
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A high-capacity, high-power organic electrode via supercritical CO2 impregnation into activated carbon micropores†
Yuta
Nakayasu‡
 *ab,
Shu
Sokabe‡
b,
Yuya
Hiraga
b and
Masaru
Watanabe
b
*ab,
Shu
Sokabe‡
b,
Yuya
Hiraga
b and
Masaru
Watanabe
b
aFrontier Research Institute for Interdisciplinary Sciences, Tohoku University, 6-3 Aoba, Aza, Aramaki, Aoba-ku, Sendai, Miyagi 980-8578, Japan. E-mail: nakayasu@tohoku.ac.jp
bResearch Center of Supercritical Fluid Technology, Graduate School of Engineering Tohoku University 6-6-11, Aoba, Aza, Aramaki, Aoba-ku, Sendai, Miyagi 980-8579, Japan
First published on 14th February 2023
Abstract
Herein, we report the impregnation of chloranil into activated carbon micropores using scCO2. The sample prepared under 105 °C and 15 MPa showed a specific capacity of 81 mAh gelectrode−1, except for the electric double layer capacity at 1 A gelectrode-Polytetrafluoroethylene (PTFE)−1. Additionally, approximately 90% of the capacity was retained even at 4 A gelectrode-PTFE−1.
The widespread use of renewable energy has increased the demand for energy-based devices such as energy storage devices, fuel cells, and solar cells. Additionally, owing to the recent global supply chain crisis, alternatives to metals and minor metals such as organic, natural, and waste-derived materials are being used to develop electrodes for energy-based devices.1,2 Among these, organic batteries using cathodes that do not require minor metals or metals are attracting attention as a novel technology, and their development has been remarkable. In recent years, several studies have been conducted on polymer-based organic storage batteries using single organic molecules.3,4
Carbon materials with a high specific surface area (SSA) have also been used as carrying and electrically conductive materials to fabricate such organic and natural product-derived electrodes.5,6 For example, quinones with conducting carbon are commonly used as a cathode in zinc-organic batteries.7 Recently, for the composite electrode synthesized using 9,10-phenanthraquinone poured and activated carbon (AC), the capacity utilization was approximately 57% at a constant current measurement of 5 A g−1, and the capacity per total weight of the cathode was 66.8 mAh g−1.8
Chloranil (CHL) is one of the key components to fabricate organic cathodes.9–11 Kundu et al. fabricated a composite of zinc foil, as the anode, and CHL (theoretical capacity: 218 mAh g−1), carbon material, and binder in a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35:5 wt % proportion as the cathode and obtained a utilization ratio of 95% with 200 mAg g−1 capacity; for total weight, a capacity of 120 mA g−1 was obtained.10 However, after 200 charge–discharge cycles at a 1 C-rate, which is the amount of current at which the theoretical capacity of a battery can be fully charged (or discharged) in one hour, the capacity utilization was approximately 70%, and the capacity per entire cathode weight was approximately 30 mAh g−1. Lin et al. used tetra amino-p-benzoquinone (TABQ) synthesized from CHL with conductive carbon as a cathode in a zinc-organic battery.12 The TABQ-based cathode showed a high capacity of 303 mAh g−1 (152 mAh gelectrode−1 including electrical double layer (EDL) capacity) at 0.1 A g−1. When the current density was increased to 5 A g−1, the specific capacity of 213 mAh g−1 (107 mAh/gelectrode including EDL capacity) was maintained after 1000 cycles. However, there are challenges in terms of the overpotential and redox capacity.
:35:5 wt % proportion as the cathode and obtained a utilization ratio of 95% with 200 mAg g−1 capacity; for total weight, a capacity of 120 mA g−1 was obtained.10 However, after 200 charge–discharge cycles at a 1 C-rate, which is the amount of current at which the theoretical capacity of a battery can be fully charged (or discharged) in one hour, the capacity utilization was approximately 70%, and the capacity per entire cathode weight was approximately 30 mAh g−1. Lin et al. used tetra amino-p-benzoquinone (TABQ) synthesized from CHL with conductive carbon as a cathode in a zinc-organic battery.12 The TABQ-based cathode showed a high capacity of 303 mAh g−1 (152 mAh gelectrode−1 including electrical double layer (EDL) capacity) at 0.1 A g−1. When the current density was increased to 5 A g−1, the specific capacity of 213 mAh g−1 (107 mAh/gelectrode including EDL capacity) was maintained after 1000 cycles. However, there are challenges in terms of the overpotential and redox capacity.
On the other hand, the production of zinc consumes a large amount of energy, emits a substantial amount of carbon dioxide (0.04 Gt of CO2), and produces a considerable amount of waste, which causes severe soil pollution and damage to human health through biological concentration.13 Organic redox supercapacitors are energy storage devices that use organic molecules to fabricate both positive and negative electrodes without any metals.14 Our research group has developed organic redox capacitors using cathodes composed of CHL- and anodes composed of 1,5-dichloroanthraquinone-impregnated in AC.15 We have also developed organic redox capacitors on a large scale.16 However, the conventional liquid-impregnation method using acetone is limited by the loading amount that can only be increased up to approximately 30%.
To address this problem, Komatsu et al. mixed acetylene black with AC and succeeded in increasing the initial capacity by several percent; however, the capacity decreased to less than the initial value when the loading amount was 30% after 300 charge–discharge cycles.17 Kanin et al. evaporated quinones into bio-derived AC; however, this process did not yield high capacity with high loading amount.18
Supercritical fluids have intermediate properties between liquid and gas such as solubility and diffusivity, enabling them to carry the solute into pores with high-concentration, compared to liquid and gas. Supercritical carbon dioxide (scCO2) is commonly used as a solvent and known as a green solvent and is superior to common organic green solvents such as ethanol and acetone in terms of recoverability and reuse.19 Metal deposition processes using supercritical carbon dioxide (scCO2) and organometallic complexes have been applied in impregnation technology, primarily for uniform embedding in high-aspect-ratio structures,20,21 solar cell thin films22 and, recently, for impregnation of organic materials in porous silica.23,24 Furthermore, scCO2 is also used to remove organic matter adsorbed on AC.25,26
In this study, we report an impregnation process of CHL into AC micropores using scCO2. The sample prepared under 105 °C_15 MPa showed a specific capacity of 81 mAh gelectrode−1, except for the EDL capacity at 1 A gelectrode-Polytetrafluoroethylene(PTFE)−1. Here, gelectrode denotes the weight of the entire electrode, and gelectrode-PTFE represents the weight of the entire electrode minus the binder, PTFE. This specific capacity is 1.41 times higher than that obtained using the liquid impregnation process. Additionally, even at 4 A gelectrode-PTFE−1, approximately 90% of the specific capacity was maintained compared with the capacity of 81 mA gelectrode−1 at 1 A gelectrode-PTFE−1 and was 1.83 times higher than the specific capacity obtained using the liquid impregnation process. The increased retention of specific capacity was caused by an increase in the loading rate of CHL into the AC pores and an increase in the π–π interactions. The specific capacity after 1000 charge–discharge cycles at 1 A gelectrode-PTFE−1 was 75.5 mAh gelectrode−1, which corresponds to a 93% retention rate when compared with the initial capacity.
ScCO2 impregnation was performed using a CHL impregnation system for AC under a scCO2 atmosphere, as shown in Fig. 1. The impregnation process was carried out at four temperatures (45, 75, 105, and 155 °C) and three pressure (10, 15, and 25 MPa) values to prepare samples under 12 temperature-pressure (T_P) combinations. Maxsorb® was used as AC. The detailed experimental method is shown in the ESI.†
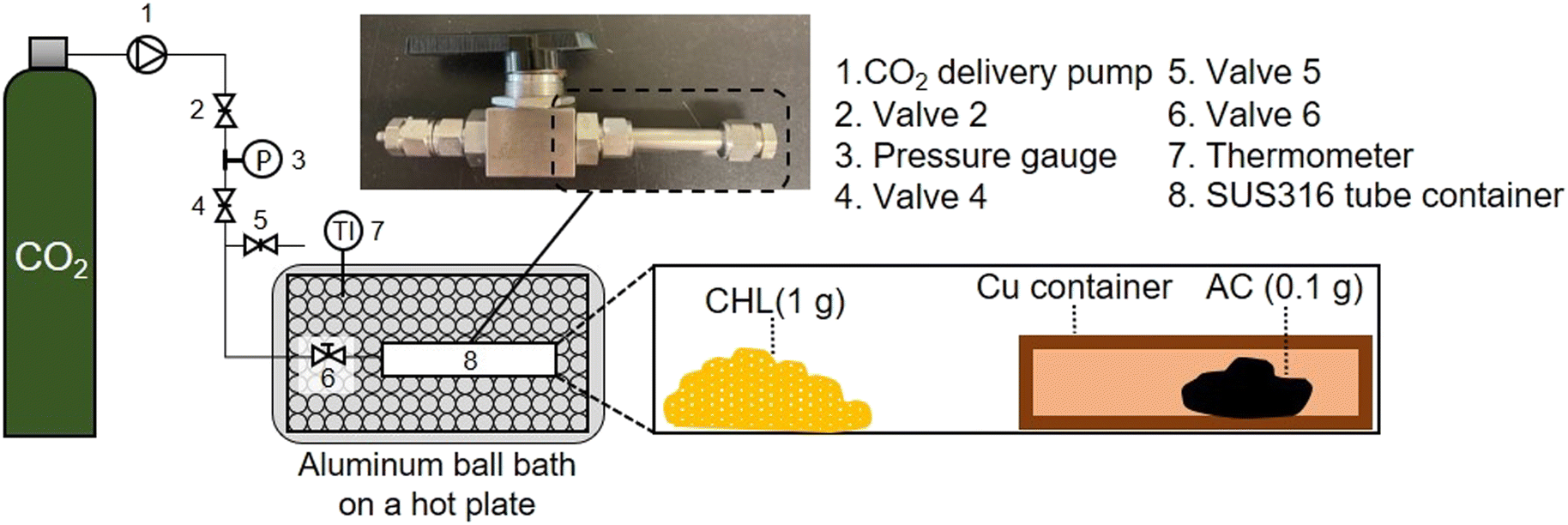 | ||
| Fig. 1 Chloranil impregnation system for AC using supercritical carbon dioxide. | ||
From the thermogravimetric (TG) results, the weight ratio of the desorbed CHL was calculated using the following formula:
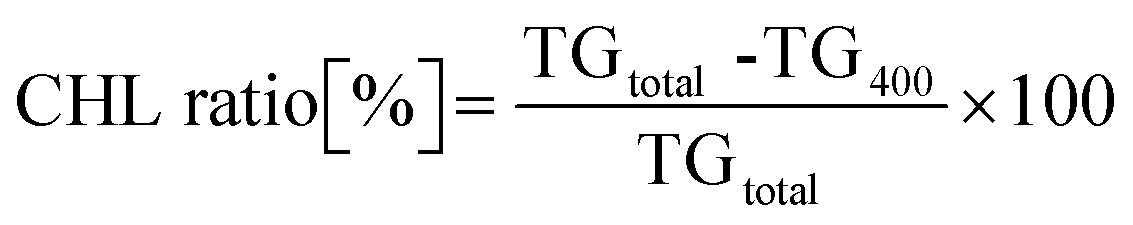 | (1) |
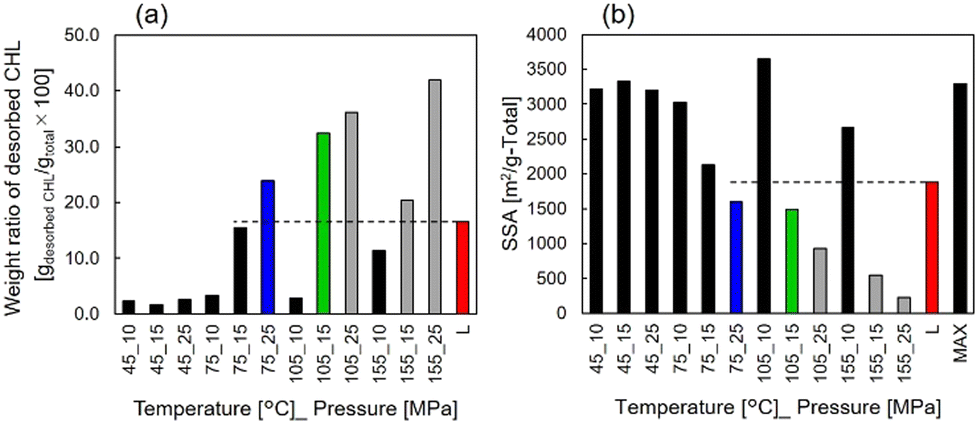 | ||
| Fig. 2 (a) Weight ratio of desorbed CHL during the TG measurement. (b) SSA after the impregnation processes. L: Liquid impregnation process, MAX: Maxsorb®. | ||
Furthermore, Fig. 3(a) shows the specific capacities except for the EDL capacity contribution, which is shown in Fig. S1 (ESI†), for samples prepared under each condition. Higher discharge capacities than the liquid-impregnated samples were obtained for scCO2-impregnated samples prepared under 75 °C_25 MPa and 105 °C_15 MPa. The specific capacity of 81 mAh gelectrode−1 at 0.1 Ah gelectrode−1, which is 1.41 times higher than that of the liquid-impregnated sample, was obtained for the sample prepared under 105 °C_15 MPa. Because of more than two-fold difference in the loading amounts and the specific capacity, it can be confirmed that the amount of loaded CHL was not completely impregnated into the AC micropores. Meanwhile, it was confirmed that the samples with higher loading amounts (i.e., samples prepared under 105 °C_25 MPa, 155 °C_15 MPa, and 155 °C_25 MPa) did not show high specific capacities. The two samples prepared under 105 °C_25 MPa and 155 °C_25 MPa exhibited high impedance, whereas the sample prepared under 155 °C_15 MPa showed almost the same impedance as other samples, as shown in Fig. S2 (ESI†). We hypothesize that this is due to the lack of conductivity in the two samples because of excessive CHL impregnation, whereas in the sample prepared under 155 °C_15 MPa, the amount of CHL impregnated in the AC micropores was high, but the amount impregnated in the AC mesopores was low. Fig. 3(b) shows the constant current measurements at 1 A gelectrode-PTFE−1 for the scCO2-impregnated sample (i.e., samples prepared under 75 °C_25 MPa and 105 °C_15 MPa) and liquid-impregnated samples. Compared with the liquid-impregnated samples, the scCO2-impregnated sample shows smaller overpotentials and a sharper transition from the plateau region to EDL capacity. Fig. 3(c) shows the rate characteristics for the scCO2-impregnated (i.e., samples prepared under 75 °C_25 MPa and 105 °C_15 MPa) and liquid-impregnated samples. It was observed that the two scCO2-impregnated samples maintained a higher specific capacity at a higher output than the liquid-impregnated samples. Even at a current density of 3 A gelectrode-PTFE−1, which corresponds to a C-rate of over 18 C, a capacity retention of 90% for 0.1 A gelectrode-PTFE−1 (approximately 0.45 C for 27 wt.% of electrodes) was observed. The specific capacity of the scCO2-impregnated sample was 1.83 times greater than that of the liquid-impregnated sample. Fig. 3(d) shows a comparison of the cycling characteristics between both the scCO2-impregnated samples and the liquid-impregnated sample at 1 A gelectrode-PTFE−1. The scCO2-impregnated sample prepared under 105 °C_15 MPa exhibited a specific capacity retention of 93% of its initial capacity. After 1000 cycles, a difference of approximately 30 mAh gelectrode−1 was observed in the specific capacity between the scCO2− and liquid-impregnated samples.
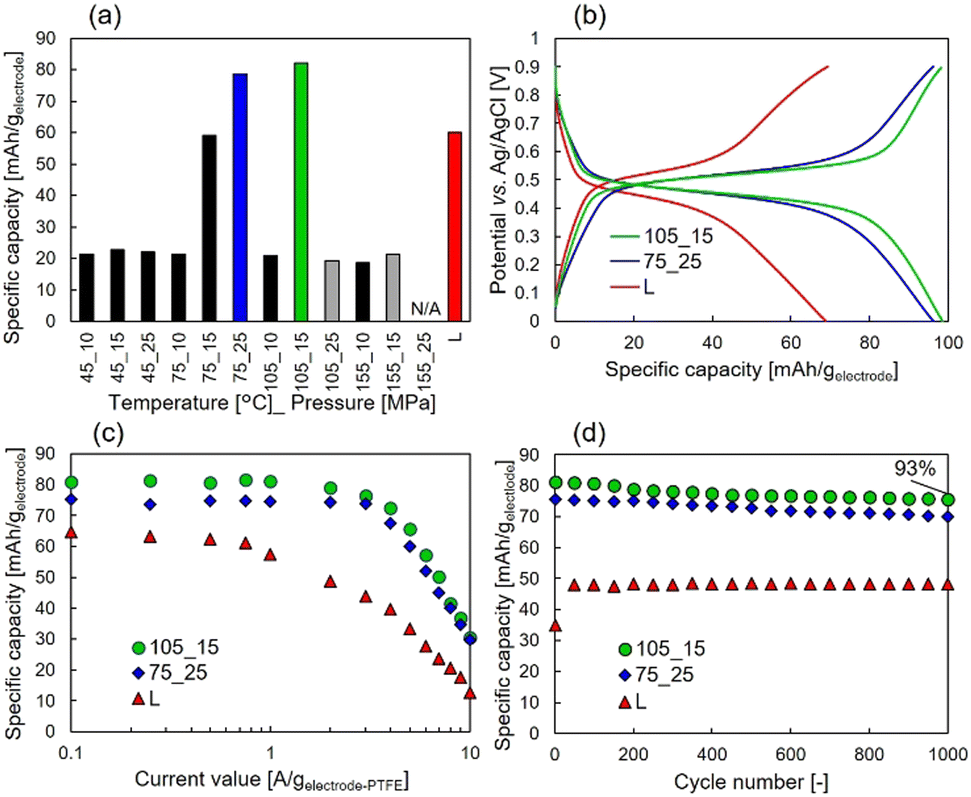 | ||
| Fig. 3 (a) Specific capacity of each sample at 0.1 A gelectrode-PTFE−1. (b) Constant current measurements for two scCO2 impregnated and liquid-impregnated samples with 1 A gelectrode-PTFE−1 of current density. (c) Rate characteristics of the three samples. (d) Cycle characteristics for two samples. | ||
The low overpotential, high rate characteristics, and high cycling characteristics indicate that strong adsorption occurs between CHL and AC during the scCO2 impregnation process. CHL competes in the π–π interaction with AC and its dispersion in the solvent; the less dispersed CHL is in the solvent, the stronger its π–π interaction with AC. In the scCO2 impregnation process, the density of supercritical carbon dioxide is lower than that of acetone used in the liquid impregnation process, resulting in a smaller partition coefficient of the solvent and stronger interaction with the AC. This phenomenon occurs in high-performance liquid chromatography (HPLC) and supercritical fluid chromatography (SFC). Pyrenylethyl (PYE) and pentabromophenyl (PBr) stationary phases exhibit strong π–π and dispersion interactions, respectively, in HPLC.27 They are found to retain solutes several times longer in SFC than in HPLC, and these forces are enhanced by SFC.
Fig. 4(a) shows micropore size distributions calculated using density functional theory (DFT) and carbon dioxide adsorption/desorption methods. The results show that the pore volumes of scCO2- and liquid-impregnated samples were smaller than that of AC in the entire micropore size range. In contrast, in the pore size range of 1–1.5 nm, the scCO2-impregnated samples exhibited a smaller pore volume than the liquid-impregnated samples. Below 1 nm, the largest pore volume was observed for the sample prepared under 75 °C_25 MPa, whereas the smallest pore volume was observed for the sample prepared under 105 °C_15 MPa. The liquid impregnation process occurred between these two volumes. Previous reports indicate that higher pressure may decrease the volume of adsorbed CHL because the amount of carbon dioxide adsorbed increases with increasing pressure.28 As shown in Fig. 4(b), below 1 nm, the pore volume of the sample prepared under 105 °C_15 MPa was 0.52 times smaller than the sample prepared under 75 °C_25 MPa; however, the discharge capacity at 0.1 A gelectrode-PTFE−1 was just 1.04 times higher (i.e., 82.1 vs. 78.7 mAh gelectrode−1). Thus, for CHL loadings on pore volumes of 1 nm or less, there is virtually no contribution to discharge capacity. This is attributed to the molecular size of CO2 being approximately 0.33 nm and that of CHL being approximately 0.5 nm, which makes the adsorption of solvated CHL on AC challenging.
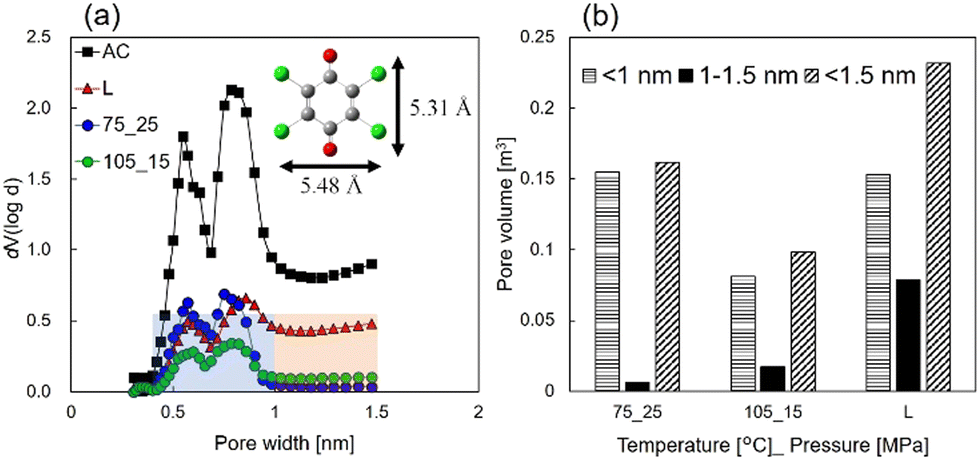 | ||
| Fig. 4 (a) Micropore size distribution calculated using DFT and carbon dioxide adsorption/desorption methods. (b) The pore volume in the CHL-impregnated AC electrodes. | ||
In this study, we demonstrated the impregnation of CHL into AC micropores using scCO2. As a result, the CHL/AC cathode prepared under 105 °C_15 MPa, showed a specific capacity of 81 mAh gelectrode−1, except for the EDL capacity at 1 A gelectrode-PTFE−1. This specific capacity is 1.41 times higher than that obtained via the liquid impregnation process. Additionally, even at 4 A gelectrode-PTFE−1, approximately 90% of the specific capacity was maintained compared with the capacity of 81 mA gelectrode−1 at 0.1 A gelectrode-PTFE−1. This was due to an increase in the loading rate of CHL into the AC pores and π–π interactions. The specific capacity after 1000 charge–discharge cycles at 1 A gelectrode-PTFE−1 was 75.5 mAh g−1, which corresponds to a retention rate of 93% compared to the initial capacity. The scCO2 impregnation process of organic molecules into porous carbons has great potential for a wide range of applications, such as the fabrication of electrodes for electrolysis, fuel cells, and other organic batteries.
This study was financially supported by the Program for Creation of Interdisciplinary Research from the Frontier Research Institute for Interdisciplinary Sciences, Tohoku University. We thank Dr. Takaaki Tomai of Institute of Multidisciplinary Research for Advanced Materials, Tohoku University for assistance with the carbon dioxide adsorption/desorption measurement.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. N. Gannett, L. Melecio-Zambrano, M. J. Theibault, B. M. Peterson, B. P. Fors and H. D. Abruña, Mater. Rep.: Energy, 2021, 1, 100008 Search PubMed.
- M. Zhang, J. Zhang, S. Ran, W. Sun and Z. Zhu, Electrochem. Commun., 2022, 138, 107283 CrossRef CAS.
- Y. Katsuyama, H. Kobayashi, K. Iwase, Y. Gambe and I. Honma, Adv. Sci. Lett., 2022, 9, 2200187 CAS.
- H. Nishide, Green Chem., 2022, 24, 4650–4679 RSC.
- P. Trogadas, T. F. Fuller and P. Strasser, Carbon, 2014, 75, 5–42 CrossRef CAS.
- W. Gu and G. Yushin, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 424–473 CAS.
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef PubMed.
- B. Yang, Y. Ma, D. Bin, H. Lu and Y. Xia, ACS Appl. Mater. Interfaces, 2021, 13, 58818–58826 CrossRef CAS PubMed.
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 CrossRef CAS.
- H. Kobayashi, K. Oizumi, T. Tomai and I. Honma, ACS Appl. Energy Mater., 2022, 5, 4707–4711 CrossRef CAS.
- J. He, X. Shi, C. Wang, H. Zhang and X. Liu, Chem. Commun., 2021, 57, 6931–6934 RSC.
- Z. Lin, H.-Y. Shi, L. Lin, X. Yang, W. Wu and X. Sun, Nat. Commun., 2021, 12, 4424 CrossRef CAS PubMed.
- C. Qi, L. Ye, X. Ma, D. Yang and J. Hong, J. Cleaner Prod., 2017, 156, 451–458 CrossRef CAS.
- T. Tomai, S. Mitani, D. Komatsu, Y. Kawaguchi and I. Honma, Sci. Rep., 2014, 4, 3591 CrossRef PubMed.
- Y. Katsuyama, Y. Nakayasu, K. Oizumi, Y. Fujihara, H. Kobayashi and I. Honma, Adv. Sustainable Syst., 2019, 3, 1900083 CrossRef CAS.
- Y. Katsuyama, T. Takehi, S. Sokabe, M. Tanaka, M. Ishizawa, H. Abe, M. Watanabe, I. Honma and Y. Nakayasu, Sci. Rep., 2022, 12, 3915 CrossRef CAS PubMed.
- D. Komatsu, T. Tomai and I. Honma, J. Power Sources, 2015, 274, 412–416 CrossRef CAS.
- N. Tisawat, C. Samart, P. Jaiyong, R. A. Bryce, K. Nueangnoraj, N. Chanlek and S. Kongparakul, Appl. Surf. Sci., 2019, 491, 784–791 CrossRef CAS.
- V. Hessel, N. N. Tran, M. R. Asrami, Q. D. Tran, N. Van Duc Long, M. Escribà-Gelonch, J. O. Tejada, S. Linke and K. Sundmacher, Green Chem., 2022, 24, 410–437 RSC.
- J. M. Blackburn, D. P. Long, A. Cabanas and J. J. Watkins, Science, 2001, 294, 141–145 CrossRef CAS PubMed.
- E. Kondoh and H. Kato, Microelectron. Eng., 2002, 64, 495–499 CrossRef CAS.
- T. Tomai, Y. Yasui, S. Watanabe, Y. Nakayasu, L. Sang, M. Sumiya, T. Momose and I. Honma, J. Supercrit. Fluids, 2017, 120, 448–452 CrossRef CAS.
- A. Patil, U. N. Chirmade, V. Trivedi, D. A. Lamprou, A. Urquart and D. Douroumis, J. Nanomed. Nanotechnol., 2011, 2, 1000111 CAS.
- W. Li-Hong, C. Xin, X. Hui, Z. Li-Li, H. Jing, Z. Mei-Juan, L. Jie, L. Yi, L. Jin-Wen, Z. Wei and C. Gang, Int. J. Pharm., 2013, 454, 135–142 CrossRef PubMed.
- I. Ushiki, M. Ota, Y. Sato and H. Inomata, Fluid Phase Equilib., 2013, 344, 101–107 CrossRef CAS.
- N. Takahashi, I. Ushiki, Y. Hamabe, M. Ota and Y. Sato, J. Supercrit. Fluids, 2016, 107, 226–233 CrossRef CAS.
- T. Hirose, D. Keck, Y. Izumi and T. Bamba, Molecules, 2019, 24, 2425 CrossRef CAS PubMed.
- X. Yang and C. T. Lira, J. Supercrit. Fluids, 2006, 37, 191–200 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06580k |
| ‡ Y. N. and S. S. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |