. García Mancheño, Halides as versatile anions in asymmetric anion-binding organocatalysis, Beilstein J. Org. Chem., 2021, 17, 2270–2286 CrossRef CAS PubMed.
- L.-M. Entgelmeier and O. García Mancheño, Activation Modes in Asymmetric Anion-Binding Catalysis, Synthesis, 2022, 3907–3927 CAS.
- For the introduction of the term anion-binding catalysis, see: M. Kotke and P. R. Schreiner, Acid-free, organocatalytic acetalization, Tetrahedron, 2006, 62, 434–439 CrossRef CAS.
- A. G. Doyle and E. N. Jacobsen, Small-Molecule H-Bond Donors in Asymmetric Catalysis, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed.
- Hydrogen Bonding in Organic Synthesis, ed. P. M. Pihko, Wiley-VCH, Weinheim, 2009 Search PubMed.
- R. R. Knowles and E. N. Jacobsen, Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed.
- A. J. Neel, M. J. Hilton, M. S. Sigman and F. Dean Toste, Exploiting non-covalent π interactions for catalyst design, Nature, 2017, 543, 637–646 CrossRef CAS PubMed.
- A. B. Weberg, R. P. Murphy and N. C. Tomson, Oriented internal electrostatic fields: an emerging design element in coordination chemistry and catalysis, Chem. Sci., 2022, 13, 5432–5446 RSC.
- M. S. Taylor, N. Tokunaga and E. N. Jacobsen, Enantioselective Thiourea-Catalyzed Acyl-Mannich Reactions of Isoquinolines, Angew. Chem., Int. Ed., 2005, 44, 6700–6704 CrossRef CAS PubMed.
- A. Reissert, Über die Einführung der Benzoyl-gruppe in tertiäre cyclische Basen, Ber. Dtsch. Chem. Ges., 1905, 38, 1603–1614 CrossRef CAS.
- M. Ahamed and M. H. Todd, Catalytic Asymmetric Additions of Carbon-Centered Nucleophiles to Nitrogen-Containing Aromatic Heterocycles, Eur. J. Org. Chem., 2010, 5935–5942 CrossRef CAS.
- A. G. Schafer, J. M. Wieting, T. J. Fisher and A. E. Mattson, Chiral silanediols in anion-binding catalysis, Angew. Chem., Int. Ed., 2013, 52, 11321–11324 CrossRef CAS PubMed.
- J. M. Wieting, T. J. Fisher, A. G. Schafer, M. D. Visco, J. C. Gallucci and A. E. Mattson, Preparation and Catalytic Activity of BINOL-Derived Silanediols, Eur. J. Org. Chem., 2015, 525–533 CrossRef CAS.
- F. Fox, J. M. Neudörfl and B. Goldfuss, Silanediol versus chlorosilanol: hydrolyses and hydrogen-bonding catalyses with fenchole-based silanes, Beilstein J. Org. Chem., 2019, 15, 167–186 CrossRef CAS PubMed.
- M. Zurro, S. Asmus, S. Beckendorf, C. Mück-Lichtenfeld and O. García Mancheño, Chiral helical oligotriazoles: New class of anion-binding catalysts for the asymmetric dearomatization of electron-deficient N-heteroarenes, J. Am. Chem. Soc., 2014, 136, 13999–14002 CrossRef CAS PubMed.
- K. Jug, S. Chiodo, P. Calaminici, A. Avramopoulos and M. G. Papadopoulos, Electronic and Vibrational Polarizabilities and Hyperpolarizabilities of Azoles: A Comparative Study of the Structure−Polarization Relationship, J. Phys. Chem. A, 2003, 107, 4172–4183 CrossRef CAS.
- R. Vargas, J. Garza, D. A. Dixon and B. P. Hay, How Strong Is the Cα-H⋯OC Hydrogen Bond?, J. Am. Chem. Soc., 2000, 122, 4750–4755 CrossRef CAS.
- K. Ohmatsu and T. Ooi, 1,2,3-Triazoles and 1,2,3-Triazolium Ions as Catalysts, in Anion-Binding Catalysis, ed. O. García Mancheño, Wiley-VCH, Weinheim, 2022, ch. 7, pp. 221–247 Search PubMed.
- M. Zurro, S. Asmus, J. Bamberger, S. Beckendorf and O. García Mancheño, Chiral Triazoles in Anion-Binding Catalysis: New Entry to Enantioselective Reissert-Type Reactions, Chem. – Eur. J., 2016, 22, 3785–3793 CrossRef CAS PubMed.
- A. Ray Choudhury and S. Mukherjee, Enantioselective dearomatization of isoquinolines by anion-binding catalysis en route to cyclic α-aminophosphonates, Chem. Sci., 2016, 7, 6940–6945 RSC.
- A. Mucha, P. Kafarski and Ł. Berlicki, Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry, J. Med. Chem., 2011, 54, 5955–5980 CrossRef CAS PubMed.
- E. Matador, J. Iglesias-Sigüenza, D. Monge, P. Merino, R. Fernández and J. M. Lassaletta, Enantio- and Diastereoselective Nucleophilic Addition of N-tert-Butylhydrazones to Isoquinolinium Ions through Anion-Binding Catalysis, Angew. Chem., Int. Ed., 2021, 60, 5096–5101 CrossRef CAS PubMed.
- C. K. De, N. Mittal and D. Seidel, A dual-catalysis approach to the asymmetric Steglich rearrangement and catalytic enantioselective addition of O-acylated azlactones to isoquinolines, J. Am. Chem. Soc., 2011, 133, 16802–16805 CrossRef CAS PubMed.
- Y. Yamaoka, H. Miyabe and Y. Takemoto, Catalytic Enantioselective Petasis-Type Reaction of Quinolines Catalyzed by a Newly Designed Thiourea Catalyst, J. Am. Chem. Soc., 2007, 129, 6686–6687 CrossRef CAS PubMed.
- T. Fischer, Q.-N. Duong and O. García Mancheño, Triazole-Based Anion-Binding Catalysis for the Enantioselective Dearomatization of N-Heteroarenes with Phosphorus Nucleophiles, Chem. – Eur. J., 2017, 23, 5983–5987 CrossRef CAS PubMed.
- Q.-N. Duong, L. Schifferer and O. García Mancheño, Nucleophile Screening in Anion-Binding Reissert-Type Reactions of Quinolines with Chiral Tetrakis(triazole) Catalysts, Eur. J. Org. Chem., 2019, 5452–5461 CrossRef CAS.
- S. Joseph, Q.-N. Duong, L. Schifferer and O. García Mancheño, Enantioselective organocatalytic synthesis of α-allylated dihydroquinolines, Tetrahedron, 2022, 114, 132767 CrossRef CAS.
- D. G. Piekarski, P. Steinforth, M. Gómez-Martínez, J. Bamberger, F. Ostler, M. Schönhoff and O. García Mancheño, Insight into the Folding and Cooperative Multi-Recognition Mechanism in Supramolecular Anion-Binding Catalysis, Chem. – Eur. J., 2020, 26, 17598–17603 CrossRef CAS PubMed.
- M. Gómez-Martínez, M. Carmen Pérez-Aguilar, D. G. Piekarski, C. G. Daniliuc and O. García Mancheño, N,N-Dialkylhydrazones as Versatile Umpolung Reagents in Enantioselective Anion-Binding Catalysis, Angew. Chem., Int. Ed., 2021, 60, 5102–5107 CrossRef PubMed.
- H. Mayr, T. Bug, M. F. Gotta, N. Hering, B. Irrgang, B. Janker, B. Kempf, R. Loos, A. R. Ofial, G. Remennikov and H. Schimmel, Reference Scales for the Characterization of Cationic Electrophiles and Neutral Nucleophiles, J. Am. Chem. Soc., 2001, 123, 9500–9512 CrossRef CAS PubMed.
- https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank/ .
- For other example of anion-binding catalyzed allylation reaction, see: A. J. Bendelsmith, S. C. Kim, M. Wasa, S. P. Roche and E. N. Jacobsen, Enantioselective Synthesis of α-Allyl Amino Esters via Hydrogen-Bond-Donor Catalysis, J. Am. Chem. Soc., 2019, 141, 11414–11419 CrossRef CAS PubMed.
- R. Lazny and A. Nodzewska, N,N-Dialkylhydrazones in Organic Synthesis. From Simple N,N-Dimethylhydrazones to Supported Chiral Auxiliaries, Chem. Rev., 2010, 110, 1386–1434 CrossRef CAS PubMed.
- T. Nakamura, K. Okuno, R. Nishiyori and S. Shirakawa, Hydrogen-Bonding Catalysis of Alkyl-Onium Salts, Chem. – Asian J., 2020, 15, 463–472 CrossRef CAS PubMed.
- C. Liu, Z. Oblak, M. N. V. Wal, A. K. Dilger, D. K. Almstead and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 2134–2137 CrossRef CAS PubMed.
- Halogen Bonding in Solution, ed. S. Huber, Wiley-VCH, Weinheim, 2021 Search PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The Halogen Bond, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- J. Bamberger, F. Ostler and O. García Mancheño, Frontiers in Halogen and Chalcogen-Bond Donor Organocatalysis, ChemCatChem, 2019, 11, 5198–5211 CrossRef CAS PubMed.
- M. Breugst and J. J. Koenig, σ-Hole Interactions in Catalysis, Eur. J. Org. Chem., 2020, 5473–5487 CrossRef CAS.
- M. Kaasik and T. Kanger, Supramolecular Halogen Bonds in Asymmetric Catalysis, Front. Chem., 2020, 8, 599064 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, The σ-hole revisited, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC.
- S. M. Huber, E. Jimenez-Izal, J. M. Ugalde and I. Infante, Unexpected trends in halogen-bond based noncovalent adducts, Chem. Commun., 2012, 48, 7708–7710 RSC.
- J. Y. C. Lim and P. D. Beer, Sigma-Hole Interactions in Anion Recognition, Chem., 2018, 4, 731–783 CAS.
- A. Bauza, A. Frontera and T. J. Mooibroek, The Bright Future of Unconventional σ/π-Hole Interactions, Chem. Phys. Chem., 2015, 16, 2496–2517 CrossRef CAS PubMed.
- A. Bruckmann, M. Peña and C. Bolm, Organocatalysis through Halogen-Bond Activation, Synlett, 2008, 900–902 CAS.
- R. L. Sutar, E. Engelage, R. Stoll and S. Huber, Bidentate Chiral Bis(imidazolium)-Based Halogen Bond Donors: Synthesis and First Applications in Enantioselective Recognition and Catalysis, Angew. Chem., Int. Ed., 2020, 59, 6806–6810 CrossRef CAS PubMed.
- F. Ostler, D. G. Piekarski, T. Danelzik, M. S. Taylor and O. García Mancheño, Neutral Chiral Tetrakis-Iodo-Triazole Halogen-Bond Donor for Chiral Recognition and Enantioselective Catalysis, Chem. – Eur. J., 2021, 27, 2315–2320 CrossRef CAS PubMed.
- A. A. Altaf, A. Shahzad, Z. Gul, N. Rasool, A. Badshah, B. Lal and E. Khan, A Review on the Medicinal Importance of Pyridine Derivatives, J. Drug Des. Med. Chem., 2015, 1, 1–11 Search PubMed.
- Y. Ling, Z.-Y. Hao, D. Liang, C.-L. Zhang, Y.-F. Liu and Y. Wang, The Expanding Role of Pyridine and Dihydropyridine Scaffolds in Drug Design, Drug Des. Devel. Ther., 2021, 15, 4289–4338 CrossRef CAS PubMed.
- Recent Developments in the Synthesis and Applications of Pyridines, ed. P. Singh, Elsevier Inc, 2022 Search PubMed.
- Heterocyclic Chemistry, in Pyridines: Reactions and Synthesis, ed. J. A. Joule and K. Mills, 5th edn, 2010, ch. 8, pp. 125–176 Search PubMed.
- O. García Mancheño, S. Asmus, M. Zurro and T. Fischer, Highly Enantioselective Nucleophilic Dearomatization of Pyridines by Anion-Binding Catalysis, Angew. Chem., Int. Ed., 2015, 54, 8823–8827 CrossRef PubMed.
- G. Bertuzzi, A. Sinisi, L. Caruana, A. Mazzanti, M. Fochi and L. Bernardi, Catalytic Enantioselective Addition of Indoles to Activated N-Benzylpyridinium Salts: Nucleophilic Dearomatization of Pyridines with Unusual C-4 Regioselectivity, ACS Catal., 2016, 6, 6473–6477 CrossRef CAS.
- K. A. Rinderspacher, 6.2-Six-membered ring systems: diazines and benzo derivatives, Prog. Heterocycl. Chem., 2021, 33, 381–439 CAS.
- T. Fischer, J. Bamberger and O. García Mancheño, Asymmetric nucleophilic dearomatization of diazarenes by anion-binding catalysis, Org. Biomol. Chem., 2016, 14, 5794–5802 RSC.
- K. Kaur, J. Humbrías-Martín, L. Hoppmann, J. A. Fernández-Salas, C. G. Daniliuc, J. Alemán and O. García Mancheño, Enantioselective vinylogous-Mukaiyama-type dearomatisation by anion-binding catalysis, Chem. Commun., 2021, 57, 9244–9247 RSC.
- J. Cossy and A. Guérinot, Chapter Five-Natural Products Containing Oxygen Heterocycles—Synthetic Advances Between 1990 and 2015, in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Advances Academic Press, 2016, vol. 119, pp. 107–142 Search PubMed.
- P. K. Singh and O. Silakari, The Current Status of O-Heterocycles: A Synthetic and Medicinal Overview, ChemMedChem, 2018, 13, 1071–1087 CrossRef CAS PubMed.
- Y. Li, H. Wang and X. Li, Over one century after discovery: pyrylium salt chemistry emerging as a powerful approach for the construction of complex macrocycles and metallo-supramolecules, Chem. Sci., 2020, 11, 12249–12268 RSC.
- T. S. Balaban and A. T. Balaban, Product Class 1: Pyrylium Salts, in Science of Synthesis: Category 2, Hetarenes and Realted Ring Systems, ed. E. J. Thomas, Thieme, Stuttgart, 2003, pp. 11–200 Search PubMed.
- N. Z. Burns, M. R. Witten and E. N. Jacobsen, Dual Catalysis in Enantioselective Oxidopyrylium-Based [5+2] Cycloadditions, J. Am. Chem. Soc., 2011, 133, 14578–14581 CrossRef CAS PubMed.
- M. R. Witten and E. N. Jacobsen, Catalytic Asymmetric Synthesis of 8-Oxabicyclooctanes by Intermolecular [5 + 2] Pyrylium Cycloadditions, Angew. Chem., Int. Ed., 2014, 53, 5912–5916 CrossRef CAS PubMed.
- A. M. Hardman-Baldwin, M. D. Visco, J. M. Wieting, C. Stern, S.-I. Kondo and A. E. Mattson, Silanediol-Catalyzed Chromenone Functionalization, Org. Lett., 2016, 18, 3766–3769 CrossRef CAS PubMed.
- A. N. Leveille, M. M. Allegrezza, K. Laybourn and A. E. Mattson, Dearomatization of benzopyrylium triflates with sulfoxonium ylides, Chem. Commun., 2022, 58, 12600–12603 RSC.
- T. Fischer, J. Bamberger, M. Gómez-Martínez, D. G. Piekarski and O. García Mancheño, Helical Multi-Coordination Anion-Binding Catalysts for the Highly Enantioselective Dearomatization of Pyrylium Derivatives, Angew. Chem., Int. Ed., 2019, 58, 3217–3221 CrossRef CAS PubMed.
- For another example of low catalytic loading in anion.binding catalysis, see: C. R. Kennedy, D. Lehnherr, N. S. Rajapaksa, D. D. Ford, Y. Park and E. N. Jacobsen, Mechanism-Guided Development of a Highly Active Bis-thiourea Catalyst for Anion-Abstraction Catalysis, J. Am. Chem. Soc., 2016, 138, 13525–13528 CrossRef CAS PubMed.
- C. S. Yeung, R. E. Ziegler, J. A. Porco, Jr. and E. N. Jacobsen, Thiourea-Catalyzed Enantioselective Addition of Indoles to Pyrones: Alkaloid Cores with Quaternary Carbons, J. Am. Chem. Soc., 2014, 136, 13614–13617 CrossRef CAS PubMed.
- Q.-H. Li, G.-S. Zhang, Y.-H. Wang, M.-S. Mei, X. Wang, Q. Liu, X.-D. Yang, P. Tian and G.-Q. Lin, A novel chiral DMAP–thiourea bifunctional catalyst catalyzed enantioselective Steglich and Black rearrangement reactions, Org. Chem. Front., 2019, 6, 2624–2629 RSC.
- M. Rueping, D. Parmar and E. Sugiono, Brønsted acid catalysis, Wiley-VCH, Weinheim, 2016 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |