
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly electron deficient diketopyrrolopyrroles†
Joshua
Humphreys‡
 a,
Ferdinando
Malagreca‡
ab,
Paul A.
Hume
c,
E. Stephen
Davies
d,
Stephen P.
Argent
d,
Tracey D.
Bradshaw
b and
David B.
Amabilino
*ae
a,
Ferdinando
Malagreca‡
ab,
Paul A.
Hume
c,
E. Stephen
Davies
d,
Stephen P.
Argent
d,
Tracey D.
Bradshaw
b and
David B.
Amabilino
*ae
aGSK Carbon Neutral Laboratories for Sustainable Chemistry, School of Chemistry, University of Nottingham, Triumph Road, Nottingham, NG7 2TU, UK
bSchool of Pharmacy, University of Nottingham, University Park, NG7 2RD, UK
cMacDiarmid Institute for Advanced Materials and Nanotechnology and School of Chemical and Physical Sciences, Victoria University of Wellington, Wellington 6010, New Zealand
dSchool of Chemistry, University of Nottingham, University Park, NG7 2RD, UK
eInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Consejo Superior de Investigaciones Científicas, Campus Universitari de Bellaterra, Cerdanyola del Vallès 08193, Spain. E-mail: amabilino@icmab.es
First published on 3rd January 2023
Abstract
The synthesis, spectroelectrochemical and structural characteristics of highly electron-accepting diketopyrrrolopyrrole (DPP) molecules with adjoining pyridinium rings is reported, along with an assessment of their toxicity, which is apparently low. The compounds show reversible electrochemistry and in one subfamily a massive increase in molar extinction coefficient upon electrochemical reduction.
Electron acceptors are attractive as components in devices related with energy storage and capture, particularly redox-flow batteries1–3 and photovoltaic solar cells.4–6 Those based on bi-pyridinium species have received significant attention, in these and other areas,7 because of their electron-deficient character and tuneable redox properties.8,9 This family of compounds is of such interest because the parent 4,4′-bipyridinium and its derivatives are relatively easy to prepare and reduce.10 Tuning their properties can be achieved by changing the N-substituent and the counter anion,11,12 as well as adding electron-withdrawing units to the pyridinium rings.13 Another approach is to modify the linker between the rings to stabilize the reduced forms of the compounds and to modify the photophysical and photochemical properties.14 In parallel, diketopyrrolopyrrole (DPP) dyes and pigments are an increasingly investigated family of organic semi-conductor materials.15 Their intermolecular interactions, most notably π–π stacking and hydrogen bonding, influence charge carrier mobility in their materials.16–20 Typically, DPPs have been used as donor materials, habitually with thiophene as the aryl substituent, despite the fact that the materials were identified as being ambipolar.21 The DPP bearing pyridyl groups has been employed as an H2 gas and acid sensor,22,23 exploiting protonation of the pyridyl moiety, resulting in distinct optical and structural changes as well as a significant decrease in resistivity.
The combination of these two structural types was appealing to us for new organic materials. While some mono-pyridinium DPP derivatives have been reported,24 we are unaware of the more highly electron deficient symmetrical structures. In this work we show the synthesis of new bipyridinium systems linked by a DPP core unit (Scheme 1). The redox properties are modulated by the electron-withdrawing character of the DPP, and lead to very different absorption characteristics, a property tuneable through access to the different redox states.
 | ||
| Scheme 1 Synthesis of 4-2.2PF6, 4-3.2PF6, 3-2.2PF6, 3-3.2PF6. | ||
The desired bipyridinium DPPs 22+ and 32+ (Scheme 1) were obtained (see ESI†) from the known 3-pyridyl and 4-pyridyl DPP compounds 1.25 The quaternization of the nitrogen atoms of the pyridine ring required a relatively harsh environment for this type of pyridine alkylation reaction, at 150 °C for 5 days in the neat alkyl halide. We believe these conditions are necessary as a result of the low solubility of the starting material and the weak nucleophilic character of the pyridine nitrogen atoms, caused by the strong electron-withdrawing character of the DPP core. This hypothesis is supported by the reported pKa of the more soluble pyridine parent.26 The products were isolated as their hexafluorophosphate salts by counter anion exchange. They have improved solubility in common polar organic solvents compared with starting materials 1, an uncommon property for DPPs without alkylation at the nitrogen atom in the lactam ring.
Single crystals were obtained for three of the compounds that are the object of this study. They were grown by slow diffusion of water and methyl–ethyl ketone for 4-2.2PF6 (isolated as a hydrate) and slow evaporation of acetonitrile for 3-2.2PF6 and 3-3.2PF6 (isolated as an acetonitrile solvate). All molecules exhibit an essentially flat π-conjugated system across the pyridinium and DPP moieties in the solid-state (Fig. 1). They display dihedral angles between the DPP core and pyridinium rings of 6.3°, 3.1° and 2.9° for 4-2.2PF6, 3-2.2PF6 and 3-3.2PF6, respectively. There are close intramolecular contacts between the lactam carbonyl group and the pyridinium hydrogen atom closest to the core (2.20–2.26 Å). For 3-2.2PF6 there are also intermolecular contacts between pyridinium hydrogen atoms and the oxygen atom (ESI†). The N–H and carbonyl of the lactam of 4-2.2PF6 are bound to water solvate molecules in the solid. A cyclic dimer is formed that results in a continuous chain of hydrogen bonded molecules. The N–H and the proton at the 2-position of the pyridinium ring in 3-2.2PF6 interact with the anion, while the carbonyl is bound to the proton at the 5-position of a neighbouring pyridinium ring. The nitrogen atoms of the pyridinium ring in 3-3.2PF6 (they are oriented to the carbonyl group) display opposite orientation with respect to the DPP core compared with 3-2.2PF6 (they are oriented to the NH group, see ESI†) in the solid state. While the DPP cores have parallel orientation, there is no significant π–π stacking.
 | ||
| Fig. 1 Views of the molecular structures of 4-2.2PF6 (A) and 3-2.2PF6 (B) from their crystal structures (anions and water molecules, in the case of the former, are omitted for clarity). | ||
The redox properties of the compounds were studied with cyclic voltammetry (CV) on a glassy carbon electrode in DMF solution with tetra-n-butyl-ammonium hexafluorophosphate (TBAPF6; 0.2M) as supporting electrolyte. Three discrete reduction processes appear for 4-2.2PF6 and 4-3.2PF6 (ESI†). The first and second one electron reduction waves at approximately −0.50 V and −0.70 V (versus Fc+/Fc) appear reversible and correspond to the formation of the mono-cation radical and neutral species, respectively. These processes are reminiscent of those in paraquat (Ered1,1/2 = −0.83 V and Ered2,1/2 = −1.24 V vs. Fc+/Fc),13 albeit at much less negative potentials. The shift to less negative potentials is attributed to the electron-withdrawing effects of the DPP core. The third process, at −2.2 V, is assigned to reduction of the DPP core itself.27 This hypothesis is supported by density functional theory (DFT) calculations where the DPP core contributes to the singly occupied molecular orbital (SOMO) (ESI†). The nature of the substituent (n-hexyl or benzyl) on the nitrogen atoms of the pyridinium rings has little effect on the potential at which the redox processes take place in these molecules.
Compounds 3-2.2PF6 and 3-3.2PF6 display quite different electrochemical behaviour compared to their 4-pyridinium analogues. Only the first reduction, at approximately −0.90 V (versus Fc+/Fc), shows behaviour that appears reversible (ESI†). The subsequent reduction has no associated oxidation wave but instead increases the re-oxidation current at the first reduction, presumably a result of the oxidation of precipitated material at the electrode surface generated upon formation of the neutral species.
The DPP family of compounds generally have distinctive absorbance maxima between 400 and 550 nm, and emission in the range of 500 and 650 nm, with fluorescence quantum yields between 0.4 and 0.9.18 By contrast, the quaternary derivatives 4-2.2PF6 and 4-3.2PF6 display a bathochromic shift in UV-visible absorption maxima and broadened emission spectra with larger Stokes shifts (ESI†). Low emission quantum yields suggest the presence of an efficient fluorescence quenching mechanism that may involve intramolecular charge transfer (ICT). In comparison, 3-2.2PF6 and 3-3.2PF6 derivatives have blue shifted UV-visible absorption maxima; their fluorescence spectra are less broadened, have larger quantum yields and give Stokes shifts approximately half those of the 4-substituted analogues. These differences suggest that the fluorescence quenching mechanism operating in 4-2.2PF6 and 4-3.2PF6 is either absent or less pronounced for 3-2.2PF6 and 3-3.2PF6, making the 3-substituted derivatives more similar in character to the broader family of DPP compounds.
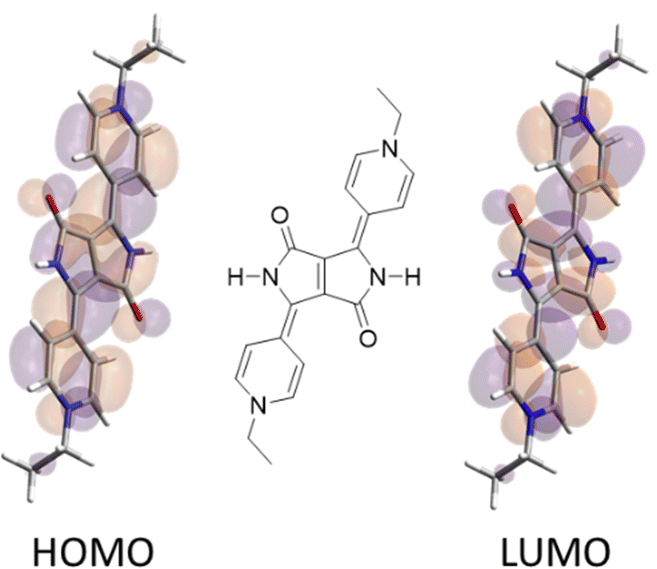
The UV-visible absorption profiles of the three redox states were investigated for 4-2.2PF6 and 4-3.2PF6 and 3-2.2PF6 and 3-3.2PF6 using spectroelectrochemistry. For the 4-substituted compounds, 4-2.2PF6 and 4-3.2PF6, the single electron reduction caused absorption bands at ∼280 nm and ∼580 nm to diminish whilst new bands at ∼640 nm and ∼775 nm emerged (Fig. 2). The only significant difference between the spectral profiles of these compounds when reduced was the relative intensity of these two bands (ESI†).
 | ||
| Fig. 2 UV-visible absorption monitoring of the first reduction process of 4-2.2PF6. Red line 4-2.2PF6, cyan line 4-2˙+·PF6. Coloured lines correspond to ε and grey lines show absorbance. Spectra were recorded in DMF containing TBAPF6 (0.2 M) as the supporting electrolyte at 273 K (A). UV-visible absorption monitoring of the second reduction process of 4-2.2PF6. Cyan line 4-2˙+·PF6, blue line 4-2. Coloured lines correspond to ε and grey lines show absorbance. Spectra were recorded in DMF containing TBAPF6 (0.2 M) as the supporting electrolyte at 273 K (B). | ||
The second reduction, generating a neutral species, caused the absorption band at ∼775 nm to diminish while the band at ∼640 nm increased significantly, together with the formation of a new band at ∼590 nm (Fig. 2). Upon reduction a large increase in extinction coefficient is noteworthy, with a value of ε that increases from ∼20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to ∼70000 M−1 cm−1 for the one-electron reduced species. The neutral 4-2 and 4-3 species have an ε value of 158600 and 276700 M−1 cm−1, respectively. This large increase in ε, we believe, is a result of the formation of the quinoidal form that leads to better overlap between ground and excited state orbitals in the DPP core, as confirmed by theoretical calculations (see below).
000 to ∼70000 M−1 cm−1 for the one-electron reduced species. The neutral 4-2 and 4-3 species have an ε value of 158600 and 276700 M−1 cm−1, respectively. This large increase in ε, we believe, is a result of the formation of the quinoidal form that leads to better overlap between ground and excited state orbitals in the DPP core, as confirmed by theoretical calculations (see below).
This kind of quinoidal state has been inferred elsewhere in another context.28 Nonetheless, the increase in absorption is quite extraordinary. It is not seen in bipyridinium derivatives, or DPPs that show the quinoidal form.28 The re-oxidation of both 4-substituted compounds generate the initial UV-visible spectral profile (ESI†) indicating chemical reversibility under these conditions.
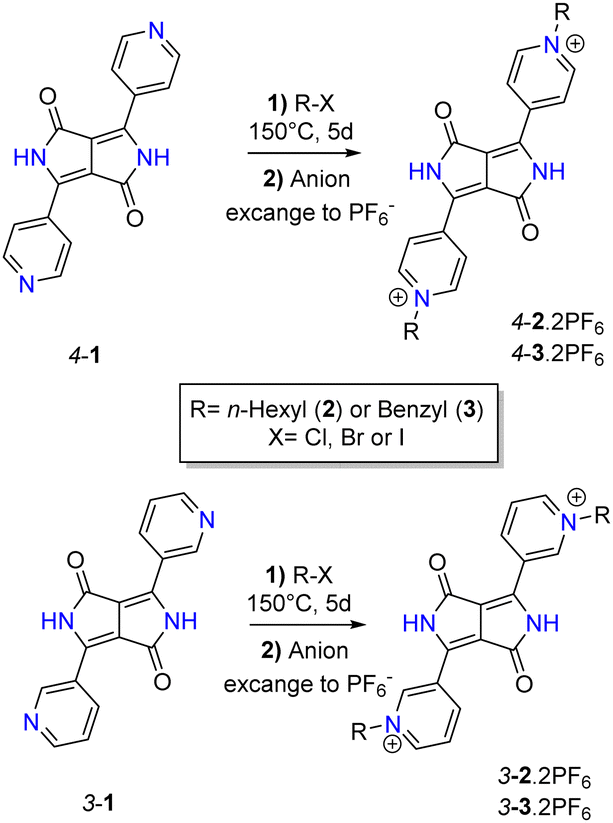
TD-DFT calculations capture the increased oscillator strength and changes in λmax on one- and two-electron reduction. The calculated excited states for the dication and neutral forms are well described by a single HOMO → LUMO transition. The close similarity between the dication LUMO and neutral HOMO suggests a quinoidal structure for the neutral (i.e. doubly reduced) form of 4-2 (Fig. 3). The low energy absorption band associated with the radical cation involves significant contributions from two transitions, the first resembling the dication transition, and the second resembling that of the neutral form.
 | ||
| Fig. 3 Calculated HOMO and LUMO of a simplified quinoidal form equivalent to the doubly reduced 4-22+ (with an ethyl substituent used for computational simplicity). | ||
The UV-visible absorbance spectra of one-electron reduced 3-2.2PF6 and 3-3.2PF6 shows a structured band, with peak maximum at 650 nm, that replaces a structured band at λmax 520 nm in the neutral compounds (Fig. 4), with this interconversion producing isosbestic points at ca. 450 and 550 nm. Re-oxidation to the original spectral profile of the dications was achieved but only slowly and after the application of a significant over-potential, which may indicate the involvement more complex electrochemical mechanisms during these longer timescale experiments.
 | ||
| Fig. 4 UV-visible absorption spectra showing the first reduction of 3-2.2PF6. Orange line 3-2.2PF6, blue line 3-2˙+·PF6. Colored lines correspond to ε and grey lines to absorbance. Spectra were recorded in DMF containing TBAPF6 (0.2 M) as the supporting electrolyte at 273 K. | ||
Electronic paramagnetic resonance (EPR) spectroscopy confirmed the presence of paramagnetic radical species for singly reduced 4-2˙+·PF6, 4-3˙+·PF6 and 3-3˙+·PF6 and the spectra show extensive hyperfine couplings (ESI†). The complexity of the EPR signals indicates delocalization of the unpaired electron over the whole π–framework which must include the pyridinium functionalities; a conclusion confirmed by MO calculations of the SOMO (ESI†).
The structural similarity of the compounds to paraquat made us concerned about their potential toxicity to researchers, as well as the compound's potential environmental impact. Therefore, we studied the effects of the more soluble 4-pyridinium compounds on two human-derived cell lines (A549 lung carcinoma and MRC-5 fibroblast), using paraquat as a standard for comparison.
MTT assays were performed at time of test agent addition and following 72 hours exposure to the compounds (see ESI† for details). While negligible activity is noted for the DPP pyridinium compounds (LC50 and GI50 values >100 μM in both cell lines), the GI50 values for paraquat are ∼5 μM against both A549 and MRC-5 cells. Therefore, growth inhibitory activity and toxicity of the title compounds is significantly less than paraquat under these conditions. Because of their characteristics, it will be of future interest to explore the effects that these compounds might have as mitochondrial probes,29 for example.
In conclusion, pyridinium DPP derivatives are synthetically accessible and are chemically and electrochemically stable, they can be reversibly converted into their radical species at potentials much higher than other bipyridinium species thanks to the presence of the DPP core, with large changes in their spectroscopic properties, making them very attractive electron acceptors. In particular, the 4-substituted analogues are excellent electron acceptors and present extraordinary increases in absorbance upon reduction, a full order of magnitude for analogous bands at around 600 nm. This feature also makes them promising candidates as electrochromic materials.30
This work was supported by the University of Nottingham through the Beacon of Excellence Propulsion Futures and the EPSRC through the LDMI DTP for funding. The Diamond Light Source Ltd (beamline I19) is thanked for access to collect diffraction data of crystals.
Conflicts of interest
There are no conflicts to declare.References
- J. F. Long, Q. Tang, Z. Lv, C. R. Zhu, X. K. Fu and C. B. Gong, Electrochim. Acta, 2017, 248, 1 CrossRef CAS.
- R. D. Pichugov, E. E. Makhaeva and M. L. Keshtov, Electrochim. Acta, 2018, 260, 139 CrossRef CAS.
- M. Kato, H. Sano, T. Kiyobayashi and M. Yao, ChemSusChem, 2020, 13, 2379 CrossRef CAS PubMed.
- T. Y. Li, C. Su, S. B. Akula, W. G. Sun, H. M. Chien and W. R. Li, Org. Lett., 2016, 14, 3386 CrossRef PubMed.
- X. Liu, D. Qin, Y. Fan, K. Li, D. Li and Q. Meng, Electrochem. Commun., 2007, 9, 1735 CrossRef CAS.
- H. Ye, X. Hu, Z. Jiang, D. Chen, X. Liu, H. Nie, S. J. Su, X. Gong and Y. Cao, J. Mater. Chem. A, 2013, 1, 3387 RSC.
- S. Sowmiah, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Org. Chem. Front., 2018, 5, 453 RSC.
- S. Hünig and H. Berneth, Top. Curr. Chem., 1980, 92, 1 CrossRef.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49 RSC.
- Y. Wang, M. Frasconi and J. F. Stoddart, ACS Cent. Sci., 2017, 3, 927 CrossRef CAS PubMed.
- (a) C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49 RSC; (b) W. W. Porter, T. P. Vaid and A. L. Rheingold, J. Am. Chem. Soc., 2005, 127, 16559 CrossRef CAS PubMed; (c) Y. Wang, S. Xu, F. Gao, Q. Chen, B.-B. Ni and Y. Ma, Supramol. Chem., 2013, 25, 344 CrossRef CAS; (d) L. Chen, F. Hartl, H. M. Colquhoun and B. W. Greenland, Tetrahedron Lett., 2017, 58, 1859 CrossRef CAS.
- L. Chen, H. Willcock, C. J. Wedge, F. Hartl, H. M. Colquhoun and B. W. Greenland, Org. Biomol. Chem., 2016, 14, 980 RSC.
- M. Berville, J. Richard, M. Stolar, S. Choua, N. Le Breton, C. Gourlaouen, C. Boudon, L. Ruhlmann, T. Baumgartner, J. A. Wytko and J. Weiss, Org. Lett., 2018, 20, 8004 CrossRef CAS PubMed.
- (a) P. R. Ashton, R. Ballardini, V. Balzani, A. Credi, M. T. Gandolfi, S. Menzer, L. Pérez-García, L. Prodi, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1995, 117, 11171 CrossRef; (b) R. Ballardini, A. Credi, M. T. Gandolfi, C. Giansante, G. Marconi, S. Serena and V. Margherita, Inorg. Chim. Acta, 2007, 360, 1072 CrossRef CAS; (c) J. C. Barnes, M. Juríček, N. A. Vermeulen, E. J. Dale and J. F. Stoddart, J. Org. Chem., 2013, 78, 11962 CrossRef CAS PubMed.
- A. Ruiz-Carretero, N. R. Ávila Rovelo, S. Militzer and P. J. Mesini, J. Mater. Chem. A, 2019, 7, 23451 RSC.
- B. Tieke, A. R. Rabindranath, K. Zhang and Y. Zhu, Beilstein J. Org. Chem., 2010, 6, 830 CrossRef PubMed.
- W. Li, L. Wang, H. Tang and D. Cao, Dyes Pigm., 2019, 162, 934 CrossRef CAS.
- M. Kaur and D. H. Choi, Chem. Soc. Rev., 2015, 44, 58 RSC.
- D. Chandran and K. S. Lee, Macromol. Res., 2013, 21, 272 CrossRef CAS.
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280 CrossRef CAS.
- J. Mizuguchi, T. Imoda, H. Takahashi and H. Yamakami, Dyes Pigm., 2006, 68, 47 CrossRef CAS.
- H. Takahashi and J. Mizuguchi, J. Appl. Phys., 2006, 100, 034908 CrossRef.
- H. Takahashi and J. Mizuguchi, J. Electrochem. Soc., 2005, 152, H69 CrossRef CAS.
- (a) J. Wang, L. Liu, W. Xu, Z. Yang, Y. Yan, X. Xie, Y. Wang, T. Yi, C. Wang and J. Hua, Anal. Chem., 2019, 91, 5786 CrossRef CAS PubMed; (b) J. Wang, W. Xu, Z. Yang, Y. Yan, X. Xie, N. Qu, Y. Wang, C. Wang and J. Hua, ACS Appl. Mater. Interfaces, 2018, 10, 3108 Search PubMed.
- (a) T. He, Y. Gao, S. Sreejith, X. Tian, L. Liu, Y. Wang, H. Joshi, S. Z. F. Phua, S. Yao, X. Lin, Y. Zhao, A. C. Grimsdale and H. Sun, Adv. Opt. Mater., 2016, 4, 746 CrossRef CAS; (b) H. Ftouni, F. Bolze and J. Nicoud, Dyes Pigm., 2013, 97, 77 CrossRef CAS.
- J. Humphreys, F. Malagreca, P. Hume, W. Lewis, E. S. Davies, S. P. Argent and D. B. Amabilino, Dyes Pigm., 2022, 197, 109836 CrossRef CAS.
- A. S. Murphy, C. E. Killalea, J. Humphreys, P. A. Hume, M. J. Cliffe, G. J. Murray, E. S. Davies, W. Lewis and D. B. Amabilino, ChemPlusChem, 2019, 84, 1413 CrossRef CAS PubMed.
- R. Rausch, M. I. S. Rohr, D. Schmidt, I. Krummenacher, H. Braunschweig and F. Wurthner, Chem. Sci., 2021, 12, 793 RSC.
- H. Crawford, M. Dimitriadi, J. Bassin, M. T. Cook, T. Fedatto Abelha and J. Calvo-Castro, Chem. – Eur. J., 2022, 51, e202202366 Search PubMed.
- D. K. Pathaka and H. C. Moon, Mater. Horiz., 2022, 9, 2949 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2160042–2160044. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc06770f |
| ‡ These authors contributed equally to the work presented here. |
| This journal is © The Royal Society of Chemistry 2023 |