
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of axial and helical chirality on circularly polarized luminescence: lessons learned from tethered twistacenes†
Anjan
Bedi‡§
*a,
Gal
Schwartz‡
b,
Uri
Hananel
b,
Amit
Manor Armon
a,
Israa
Shioukhi
a,
Gil
Markovich
 b and
Ori
Gidron
*a
b and
Ori
Gidron
*a
aInstitute of Chemistry, Center for Nanoscience and Nanotechnology and the Cazalli Institute, The Hebrew University of Jerusalem, Jerusalem 9190401, Israel. E-mail: ori.gidron@mail.huji.ac.il; gilmar@post.tau.ac.il
bSchool of Chemistry, Raymond and Beverly Sackler Faculty of Exact Sciences, Tel Aviv University, Tel Aviv 6997801, Israel
First published on 25th January 2023
Abstract
The effect of axial and helical twisting on the circularly polarized luminescence of acenes was studied both experimentally and computationally, using four series of tethered twisted acenes. We find that the combination of axial and helical chirality yields the highest anisotropy factors, and that the ratio between the absorption and emission anisotropy factors is an intrinsic property for twistacenes.
Circularly polarized luminescence (CPL) lies at the heart of several cutting-edge photonic technologies, including enantioselective biosensors,1 optical information storage and encryption devices, and security inks,2 as well as CPL emitters for CP-OLED,3 bio-imaging and chiral sensing applications.4 CPL is often quantified by the emission anisotropy factor, glum, with higher glum values indicating larger difference between left and right circularly polarized emission. However, in small organic molecules, the glum values are low (10−5–10−2), limiting their applications.5,6 In order to design small organic molecules with higher glum values, there is a need for clearer understanding of the factors affecting the glum values. Undertaking an intelligent search for such molecules relies on first obtaining a profound understanding of excited-state chirality, how chiral molecules interact with their surroundings, and of the structural underpinnings of CPL.7
Several studies have attempted to identify the structural features expected to increase CPL.7 The axial angle between two chromophores was found to be a crucial factor. For example, two binaphthyl derivatives that differ only with respect to degree of axial twist (70° versus 90°) show CPL with opposite signals.8 The CPL of helicenes has also been extensively studied,9,10 and was found to be significantly affected by length, orientation (“s” or “c”), and substituents.11 The nature of the substituents also has substantial effects when using a helicene scaffold.12,13 Several works highlighted the effect of extended conjugation of the dissymmetry factor.14 The addition of substituents on various position of an intrinsically-chiral backbone was also reported to increase glum.15 While helical chirality, axial chirality and extended conjugation were all mentioned as factors affecting the CPL, it is often difficult to draw clear conclusions from the underlying trends since they do not reflect systematic structural change in a single series of compounds, because of the unavailability of such series.7 In addition, whereas CPL was extensively studied in helicenes,10,16–18 the CPL of their para-fused analogues, twisted acenes (twistacenes), has not been investigated.
We recently introduced tethered twistacenes, in which tether length determines the degree of end-to-end twisting of the acene backbone.19 This allows various properties to be studied systematically. We found that twisting affects electronic, optical and magnetic properties.20–22 We also found that maximal ellipticity increases linearly with increasing twist.23 Extension of the twistacenes resulted in a non-linear increase in their chiroptical properties, an advantage that was explained by their helical secondary structure.24
In this work, we experimentally and computationally study the geometric factors affecting CPL in acenes. For this purpose, we investigate four series of twistacenes with varying degrees of helical and axial chirality, and extended conjugation. In the first series, we measure CPL as helical chirality varies (1-Cn and 2-Cn, Fig. 1a). In the second series, consisting of phenyls rather than alkynes in the 9,10-positions of anthracene, we study the effect of varying both axial and helical chirality (3-Cn, Fig. 1b). Finally, we study the combined effect of helicity and extended conjugation by introducing a new family of compounds composed of tethered twistacenes connected by a diacetylene linker (4-Cn, Fig. 1c). Our results indicate that CPL is similarly affected by increasing twist in all twistacenes, but that the combination of axial and helical chirality contributes significantly to increased CPL.
 | ||
| Fig. 1 Structures of the compounds studied in this work. | ||
Compounds 1-Cn, 2-Cn and 3-Cn were synthesized according to previous reports.19,24 For the synthesis of compound 4-Cn, the precursor 5-Cn underwent Pd-catalysed homocoupling to yield the respective products with 96% yield (see ESI† for synthetic details). All compounds were synthesized from their enantiopure precursors (separated by preparative chiral HPLC).
We began our investigation by calculating the effects of conformational changes (i.e., backbone twist and dihedral angles) on chiroptical properties as expressed in both the absorption and emission spectra. The electronic circular dichroism (ECD) or CPL intensity of a given transition is proportional to its rotational strength (R): R = |μ||m|·cos(Θ), where μ and m are the electric and magnetic transition dipole moment vectors, respectively, and Θ is the angle between them.5,25 To understand how the abovementioned parameters are affected by a systematic deviation from planarity, we used time-dependent density-functional theory (TD-DFT) to calculate the properties and dynamics of each twistacene skeleton without the tethers (Fig. 2), following the lowest energy transition (the 1La transition in anthracene, which is a π–π* transition). Since the backbone is rigid, the excited state (S1) shows a trend similar to that observed for the ground-state (S0); that is, increasing twist correlates with a systematic increase in the absolute value of the rotational strength and cos(Θ).
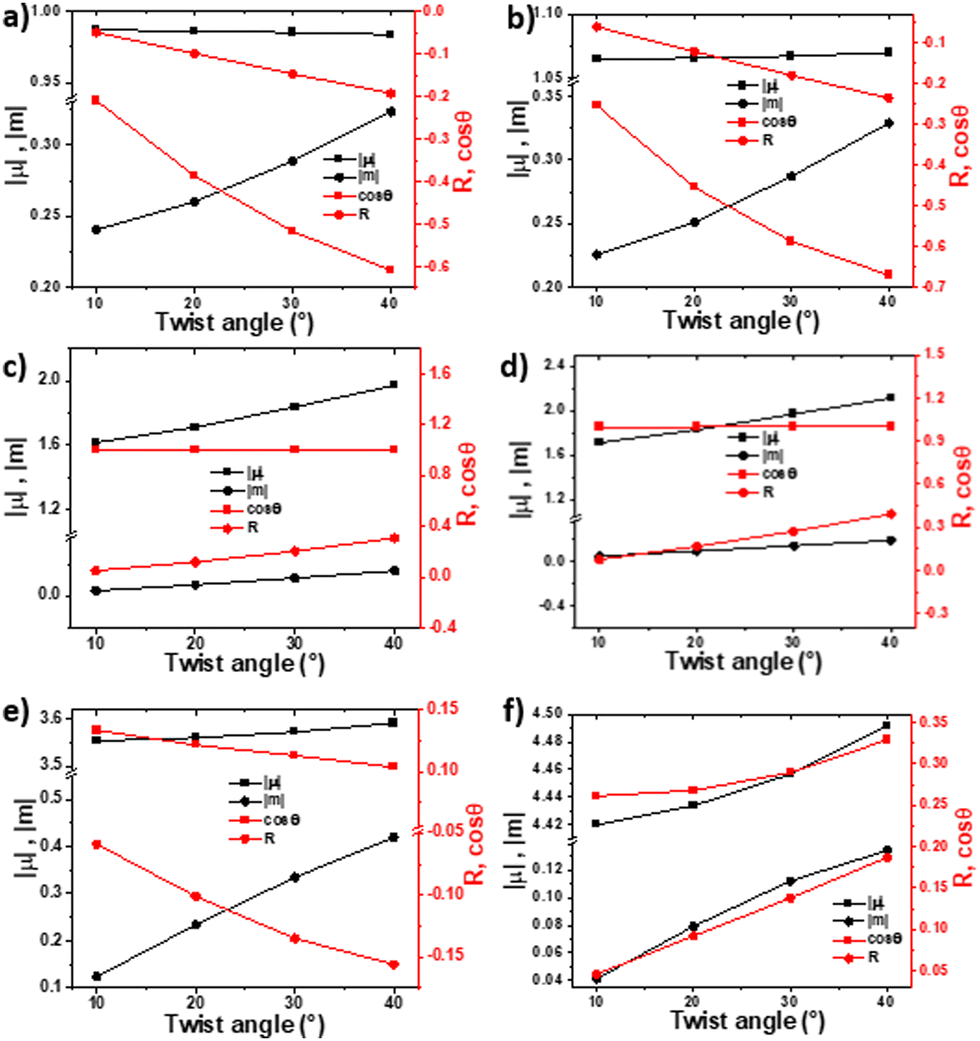 | ||
| Fig. 2 Computed (TDDFT-CAMB3LYP-6-31G(d)) transition dipole moments (left axis) and the rotational strength (R) and cos(θ) (right axis) at the optimized (a) S0 of 1-Cn, (b) S1 of 1-Cn (c) S0 of 3-Cn and (d) S1 of 3-Cn, (e) S0 of 4-Cn and (f) S1 of 4-Cn. | ||
For helically chiral 1-Cn, the increase in R is non-linear but becomes more significant as the degree of twist increases. In contrast, for 3-Cn, which exhibits both axial and helical chirality, R increases linearly with increasing twist. This can be explained by two opposite causes: the increase in the end-to-end anthracene twist, and decrease in the axial twist of the central phenyl-rings. However, for any given end-to-end twist, the value of R is larger for 3-Cn compared with 1-Cn. This can be explained by the combined contribution of axial and helical twisting on the overall value of R. Despite larger conjugation length, the dimer, 4-Cn is expected to show low rotational strengths, since the angle between μ and m is larger, resulting in relatively small cos(Θ).
Fig. 3 displays the photophysical properties of representative members of 1,3,4-Cn (for the entire series, see ESI†). The absorption and emission spectra are presented in the upper panels, while the ECD and CPL spectra are presented in the lower panels of each pair. Both 1-Cn and 2-Cn produce similar absorption spectra, with the typical vibronic structure of the 1La band of acenes in the 420–490 nm range (Fig. 3a, upper panels). For 3-Cn, the vibronic structure is slightly broader, which can be ascribed to the additional degrees of freedom provided by the axial twisting (Fig. 3b, upper panel). Finally, 4-Cn (Fig. 3c, upper panel) shows a bathchromically-shifted 1La band extending up to 520 nm, which can be attributed to the extended conjugation provided by the diacetylene spacer.
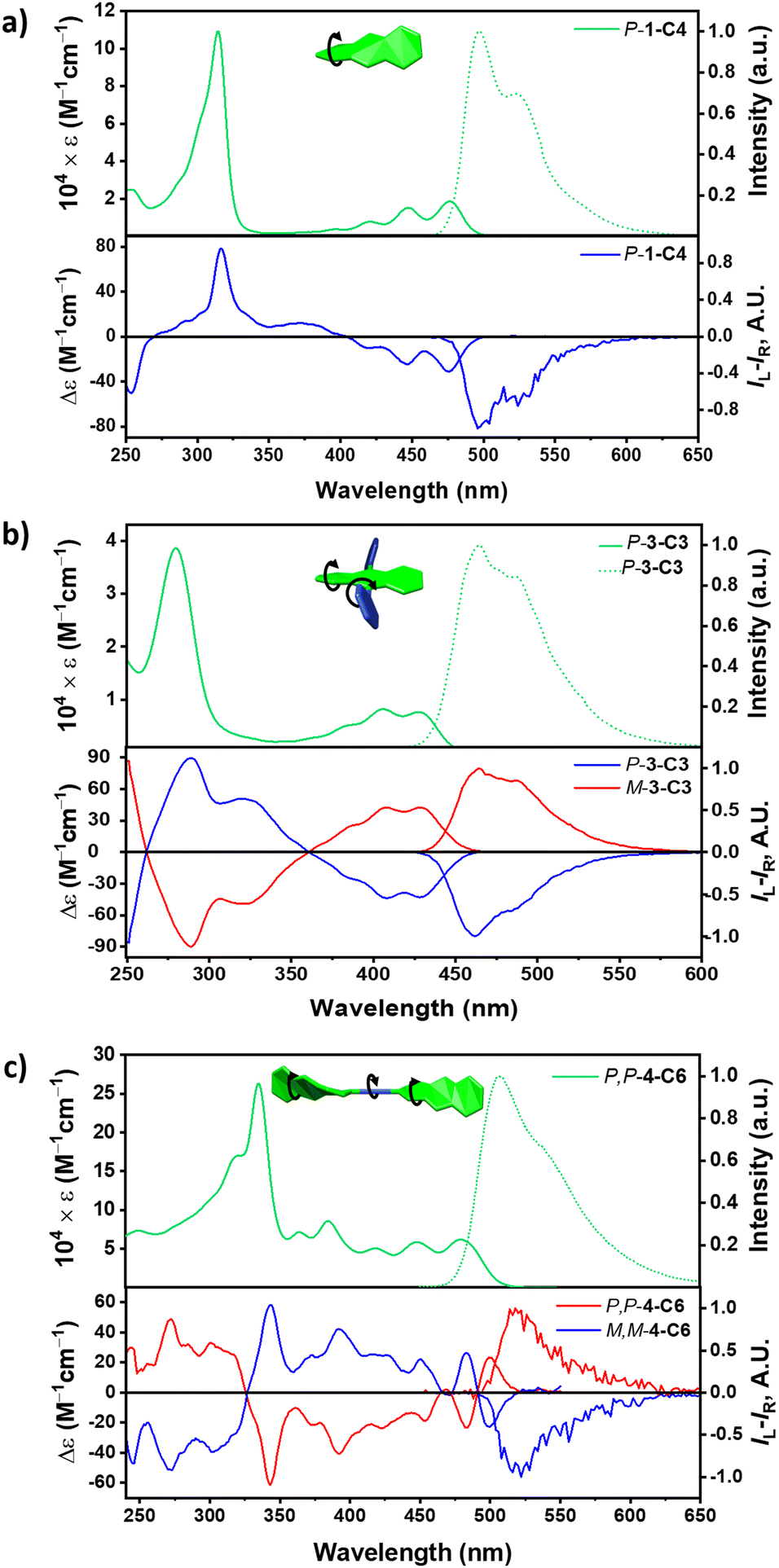 | ||
| Fig. 3 Absorption and emission spectra (upper panel of each pair) and ECD and CPL spectra (lower panel of each pair) for (a) 1-C4, (b) 3-C3 and (c) 4-C6. | ||
The emission spectra are similar for all the compounds, and the differences between members of the series are similar to those observed in the absorption spectra, namely sharper vibronic shoulders for shorter tethers and a bathochromic shift with twisting. Small Stokes shift (0.1–0.3 eV) indicates that no significant structural reorganization takes place upon excitation. This also applies to the dimer 4-Cn, which has a rotatable diacetylene linker between the two chromophores. We note that for all the compounds, the fluorescence quantum efficiency decreases with twisting, as has previously been explained by faster intersystem crossing.21 Being the most rigid members, 1-Cn and 2-Cn display the most pronounced vibronic structures.
We have previously observed that the maximal molar circular dichroism (Δεmax) increases linearly with increasing twist angle for 1-Cn, 2-Cn and 3-Cn.19,24 We note that a similar trend is observed for the longer dimeric 4-Cn. It is interesting to note that while values of Δεmax are higher for 4-Cn, this increase correlates with the increased number of chromophores, thus its anisotropy factor, gabs, is similar to the values found for 1-Cn and 2-Cn. In contrast to the 1–3-Cn series, the ECD spectrum of 4-Cn displays a distinct Cotton effect, with two opposite signs at 504 and 487 nm, which can result from the excitonic coupling between the two twistacenes connected by the diacetylene bridge.17 Upon cooling to −40 °C, 4-Cn shows increase in the intensity of Δε, which can be mostly associated with the rotation around the diacetylene bond.
The CPL for all compounds is displayed in the lower right panels of Fig. 3a–c, in the lower panel of each pair. As expected, the CPL signals of all the compounds are identical with those obtained from the lowest energy transition, indicating that the S1 → S0 transition is of the same nature as the S0 → S1 transition, as expected for rigid chromophores. This is true also for the dimer, 4-Cn, which indicates that despite the flexible diacetylene linker, the geometry is similar for both S0 and S1 states. We note that 4-Cn shows the most intense brightness as it consists of the largest ε and relatively strong quantum efficiency (Table S1, see ESI†).
The degree of chirality is often expressed by the absorption and emission anisotropy factors, gabs and glum, respectively, where gabs = Δε/ε and glum = 2(IL − IR)/(IL + IR), where IL and IR are the emission intensities of left- and right-handed circularly polarized light, respectively. Fig. 4a and b display the gabs and glum values for 1–4-Cn as a function of backbone twist. For all cases, the anisotropy factors increase with backbone twisting. The lowest gabs values are for 2-Cn, which has triazole linkers. This can be explained by 2-Cn having the shortest conjugation length, given the relation between effective conjugation length and anisotropy factor.7,10 In particular, it is interesting to note that 4-Cn, despite having the longest effective conjugation, has anisotropy factors similar to those of 1-Cn and 2-Cn. This can be explained by the angles of the calculated transition dipole moments for the lowest energy transition of 4-Cn (71–75°) being unable to yield maximal dissymmetry factors because they are neither parallel nor antiparallel. In contrast, in parent acenes these transition dipole moments are antiparallel regardless of the twisting angle, which should yield the maximal value.23
 | ||
| Fig. 4 Anisotropy factors (a) glum and (b) gabsversus backbone twist in 1–4-Cn, and (c) their glumversus gabs. | ||
Compound 3-Cn, possessing both axial and helical chirality, stands out with significantly higher gabs (3–5 × 10−3) and glum (1.8–2.5 × 10−3) values, although these values show a dependence on backbone twist similar to that observed for the other compounds. Fig. 4c displays the ratio glum/gabs that was previously formulated by Mori as indicating the nature of the specific class of molecules.26 Plotting the value for all twistacene members on the same graph indicates good correlation (r2 = 0.97) and shows that the average ratio of glum/gabs is 0.52 for the whole series. This value is slightly lower than that observed for helicenes (0.61). A potential reason for the lower ratio in our twistacene series is the increased degrees of freedom arising from the phenyl side groups, which induce larger conformational change upon excitation. However, overall, the good correlation between all members of the 1–4-Cn series suggests that the glum/gabs value of 0.52 is an intrinsic property of twisted acenes.
In summary, this work presents the first computational and experimental study of the effect of helical and axial chirality on CPL in twisted acenes. The glum values range between 10−5 and 5 × 10−3, depending on the degree of end-to-end twisting, conjugation, and whether there is a combination of axial and helical chirality. We find that increasing the end-to-end twist increases CPL in all cases, with the increase arising mostly from a steady increase in the magnetic transition dipole moment. Extending conjugation by using a diacetylene linker, while producing a Cotton effect, does not result in a noticeable strengthening of the chiroptical properties, which was explained by the angle formed between the electric and magnetic dipole moments. In contrast, combined axial and helical chirality significantly increases glum. We can conclude that inducing a backbone twist and addition axial chirality elements are a highly effective method for increasing the chiroptical properties of acenes. In contrast, extending the effective conjugation length does not necessarily leads to this result. As we observed that the rotational strength is highly dependent on the substituent position, it seems that the diagonal extension from the 1,5-position is not necessarily the optimal pathway, and alternative extension, for example from 9,10-positions might result in stronger rotational strength. The glum/gabs ratio is consistent for all the twistacenes studied, and was found to be 0.52, which is slightly lower than the value observed for helicenes. Overall, this study provides the first design principles for increasing CPL activity in twisted acenes.
O. G. acknowledges the support of European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement No. 850836, ERC Starting Grant “PolyHelix”) and by The Israel Science Foundation – FIRST Program (grant No. 1453/19). G.M. acknowledges the Israel Science Foundation (grant No 1257/22).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 1–12 CrossRef.
- L. E. MacKenzie and R. Pal, Nat. Rev. Chem., 2021, 5, 109–124 CrossRef CAS.
- L. Wan, J. Wade, X. Shi, S. Xu, M. J. Fuchter and A. J. Campbell, ACS Appl. Mater. Interfaces, 2020, 12, 39471–39478 CrossRef CAS PubMed.
- Y. He, S. Lin, J. Guo and Q. Li, Aggregate, 2021, 2, e141 Search PubMed.
- Circularly Polarized Luminescence of Isolated Small Organic Molecules, ed. T. Mori, Springer, Singapore, 2020 Search PubMed.
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem. – Eur. J., 2015, 21, 13488–13500 CrossRef PubMed.
- J. L. Greenfield, J. Wade, J. R. Brandt, X. Shi, T. J. Penfold and M. J. Fuchter, Chem. Sci., 2021, 12, 8589–8602 RSC.
- T. Kimoto, N. Tajima, M. Fujiki and Y. Imai, Chem. – Asian J., 2012, 7, 2836–2841 CrossRef CAS PubMed.
- W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Chem. Commun., 2019, 55, 13793–13803 RSC.
- Y. Nakai, T. Mori and Y. Inoue, J. Phys. Chem. A, 2012, 116, 7372–7385 CrossRef CAS PubMed.
- H. Tanaka, M. Ikenosako, Y. Kato, M. Fujiki, Y. Inoue and T. Mori, Commun. Chem., 2018, 1, 1–8 CrossRef CAS.
- B. C. Baciu, P. J. Bronk, T. de Ara, R. Rodriguez, P. Morgante, N. Vanthuyne, C. Sabater, C. Untiedt, J. Autschbach, J. Crassous and A. Guijarro, J. Mater. Chem. C, 2022, 10, 14306–14318 RSC.
- P. Sumsalee, L. Abella, T. Roisnel, S. Lebrequier, G. Pieters, J. Autschbach, J. Crassous and L. Favereau, J. Mater. Chem. C, 2021, 9, 11905–11914 RSC.
- J. L. Greenfield, E. W. Evans, D. Di Nuzzo, M. Di Antonio, R. H. Friend and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 10344–10353 CrossRef PubMed.
- H. Kubo, T. Hirose, T. Nakashima, T. Kawai, J. Hasegawa and K. Matsuda, J. Phys. Chem. Lett., 2021, 12, 686–695 CrossRef CAS PubMed.
- T. Hasobe, in Circularly Polarized Luminescence of Isolated Small Organic Molecules, ed. T. Mori, Springer, Singapore, 2020, pp. 99–116 Search PubMed.
- K. Dhbaibi, L. Favereau, M. Srebro-Hooper, C. Quinton, N. Vanthuyne, L. Arrico, T. Roisnel, B. Jamoussi, C. Poriel, C. Cabanetos, J. Autschbach and J. Crassous, Chem. Sci., 2020, 11, 567–576 RSC.
- J. Crassous, in Circularly Polarized Luminescence of Isolated Small Organic Molecules, ed. T. Mori, Springer, Singapore, 2020, pp. 53–97 Search PubMed.
- A. Bedi, L. J. W. Shimon and O. Gidron, J. Am. Chem. Soc., 2018, 140, 8086–8090 CrossRef CAS PubMed.
- A. Bedi and O. Gidron, Acc. Chem. Res., 2019, 52, 2482–2490 CrossRef CAS PubMed.
- P. Malakar, V. Borin, A. Bedi, I. Schapiro, O. Gidron and S. Ruhman, Phys. Chem. Chem. Phys., 2022, 24, 2357–2362 RSC.
- A. Bedi, R. Carmieli and O. Gidron, Chem. Commun., 2019, 55, 6022–6025 RSC.
- A. Bedi and O. Gidron, Chem. – Eur. J., 2019, 25, 3279–3285 CrossRef CAS PubMed.
- A. Bedi, A. Manor Armon, Y. Diskin-Posner, B. Bogosalvsky and O. Gidron, Nat. Commun., 2022, 13, 451 CrossRef CAS PubMed.
- N. Berova, L. D. Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc07074j |
| ‡ Authors contributed equally. |
| § Present address: Department of Chemistry, SRM Institute of Science and Technology, Kattankulathur, 603203, Tamil Nadu, India. |
| This journal is © The Royal Society of Chemistry 2023 |