
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of functionalized spirocyclic oxetanes through Paternò–Büchi reactions of cyclic ketones and maleic acid derivatives†
Weronika Z.
Michalska
 ,
Nathan R.
Halcovitch
and
Susannah C.
Coote
*
,
Nathan R.
Halcovitch
and
Susannah C.
Coote
*
Department of Chemistry, Lancaster University, Bailrigg, LA1 4YB, UK. E-mail: w.michalska@lancaster.ac.uk; n.r.halcovitch@lancaster.ac.uk; s.coote@lancaster.ac.uk
First published on 20th December 2022
Abstract
A telescoped three-step sequence to functionalised spirocyclic oxetanes is reported, involving Paternò–Büchi reactions between maleic acid derivatives and cyclic ketones. p-Xylene suppresses the competing alkene dimerization that has plagued previous work, allowing access to 35 novel spirocyclic oxetanes that cannot be prepared using existing methodologies, and which represent versatile intermediates for further elaboration.
Oxetanes (four-membered cyclic ethers) enjoy considerable interest as motifs within drug molecules and as valued synthetic intermediates.1,2 For example, oxetanes have been used as “replacement groups” for metabolically vulnerable groups such as gem-dimethyl and carbonyl groups,3 and spirocyclic oxetanes are valuable in studying hitherto inaccessible areas of chemical space.4 As a result, the expedient synthesis of diverse oxetanes bearing varied substitution patterns is of broad interest.
Of the main synthetic approaches to oxetanes,2 the Paternò–Büchi reaction5 ([2+2] photocycloaddition of an aldehyde or a ketone with an alkene) is arguably the most attractive, as the oxetane ring is constructed in a single step. However, despite recent advances,6 the substrate scope of the Paternò–Büchi reaction remains limited; typically, aromatic aldehydes/ketones and electron-rich alkenes are required, as exemplified by the original Paternò–Büchi reaction involving benzaldehyde and 2-methyl-2-butene (Fig. 1A).7,8 The oxetane products thus obtained are often restricted in terms of diversification opportunities (especially when retention of the oxetane ring is desired), the control of regioselectivity is often poor, and levels of diastereoselectivity are typically low. Recently, Flores and Schmidt disclosed an impressive Cu-catalyzed carbonyl-olefin photocycloaddition that is mechanistically distinct from the Paternò–Büchi reaction, and which tolerates aliphatic ketones but not aromatic ketones.9 Although this work extended the ketone scope of Paternò–Büchi-type reactions, only norbornene was employed as the alkene partner, limiting the prospects for further derivatization of the resulting oxetane products.
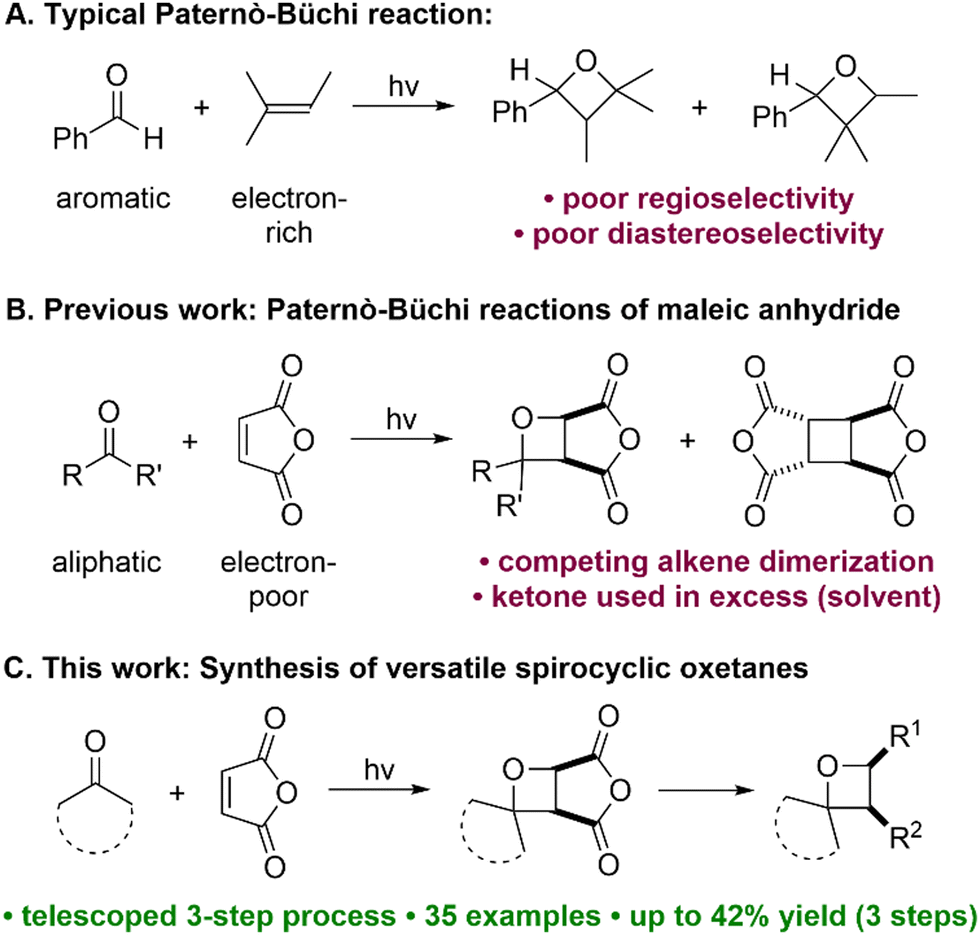 | ||
| Fig. 1 Limitations of current Paternò–Büchi methods (A and B). A new, telescoped approach to spirocyclic oxetanes (C). | ||
Relatively few reports of Paternò–Büchi reactions employing electron-deficient alkenes have appeared,10 but these reactions are generally less well understood, and yields of oxetane products are usually low. The combination of an aliphatic ketone with an electron-deficient alkene is rare, but would offer complementary substrate scope and access to novel oxetanes that cannot currently be generated. Previously, Turro and Wriede reported Paternò–Büchi reactions of maleic anhydride with simple ketones (e.g. acetone), using the ketones as both reagent and solvent (Fig. 1B).10a,c The desired oxetane products were formed in low yields since competing dimerization and polymerization of maleic anhydride were also observed, and the authors concluded that the reactions were not synthetically useful. Inspired by this seminal work, we describe the Paternò–Büchi reactions of cyclic aliphatic ketones with maleic anhydride derivatives, giving versatile cycloadducts that easily undergo further diversification (Fig. 1C). Our approach employs two inexpensive, essentially flat starting materials to give three-dimensional functionalized oxetanes as single isomers, via a telescoped three-step synthesis.
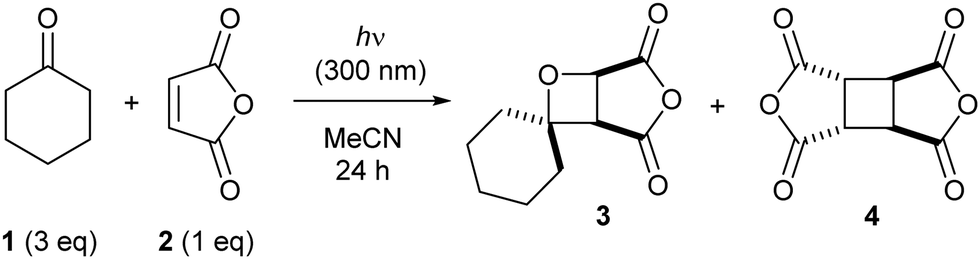
Firstly, the Paternò–Büchi reaction of cyclohexanone (1) with maleic anhydride (2) was investigated at 300 nm. Both starting materials absorb light of this wavelength, potentially leading to a complex mechanistic picture. Irradiation of a solution of 1 and 2 in acetonitrile generates a complex mixture comprising oxetane 3, dimer 4 (from dimerization of 2 through [2+2] photocycloaddition), 5-hexenal (from initial Norrish Type 1 fragmentation of 1) and polymeric material (polymerization of 2). Since dimer 4 is the major side product, we focused on suppressing this dimerization to increase the yield of oxetane 3. Moreover, dimer 4 (or its derivatives) could not be separated from oxetane 3 (or its derivatives), whereas 5-hexenal and the polymeric material could be easily removed chromatographically from the oxetane product. We sought to optimize the Paternò–Büchi reaction through the addition of additives that were expected to influence the singlet/triplet excited state populations of each reagent through triplet–triplet energy transfer processes (TTET). Specifically, a triplet sensitizer (with a triplet energy higher than the reagent) should directly populate the reagent's triplet excited state, promoting triplet-mediated reactions. Conversely, a triplet quencher (with a triplet energy lower than the reagent) should promote singlet-mediated reactions, by depleting the triplet excited state. The triplet energy of 1 is approximately 69 kcal mol−1 (see the ESI† for the phosphorescence spectrum), and the triplet energy of 2 is reported as 72 kcal mol−1 (in benzene),11 thus a range of inexpensive, readily available additives with varying triplet energies was screened (Table 1). Since oxetane 3 is unstable in air (the anhydride is readily opened by water), 1H NMR spectroscopy with solvent suppression was used to follow the reaction progress. Each reaction was irradiated for 24 h, then the relative amounts of unreacted maleic anhydride (2), oxetane 3 and dimer 4 were calculated through analysis of an aliquot from the reaction mixture (Table 1). With no additive present, full consumption of 2 was observed, with oxetane 3 obtained as the major product (entry 1), but with significant formation of dimer 4. A similar amount of dimer 4 was produced in the absence of 1 (entry 2), but the rate of consumption of maleic anhydride was much slower. One equivalent of benzophenone slowed both the Paternò–Büchi reaction and the dimerization, with mainly unreacted 2 present (entry 3). Reducing the benzophenone loading tenfold partially restored the rate of conversion of 2, and the selectivity for oxetane 3 over dimer 4 was improved (entry 4). Notably, benzophenone did not react with maleic anhydride, showing that aliphatic ketones are required for productive Paternò–Büchi reaction under these conditions. The remaining additives (ET > 72 kcal mol−1) gave broadly similar results (entries 6–8 and 11–12); conversion of 2 was significantly slowed compared to experiments in the absence of additive, but crucially, the selectivity for oxetane 3 over dimer 4 was much improved. Conversely, employing anisole led to preferential formation of dimer 4 rather than oxetane 3 (entry 10); the electron-rich nature of anisole compared to the other additives may enable other processes (e.g. electron transfer).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Change to standard conditions | Triplet energy of additivea (kcal mol−1) | Composition of reactionbc | ||
| 2 | 3 | 4 | |||
| a Reported triplet energies.12 b Relative composition of reaction mixture determined by 1H NMR spectroscopy (with solvent suppression) of an aliquot of the reaction mixture. c Quantity of polymeric material was not determined. | |||||
| 1 | — | — | 0 | 75 | 25 |
| 2 | No cyclohexanone | — | 79 | 0 | 21 |
| 3 | + dicyanobenzene (1 eq.) | 70.1 | 60 | 31 | 9 |
| 4 | + benzophenone (1 eq.) | 72.3 | 94 | 6 | 0 |
| 5 | + benzophenone (0.1 eq.) | 72.3 | 60 | 26 | 4 |
| 6 | + p-tolunitrile (1 eq.) | 75.7 | 59 | 39 | 2 |
| 7 | + Ph-1H-tetrazole (1 eq.) | 79.4 | 62 | 36 | 2 |
| 8 | + p-xylene (1 eq.) | 80.4 | 65 | 34 | 1 |
| 9 | + p-xylene (1 eq.), 72 h | 80.4 | 0 | 97 | 3 |
| 10 | + anisole (1 eq.) | 80.8 | 45 | 12 | 43 |
| 11 | + toluene (1 eq.) | 82.5 | 66 | 33 | 1 |
| 12 | + fluorobenzene (1 eq.) | 84.0 | 64 | 35 | 1 |
Ultimately, p-xylene was chosen as the optimal additive, owing to its ready availability, easy removal, and the clear separation of its signals and the product signals during 1H NMR analysis. Thus, extended irradiation (72 h) with one equivalent of p-xylene resulted in complete consumption of 2 and near-complete suppression of the dimerization (entry 9).
Having optimized the reaction conditions, the mode of action of the additives remained unclear. As the rates of the dimerization and the Paternò–Büchi reaction were slowed by the addition of additives (which would be expected to be capable of sensitizing both 1 and 2), we propose that dimer 4 forms primarily via the singlet excited state of 2, and that the Paternò–Büchi reaction occurs primarily from the singlet excited state of 1 (increased triplet populations of 1/2 would therefore reduce the rate of these processes). This proposal is in line with previous work, in which aliphatic ketones were reported to undergo singlet-mediated Paternò–Büchi reactions.5 The effect of molecular oxygen (a triplet quencher) on the rate of the Paternò–Büchi reaction was also studied. Thus, several pairs of otherwise identical reactions were performed, one under inert atmosphere, the other under air (see the ESI†). There were no significant differences in the rates of dimerization or Paternò–Büchi reaction, suggesting that triplet excited states are not key intermediates in these processes, reinforcing our proposal that the Paternò–Büchi reaction is driven by the singlet excited state of 1. Nevertheless, since both 1 and 2 absorb the incident light, and with the potential for cross-sensitization, mechanistic studies are complicated. To simplify the system, maleic anhydride was replaced with N-Boc-maleimide (5), the absorption of which extends to longer wavelengths than those of 2 and 1. Irradiation of a mixture of 5 and 1 at 350 nm (5 absorbs but 1 does not) results in dimer 6 but no oxetane 7, whilst irradiation at 300 nm (both 1 and 5 absorb) gives oxetane 7 and dimer 6 (Scheme 1). Assuming that the reaction between 1 and 2 behaves similarly to that between 1 and 5, we suggest that the excited state of 1 is required for productive Paternò–Büchi reaction, although more detailed investigations are clearly required to gain a full mechanistic picture.
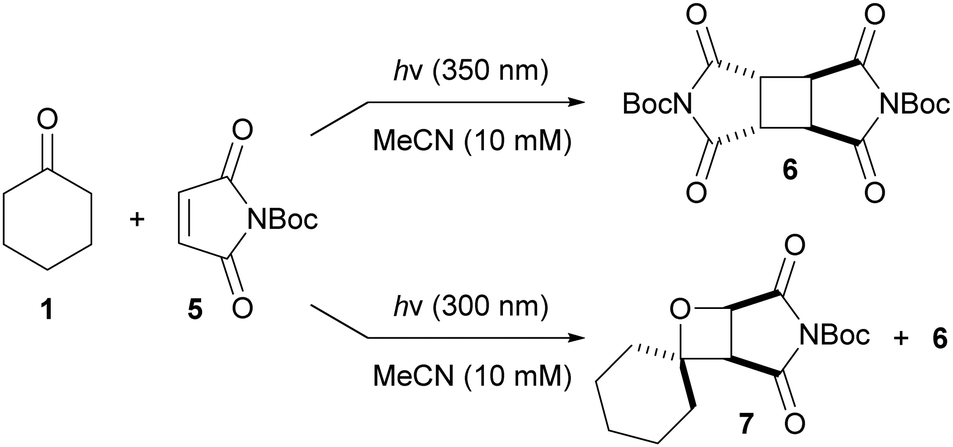 | ||
| Scheme 1 Paternò–Büchi reaction occurs at 300 nm, but not at 350 nm (1 does not absorb at 350 nm, 5 does absorb at 350 nm; both 1 and 5 absorb at 300 nm). | ||
Derivatizations of oxetanes 8 were next studied, via nucleophilic opening of the anhydride (Scheme 2). The resulting carboxylic acids 9 were difficult to purify, so a telescoped three-step procedure was developed, involving the Paternò–Büchi reaction, nucleophile addition and coupling of the resulting carboxylic acid. Functionalized spirocyclic oxetanes 10–30 were obtained as single isomers, after a single chromatographic purification. Considering the simplicity of the procedure and the complexity of the products, reasonable yields of oxetanes were obtained (up to 42% over three steps), with the rest of the mass balance attributed to polymerization of maleic anhydride (which could not be suppressed). The Paternò–Büchi reaction was also studied in a commercial flow photoreactor, but reactor fouling was observed at the very slow flow rates required, even using very dilute solutions. Thus, batch conditions were judged more convenient and capable of delivering multigram quantities of product (e.g.14 was synthesized on 30 mmol scale in comparable yield to that obtained on 1 mmol scale). Pleasingly, nucleophilic opening of the anhydride ring took place with complete regioselectivity, likely due to avoiding the steric hindrance imposed by the cyclohexyl ring. A range of primary alcohols was employed for ring-opening, upon heating for 24–72 hours. Conversely, secondary alcohols (e.g. 2-propanol) did not open the anhydride even upon extended heating, but could be employed in the final coupling step. Ring-opening of the anhydride with amines took place at room temperature, including less nucleophilic examples (e.g. aniline), plus more bulky amines (morpholine, thiomorpholine, benzhydrylamine). Several derivatives of maleic anhydride were also compatible with the optimized conditions – citraconic anhydride gave oxetane 28, with complete regioselectivity observed in the Paternò–Büchi reaction, and N-Boc-maleimide (5) and maleimide itself also produced the expected oxetanes, although chromatographic purification was more difficult than for oxetanes 10–28, resulting in reduced isolated yields of 29/30.
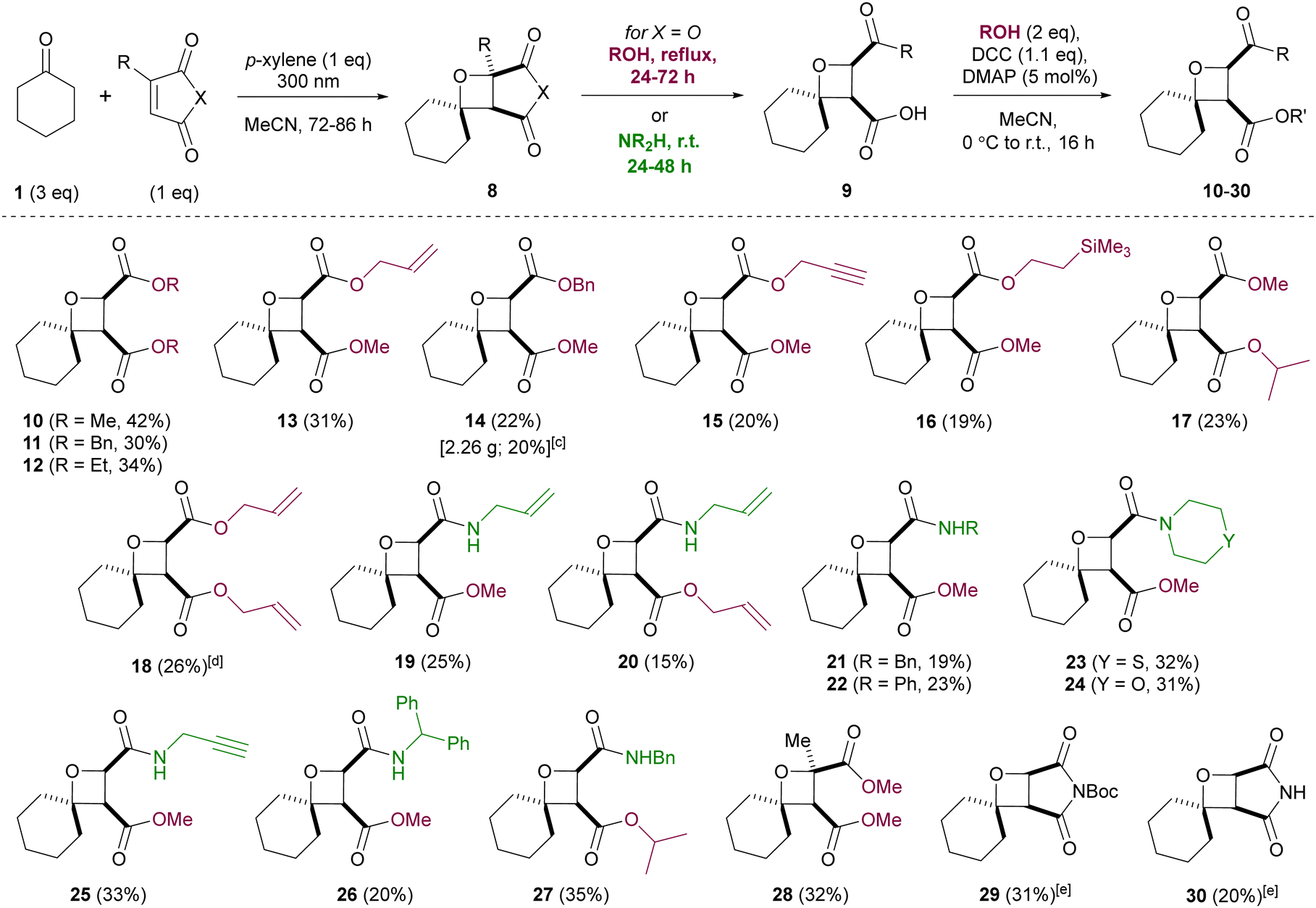 | ||
Scheme 2 Telescoped three-step synthesis of spirocyclic oxetanes. Via Paternò–Büchi reaction, nucleophilic ring-opening and DCC-mediated esterification.a,b a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Isolated yields. bPaternò–Büchi reactions were performed using 1 mmol of 2, 1 mmol p-xylene and 3 mmol of ketone; 0.1 M solution in MeCN with respect to maleic anhydride. cScale-up: 30 mmol of 2, 30 mmol p-xylene and 90 mmol of ketone. dScale-up: 5 mmol of 2, 5 mmol p-xylene and 15 mmol of ketone. eExtra equivalents of p-xylene employed (see the ESI†). Isolated yields. bPaternò–Büchi reactions were performed using 1 mmol of 2, 1 mmol p-xylene and 3 mmol of ketone; 0.1 M solution in MeCN with respect to maleic anhydride. cScale-up: 30 mmol of 2, 30 mmol p-xylene and 90 mmol of ketone. dScale-up: 5 mmol of 2, 5 mmol p-xylene and 15 mmol of ketone. eExtra equivalents of p-xylene employed (see the ESI†). | ||
The methodology was also extended to substituted and heteroatom-containing cyclic ketones, giving oxetane products with synthetic handles for further manipulation (Scheme 3). Cyclopentanone could also be employed, albeit consistently lower-yielding), but four-membered ketones (cyclobutanone, 3-oxetanone, 3-azetidinones) were not successful due to their fast decarbonylation upon irradiation.13 Finally, the expected relative cis configuration of the two substituents on the oxetane ring was confirmed by X-ray diffraction of oxetane 36.
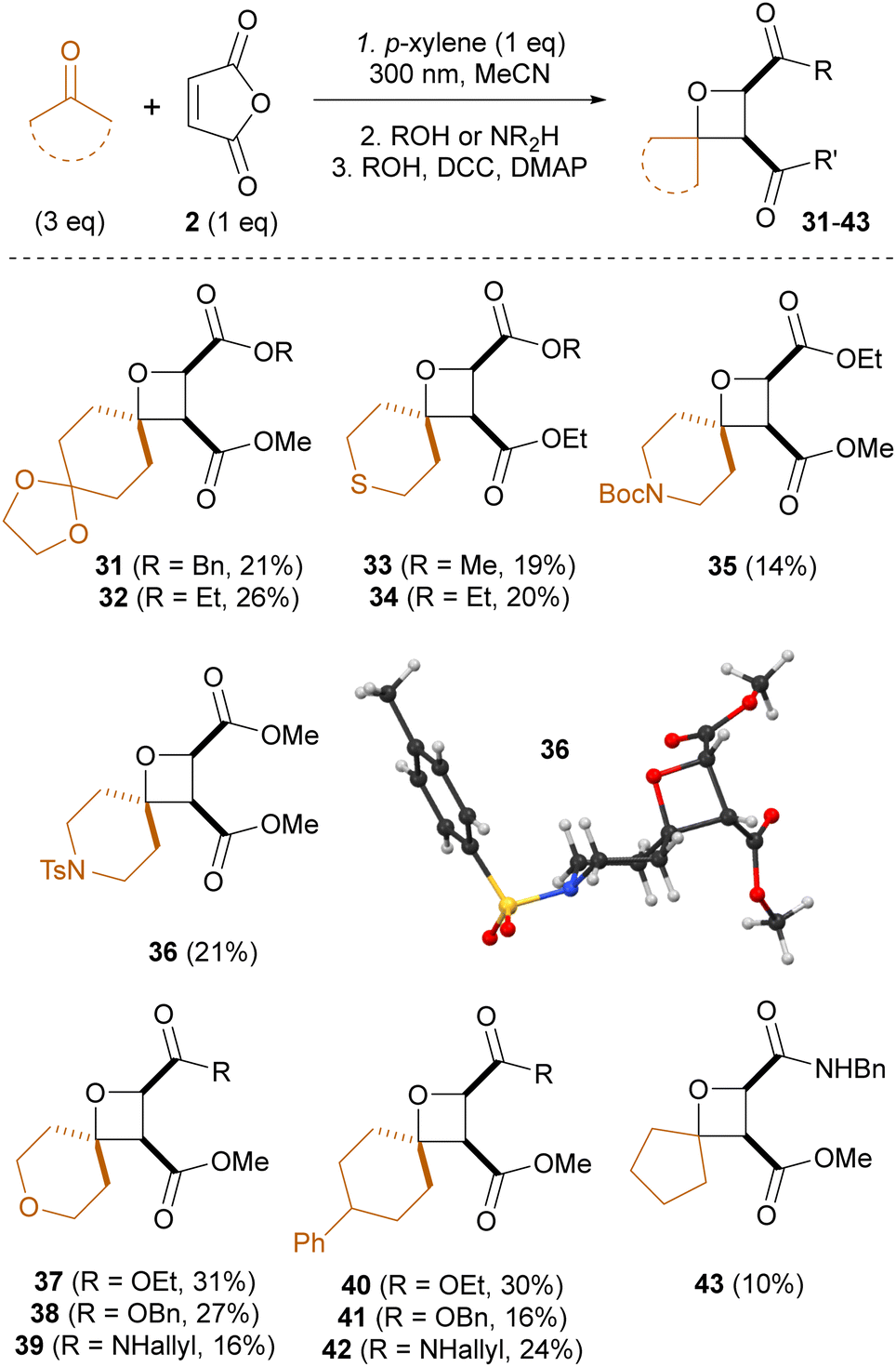 | ||
| Scheme 3 Telescoped three-step synthesis of spirocyclic oxetanes using maleic anhydride and a range of cyclic ketones. | ||
In conclusion, we have developed an efficient route to spirocyclic oxetane products that expands the typical scope of the Paternò–Büchi reaction to substrates that are usually avoided. The combination of cyclic aliphatic ketones with simple maleic anhydride derivatives in a telescoped three-step sequence allows the generation of single isomers of complex three-dimensional oxetanes after a single chromatographic purification, which were obtained in satisfactory yields considering their complexity and the brevity of the sequence, and which represent versatile intermediates for further diversification. Further studies aim to elucidate the probable mechanism of this intriguing Paternò–Büchi reaction.
The authors thank Lilly and Lancaster University for the award of a CASE doctoral studentship (to W. Z. M.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052 CrossRef CAS PubMed.
- (a) J. J. Rojas and J. A. Bull, in Comprehensive Heterocyclic Chemistry IV, ed. D. StC Black, J. Cossy and C. V. Stevens, Elsevier Ltd, 2002, pp. 212–256 Search PubMed; (b) K. P. Malarney, K. C. Shekhar and V. A. Schmidt, Org. Biomol. Chem., 2021, 19, 9825 RSC; (c) J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150 CrossRef CAS PubMed.
- (a) M. R. Bauer, P. Di Fruscia, S. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448 RSC; (b) G. Wuitschik, E. M. Carreira, B. Wagner, H. Fischer, I. Parrilla, F. Schuler, M. Rogers-Evans and K. Müller, J. Med. Chem., 2010, 53, 3227 CrossRef CAS PubMed.
- E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257 CrossRef CAS PubMed.
- (a) M. D’Auria, Photochem. Photobiol. Sci., 2019, 18, 2297 CrossRef PubMed; (b) M. Fréneau and N. Hoffmann, J. Photochem. Photobiol., C, 2017, 33, 83 CrossRef; (c) M. D’Auria, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Elsevier Ltd, 2014, pp. 159–199 Search PubMed; (d) M. D’Auria and R. Racioppi, Molecules, 2013, 18, 11384 CrossRef PubMed; (e) M. Abe, in Handbook of Synthetic Photochemistry, ed. A. Albini and M. Fagnoni, Wiley-VCH GmbH & Co. KGaA, 2010 Search PubMed.
- (a) D. Zhang, C. Wu, H. Zhou, Y. Ma and Y. Zhu, Asian J. Org. Chem., 2022, 11, e202200561 CAS; (b) J. Mateos, F. Rigodanza, P. Costa, M. Natali, A. Vega-Peñaloza, E. Fresch, E. Collini, M. Bonchio, A. Sartorel and L. Dell’Amico, Nat. Synth., 2022 DOI:10.1038/s44160-022-00191-5; (c) M. A. Gomez Fernandez, C. Lefebvre, A. Sudau, P. Genix, J.-P. Vors, M. Abe and N. Hoffmann, Chem. – Eur. J., 2021, 27, 15722 CrossRef CAS PubMed; (d) J. Zhao, X. Xie, Q. Lin, X. Ma, P. Su and Y. Xia, Anal. Chem., 2020, 92, 13470 CrossRef CAS PubMed; (e) P. Franceschi, J. Mateos, A. Vega-Peñaloza and L. Dell-Amico, Eur. J. Org. Chem., 2020, 6718 CrossRef CAS; (f) J. Zheng, X. Dong and T. P. Yoon, Org. Lett., 2020, 22, 6520 CrossRef CAS PubMed; (g) K. A. Rykaczewski and C. S. Schindler, Org. Lett., 2020, 22, 6516 CrossRef CAS PubMed; (h) J. Mateos, A. Vega-Peñaloza, P. Francheschi, F. Rigodanza, P. Andreetta, X. Companyó, G. Pelosi, M. Bonchio and L. Dell’Amico, Chem. Sci., 2020, 11, 6532 RSC.
- E. Paternò and G. Chieffi, Gazz. Chim. Ital., 1909, 39, 341 Search PubMed.
- G. Büchi, C. G. Inman and E. S. Lipinsky, J. Am. Chem. Soc., 1954, 76, 4327 CrossRef.
- D. M. Flores and V. A. Schmidt, J. Am. Chem. Soc., 2019, 141, 8741 CrossRef CAS PubMed.
- (a) N. J. Turro, P. Wriede, J. C. Dalton, D. Arnold and A. Glick, J. Am. Chem. Soc., 1967, 89, 3950 CrossRef CAS; (b) J. A. Barltrop and H. A. J. Carless, Tetrahedron Lett., 1968, 9, 3901 CrossRef; (c) N. J. Turro and P. A. Wriede, J. Org. Chem., 1969, 34, 3562 CrossRef CAS; (d) J. C. Dalton, P. A. Wriede and N. J. Turro, J. Am. Chem. Soc., 1969, 92, 1318 CrossRef.
- W. M. Hardham and G. S. Hammond, J. Am. Chem. Soc., 1967, 89, 3200 CrossRef CAS.
- S. L. Murov, Handbook of Photochemistry, Marcel Dekker Inc., 1973 Search PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2180492. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc06459f |
| This journal is © The Royal Society of Chemistry 2023 |