
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anion–π interaction inside the polyanionic Mo132O372 cage with hydrophobic inner space†
Chinatsu
Murata
a,
Jaesob
Shin
a and
Katsuaki
Konishi
 *ab
*ab
aGraduate School of Environmental Science, Hokkaido University, North 10 West 5, Sapporo 060-0810, Japan. E-mail: konishi@ees.hokudai.ac.jp
bFaculty of Environmental Earth Science, Hokkaido University, North 10 West 5, Sapporo 060-0810, Japan
First published on 27th January 2023
Abstract
In this paper, we provide experimental evidence to indicate that the polyanionic Mo132O372 cage with a hydrophobic inner nanospace has a unique capability to participate in anion–π interactions by showing a preference for electron-deficient mono-substituted benzenes over non-electron-deficient guests in inclusion.
It is well known that π-mediated intermolecular forces involving aromatic moieties play critical roles in many chemical/biological processes.1 Aromatic rings are generally π-electron rich, which facilitates the attractive interactions with cations and H atoms of δ+ character, which are respectively known as cation–π and X–H⋯π interactions (X = C, N).2–6 Besides, it has been also shown that highly electron-deficient aromatic rings, such as perfluorinated triazines, have δ+ character to interact with anions, which has been identified by the SCXRD structures coupled with theoretical consideration. However, for such anion–π interactions, examples of interacting counterparts have been limited to conventional monoanions, such as halide anions.7–12 In this respect, the use of polyvalent anions is interesting as it could modulate or enhance the anion–π interactions, but so far such aspects have been unexplored. In this paper, we report the first experimental evidence of anion–π interactions within the caged Mo132O372 polyoxometalate polyanion.
Discrete cage architectures have attracted continuing attention since the confined environment inside endows unusual chemical properties and behaviours that are not found in open bulk space.13,14 The inorganic host molecule we used here is Keplerate-type Mo oxide polyoxometalate anion [Mo132O372(L)30(H2O)72]42− ({Mo132}) (Fig. 1),15,16 which has a nanoscale inner void space within the cage framework composed of 12 pentagonal {(Mo)Mo5O21} units and 30 bridging Mo2O4 units. To note, the inner space can communicate with the exterior environment through the Mo9O9 macrocyclic pores in the inorganic framework (Fig. 1a), through which small molecules can penetrate the inner void. In addition, the interior surface is mostly covered by coordinating ligands (L), and hence the inner environment can be tailored by the choice of the L ligands. The modification with alkylcarboxylate ligands offers a micelle-like feature with a hydrophilic outer surface and hydrophobic interior surface, allowing the incorporation of organic guests in aqueous solution.17–22 Examples of the guests range from quaternary ammonium ions,19 benzenes,20 and alkanols,21 to alkanethiols,22 but these examples are case-studies and no comprehensive elucidation has been established. In this work, we investigated the binding affinities of the acetate-type {Mo132} cluster (1) to a series of mono-substituted benzenes and found notable positive effects of the electron-withdrawing substituents conjugated with the aromatic system in accommodation, demonstrating the possibility of the involvement of anion–π interactions.
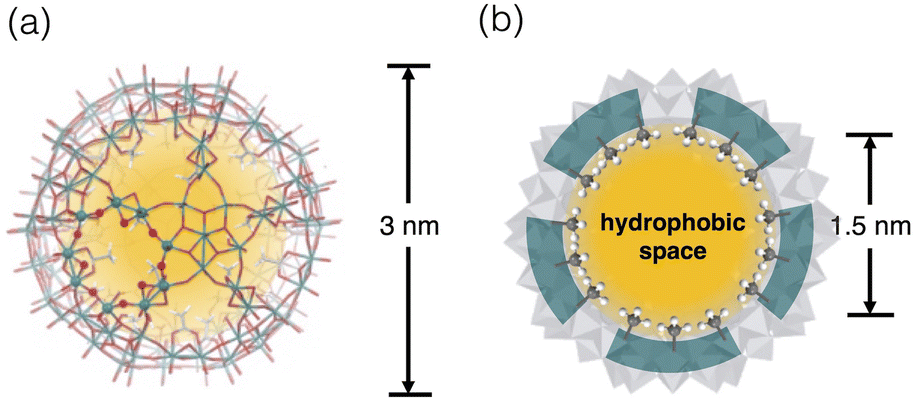 | ||
| Fig. 1 (a) X-Ray structure of [Mo132O372(OCOCH3)30(H2O)72]42− (1) highlighting one Mo9O9 macrocycle shown in ball and stick presentation. (b) Schematic presentation of the Me-rich hydrophobic inner space of 1. | ||
As mentioned, [Mo132O372(OCOCH3)30(H2O)72]42− (NH4 salt, 1) can extract benzene into its inner space in water. For example, the 1H NMR spectrum of 1 (as-crystallized form) dissolved in D2O after the treatment with excess benzene gave a signal at δ 5.73, together with that of free benzene extracted into the bulk water phase (δ 7.39) (Fig. S1a, ESI†). DOSY measurements showed that the diffusion coefficient of the former signal was comparable to that of the acetate ligand but was smaller than that of free benzene, indicating that it is assignable to benzene included within the inner space of 1. Here we evaluated the affinities of a series of mono-substituted benzenes to 1 in water based on the amounts of the trapped guests under thermodynamic conditions. Since most of the guests examined have limited solubilities in water, the NMR spectra were measured at 96 h after mixing with the guests to ensure the attainment of the equilibrium state (Fig. S1, ESI†). As an example, time-course monitoring experiments showed that the inclusion of benzene almost reached saturation after 12 h (Fig. S2, ESI†). Thus, it would be reasonable to compare the amounts of the trapped guests after 96 h to assess the static environment of the internal space of 1.
Fig. 2 summarizes the results when common mono substituted benzenes were utilized as guests. The trapped amounts vary significantly, where notably high affinities are observed when the substituent has a strongly electron-withdrawing character (Fig. 2b). For example, for the benzenes modified with a F, COCH3, CN, or NO2 group, the uptake amounts per host (r = [G]t/[1]0) were in the range of 9–14, and were obviously larger than and that for simple benzene (r = 5.6). On the other hand, the other guests with an electron-donating or less electron-withdrawing group were trapped comparably (r = 4–7) to benzene. From the volume estimated from the crystal structure (Fig. S3, ESI†), benzonitrile and nitrobenzene were fairly packed to occupy nearly 90% of the inner void sphere. The inclusion of these aromatic guests may be driven by the hydrophobic effect of the methyl-rich interior surface of 1, but it does not appear a primary factor considering that the high-affinity guests are less lipophilic than benzene, as shown in the octanol/water partition coefficients (Table S1, ESI†).23 Thus, other factors must be considered to account for the difference.
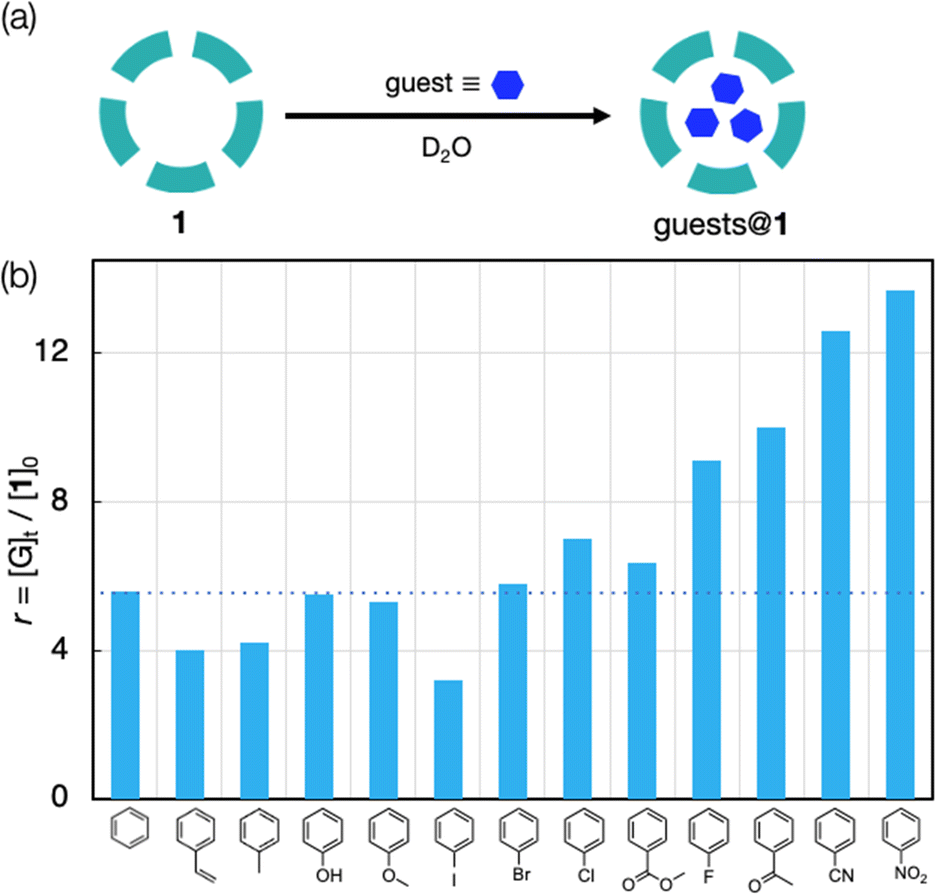 | ||
| Fig. 2 (a) Schematic illustration of guest inclusion within the inner space of 1. (b) Amounts of mono-substituted benzenes included within the inner space (r = [G]t/[1]0) after 1 (0.1 mM in D2O) was treated with neat guests (200 molar equiv.) for 96 h at room temperature. | ||
It should also be addressed that the observed r values and the trend of the guest preference are obviously different from those obtained by Neumann et al. in a similar extraction system with the {Mo132} host.20 For example, the r value for benzene in their system (∼7.8) is apparently larger than that in the present study (5.6, Fig. 2b), while those for toluene and chlorobenzene (0.5 and 3.6) are substantially smaller than ours (4.2 and 7.0, Fig. 2b). This discrepancy may be due to the experimental conditions, which differ in several points. Among them, the counter cation seems the most likely factor: they used the Na+ salt, while our system used the NH4+ salt. At the present stage, it is not easy to elucidate how the counter cations are involved in the guest inclusion processes. Nevertheless, the effects of counter cations are interesting and would be worthy of future investigation.
As mentioned, the high-affinity guests commonly have a strongly electron-withdrawing substituent, which leads to highly polar character. To obtain further insights, we next investigated the relationship between the dipole moments and inclusion activities of the guests,23 where the guest-occupying volumes (Table S1, ESI†) were used to assess the affinities considering the size difference of the guest molecules. As shown in Fig. 3, the plots give a good correlation where the inclusion activities are enhanced with the increase of the dipole moment. The overview of the plots reveals that the guests can be categorized into two groups, A and B. The guests in category A have relatively small dipole moments, where a transition from categories A to B was observed between μ = 2 and 3 D. Thus, the substituents of the high-affinity guests in category B can be characterized by their highly polar characters. With respect to the attractive forces contributing to the accommodation of these aromatic guests, it was claimed that π-electrons can interact with the Me-rich interior surface via dispersion-based CH⋯π interactions.20 Accordingly, the enhanced affinities of the category B guests may be due to the additional interactions associated with their polarity. In this relation, we further set up several control systems to investigate the effects of the polar substituents. Here, benzonitrile (PhCN) was chosen as the representative and was compared with related nitrile guests, araliphatic 2-phenylacetonitrile (BnCN) and aliphatic cyclohexanecarbonitrile (CyCN). They both are highly polar to present large dipole moments (μ = 3.5 D and 3.8 D, respectively) similarly to PhCN.23 If the guest polarity is the predominant factor to govern the incorporation, they should be trapped similarly to PhCN. However, they showed much lower affinities than PhCN (Fig. 4a). The considerably low affinity to CyCN (r = 1.2) implies that the contribution of the polar C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group alone is not significant, and the cooperation with the aromatic moiety is critically involved in the interaction with the interior surface. It should also be noted that BnCN, though having both an aromatic ring and polar CN group, was also less active (r = 3.0), indicating that the scission of π-electronic communication by an sp3 spacer causes a considerable drop of the binding affinity. Thus, the extended π-system resulting from the conjugation between the CN group and the aromatic ring is likely to contribute to the efficient inclusion of PhCN. The polar nature seems also an important factor to facilitate the interaction with the interior surface. In fact, phenylacetylene (PA), a less polar analogue (μ = 0.7 D) of PhCN (Fig. 4a), and styrene with a π-conjugated vinyl group (Fig. 2) behaved similarly to benzene.
N group alone is not significant, and the cooperation with the aromatic moiety is critically involved in the interaction with the interior surface. It should also be noted that BnCN, though having both an aromatic ring and polar CN group, was also less active (r = 3.0), indicating that the scission of π-electronic communication by an sp3 spacer causes a considerable drop of the binding affinity. Thus, the extended π-system resulting from the conjugation between the CN group and the aromatic ring is likely to contribute to the efficient inclusion of PhCN. The polar nature seems also an important factor to facilitate the interaction with the interior surface. In fact, phenylacetylene (PA), a less polar analogue (μ = 0.7 D) of PhCN (Fig. 4a), and styrene with a π-conjugated vinyl group (Fig. 2) behaved similarly to benzene.
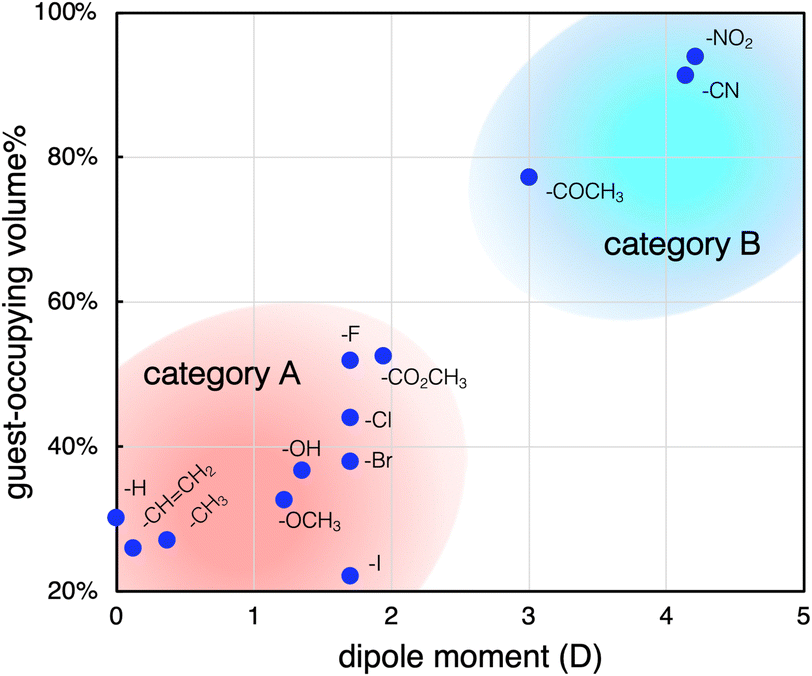 | ||
| Fig. 3 Plots of the amounts of guest-occupying volume vs. the dipole moment of the guests. Conditions are given in the caption of Fig. 2. | ||
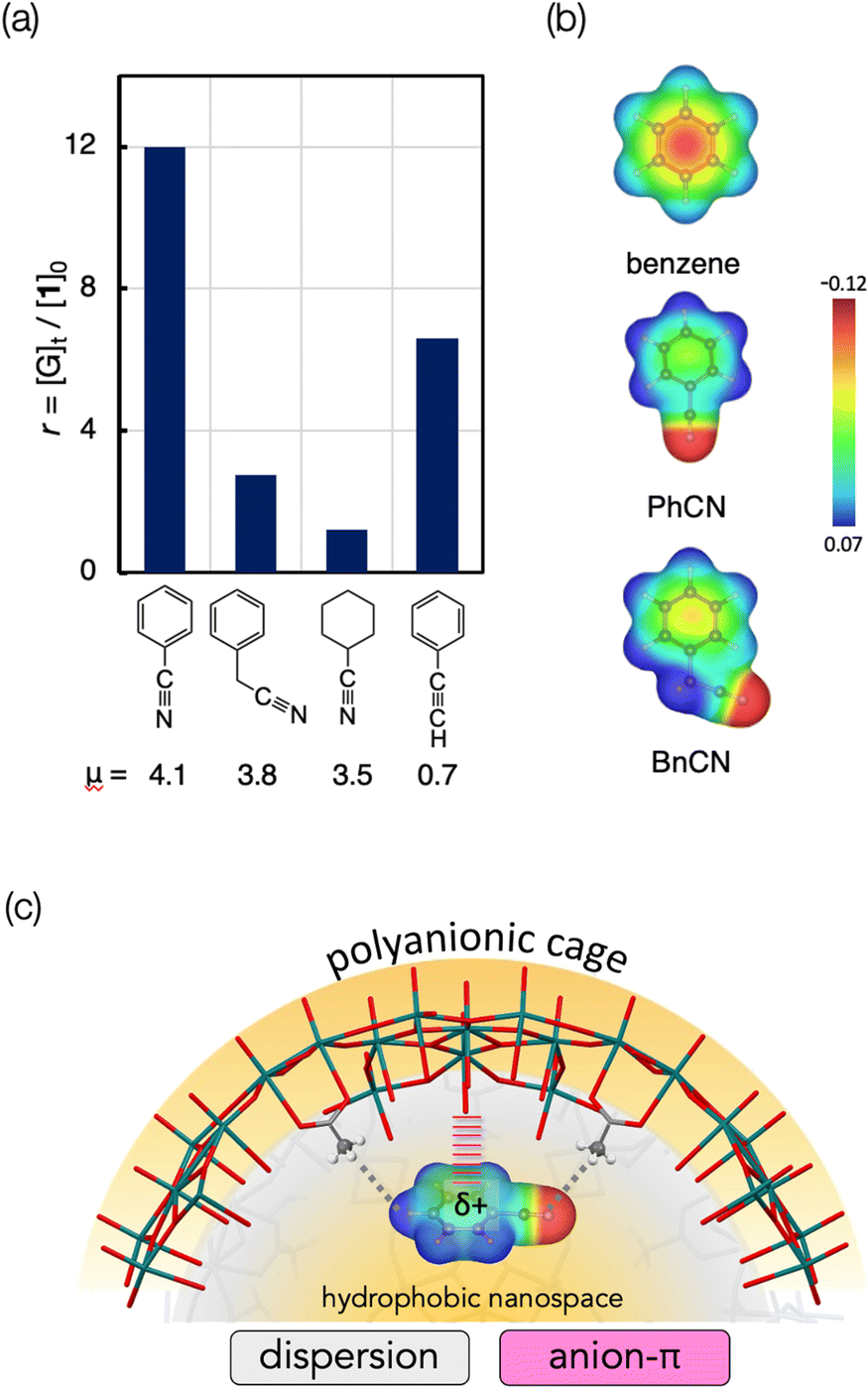 | ||
| Fig. 4 (a) Inclusion of nitrile guests (PhCN, BnCN, CyCN) and phenylacetylene (PA) by 1 under the conditions given in the caption of Fig. 2. (b) Plot of electrostatic potentials of benzene, PhCN and BnCN mapped on electron density isosurfaces (0.05 e a.u.−3) (c) Schematic illustration of the interaction of the interior surface of 1 with PhCN. | ||
From the above results, the high affinities to the category B guests can be ascribed to the unique capability of polar π-electron systems conjugated with CN, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and NO bonds of the substituents. As mentioned, the Me-rich inner environment surrounded by bulk water may facilitate the capture of benzenes through London dispersion interactions assisted by hydrophobic effects. As the additional interactions, ion–dipole and dipole–dipole interactions may be considered, because the {Mo132} host has many polar Mo–O and MoO bonds and a massive minus charge (42−). However, as seen in the cases of BnCN and CyCN, the polar groups alone were not so effective to boost the inclusion, implying that the dipole-based van der Waals force is not predominant.
O, and NO bonds of the substituents. As mentioned, the Me-rich inner environment surrounded by bulk water may facilitate the capture of benzenes through London dispersion interactions assisted by hydrophobic effects. As the additional interactions, ion–dipole and dipole–dipole interactions may be considered, because the {Mo132} host has many polar Mo–O and MoO bonds and a massive minus charge (42−). However, as seen in the cases of BnCN and CyCN, the polar groups alone were not so effective to boost the inclusion, implying that the dipole-based van der Waals force is not predominant.
Instead, anion–π interaction can be considered as a plausible additional force to account for the high affinities of the category B guests. Upon conjugation with the electron-withdrawing groups, the π-electron density of the benzene ring is decreased, affording weak δ+ feature in the aromatic system. In fact, as shown in the electrostatic potential profiles (Fig. 4b), the electron density of the aromatic ring of PhCN is rather sparse when compared with benzene and BnCN, reflecting the effective electron-withdrawing ability of the π-conjugated CN group of PhCN. The π surface energies estimated from the literature are 4 and −15 kcal mol−1 for benzene and PhCN, respectively.24 Accordingly, the benzene ring of PhCN is substantially electron deficient to display a slightly positive character, thus being eligible to interact with anionic species. Consequently, as illustrated in Fig. 4c, anion-π interactions may be involved as the attractive interactions in addition to the inherent dispersion interactions, leading to the efficient inclusion of the category B guests. Among the category A guests, the order of the inclusion preference for the halogenated guests (F > Cl > Br > I, Fig. 3) may reflect the involvement of the anion–π interaction.
It should be noted that anion–π interactions have been reported mostly for highly electron deficient aromatic rings with multiple electron-withdrawing groups (e.g., hexafluorobenzene). However, the aromatic rings of the category B guests (e.g., PhCN, PhNO2) are not so electron-deficient when compared with the above anion–π-active guests, and hence the interactions with conventional anions would be very weak. In fact, the examples reported so far have been very limited.25,26 One exception is the macrocycle containing multiple nitro-substituted arenes, whose anion complex has been identified by SCXRD. In this respect, it would be reasonable to assume that the polyanionic feature (42− anion) of the inorganic framework plays a critical role, which may facilitate anion–π interactions even with slightly electron-deficient aromatic guests.
Since the first discovery of anion–π interactions traced back to several decades ago, they have attracted continuing attention in supramolecular chemistry but have not yet been fully elucidated. In this paper, we systematically studied the inclusion of mono-substituted benzenes into the inner hydrophobic nanospace of a polyanionic caged framework of Mo132O372. Our results clearly show the preference for electron-deficient aromatic guests with a polar electron-withdrawing group, which implies the involvement of anion–π interactions in the host–guest interaction within the inner space. Considering that the degree of electron deficiency of the guests is not high, this work presents an example of a novel type of anion–π interaction, which is offered by a polyanionic host. The SCXRD structural determination to identify the interaction at the atomic level as well as theoretical studies are worthy of further investigation.
This research was partly supported by the Photo-excitonix Project in Hokkaido University, MEXT/JSPS Grants-in-Aid to KK (KAKENHI 21H0468301), and JST SPRING to CM (JPMJSP2119).
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808 CrossRef CAS PubMed.
- M. Nishino, Y. Umezawa, M. Hirota and Y. Takeuchi, Tetrahedron, 1995, 51, 8665 CrossRef.
- C. M. Jennifer and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef PubMed.
- S. Tsuzuki, Annu. Rep. Sect. C: Phys. Chem., 2012, 108, 69 RSC.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100 CrossRef CAS PubMed.
- S. Yamada, Chem. Rev., 2018, 118, 11353 CrossRef CAS PubMed.
- B. L. Schottel, H. T. Chifotides, M. Shatruk, A. Chouai, L. M. Perez, J. Basca and K. R. Dunbar, J. Am. Chem. Soc., 2006, 128, 5895 CrossRef CAS PubMed.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68 RSC.
- S. Guha, F. S. Goodson, L. J. Corson and S. Saha, J. Am. Chem. Soc., 2012, 134, 13679 CrossRef CAS PubMed.
- D. X. Wang and M. X. Wang, J. Am. Chem. Soc., 2013, 135, 892 CrossRef CAS PubMed.
- M. Giese, M. Albrecht and K. Rissanen, Chem. Commun., 2016, 52, 1778 RSC.
- B. Jiang, W. Wang, Y. Zhang, Y. Lu, C. W. Zhang, G. Q. Yin, X. L. Zhao, L. Xu, H. Tan, X. Li, G. X. Jin and H. B. Yang, Angew. Chem., Int. Ed., 2017, 56, 14438 CrossRef CAS PubMed.
- H. Takezawa and M. Fujita, Bull. Chem. Soc. Jpn., 2021, 94, 2351 CrossRef CAS.
- R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244 CrossRef CAS PubMed.
- A. Müller, E. Krickemeyer, H. Bogge, M. Schmidtmann and F. Peters, Angew. Chem., Int. Ed., 1998, 37, 3360 Search PubMed.
- A. Müller and P. Gouzerh, Chem. Soc. Rev., 2012, 41, 7431 RSC.
- W. Deng, Q. Zhang and Y. Wang, Dalton Trans., 2012, 41, 9817 RSC.
- R. W. Pow, Z. L. Sinclair, N. L. Bell, N. Watfa, Y. M. Abul-Haija, D. L. Long and L. Cronin, Chem. – Eur. J., 2021, 27, 12327 CrossRef CAS PubMed.
- N. Watfa, D. Melgar, M. Haouas, F. Taulelle, A. Hijazi, D. Naoufal, J. B. Avalos, S. Floquet, C. Bo and E. Cadot, J. Am. Chem. Soc., 2015, 137, 5845 CrossRef CAS PubMed.
- B. B. Sarma, L. Avram and R. Neumann, Chem. – Eur. J., 2016, 22, 15231 CrossRef PubMed.
- C. Schaffer, A. M. Todea, H. Bogge, O. A. Petina, D. Rehder, E. T. Haupt and A. Müller, Chem. – Eur. J., 2011, 17, 9634 CrossRef PubMed.
- R. W. Pow, W. Xuan, D. L. Long, N. L. Bell and L. Cronin, Chem. Sci., 2020, 11, 2388 RSC.
- CRC handbook of chemistry and physics, ed. D. R. Lide, CRC Press, 81st edn, 2000 Search PubMed.
- S. E. Wheeler, J. Am. Chem. Soc., 2011, 133, 10262 CrossRef CAS PubMed.
- G. Gil-Ramírez, E. C. Escudero-Adán, J. Benet-Buchholz and P. Ballester, Angew. Chem., Int. Ed., 2008, 47, 4114 CrossRef PubMed.
- L. Adriaenssens, C. Estarellas, A. Vargas Jentzsch, M. Martinez Belmonte, S. Matile and P. Ballester, J. Am. Chem. Soc., 2013, 135, 8324 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06875c |
| This journal is © The Royal Society of Chemistry 2023 |