
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How long are Ga⇆Ga double bonds and Ga–Ga single bonds in dicationic gallium dimers?†
Antoine
Barthélemy
 ,
Harald
Scherer
,
Hanna
Weller
and
Ingo
Krossing
*
,
Harald
Scherer
,
Hanna
Weller
and
Ingo
Krossing
*
Institut für Anorganische und Analytische Chemie and Freiburger Materialforschungszentrum (FMF), Universität Freiburg, Albertstr. 21, Freiburg 79104, Germany. E-mail: krossing@uni-freiburg.de
First published on 20th December 2022
Abstract
Syntheses and characterization of two salts [(L)GaGa(L)][pf]2 ([pf]− = [Al(ORF)4]−; RF = C(CF3)3) are reported. They include the first dicationic digallene [(L)Ga⇆Ga(L)]2+ (L = CDPPh = C(PPh3)2) and a digallane [(L)Ga–Ga(L)]2+ (L = [NacNacMes]−). The CDPPh-supported digallene dication includes a trans-bent [L–GaGa–L]2+ bond that is analogous to neutral R–GaGa–R molecules and related to Robinson's famous “Digallyne” [R–GaGa–R]2−. The dicationic digallane [(L)Ga–Ga(L)]2+ is analogous to the widely used “Jones magnesium dimer”, but includes a very short GaII–GaII single bond.
An understanding of the nature and strength of covalent bonds is at the heart of chemistry. Using the standard notion of bond strengths and lengths, a single bond should be longer and weaker than a double or triple bond. Yet, more generally and following the Carter–Goddard–Malrieu–Trinquier model, the different binding modes in formally doubly bonded RxE
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ERx systems depend on the energy difference between singlet and triplet state of the two fragments: ERx.1,2 For most substituents R and elements E, the singlet state is energetically more favourable than the respective triplet state.3 Thus, formal double bonds between heavier elements are better described as two dative bonds “RxE⇆ERx” between filled s-orbitals and empty p-orbitals of two singlet carbene-analogue fragments.1,4–6 In this respect, group 13 elements are somewhat notorious.7,8 Several compounds exhibit unusual GaGa bonds and have triggered fundamental discussions about the nature of covalent bonds and the concept of bond orders in general.7–11 The digallium bonding in such compounds is illustrated in Fig. 1, i.e. digallanes, formal digallenes and digallynes. Note that only the incorporation of Na+ ions into the cluster core in Robinson's formal digallyne Na2[Ar*GaGaAr*], one of the most discussed organometallic compounds,7,8,11,12 impedes Coulomb explosion into two [Ar*Ga]− anions.
ERx systems depend on the energy difference between singlet and triplet state of the two fragments: ERx.1,2 For most substituents R and elements E, the singlet state is energetically more favourable than the respective triplet state.3 Thus, formal double bonds between heavier elements are better described as two dative bonds “RxE⇆ERx” between filled s-orbitals and empty p-orbitals of two singlet carbene-analogue fragments.1,4–6 In this respect, group 13 elements are somewhat notorious.7,8 Several compounds exhibit unusual GaGa bonds and have triggered fundamental discussions about the nature of covalent bonds and the concept of bond orders in general.7–11 The digallium bonding in such compounds is illustrated in Fig. 1, i.e. digallanes, formal digallenes and digallynes. Note that only the incorporation of Na+ ions into the cluster core in Robinson's formal digallyne Na2[Ar*GaGaAr*], one of the most discussed organometallic compounds,7,8,11,12 impedes Coulomb explosion into two [Ar*Ga]− anions.
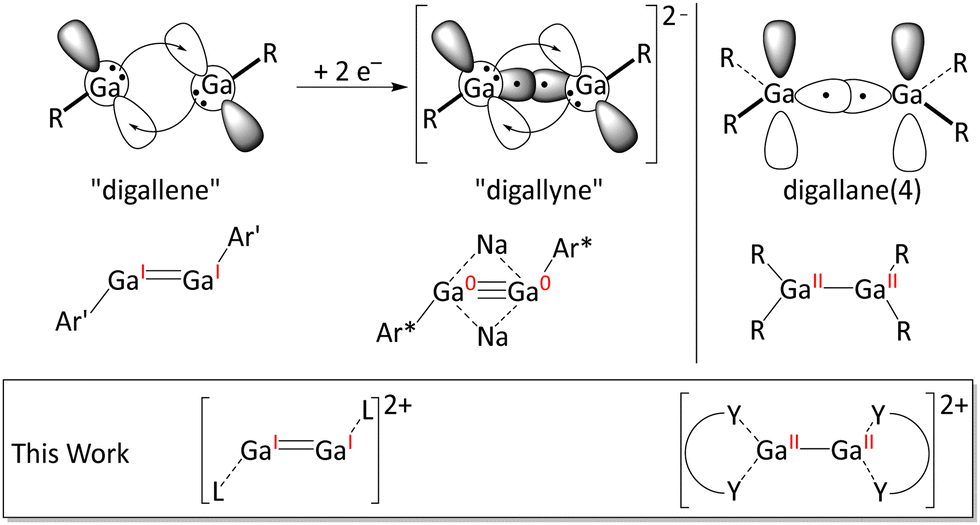 | ||
| Fig. 1 Non-classical formal double bonds between two R–GaI fragments (digallene; top left), two-electron reduction, giving a formal digallyne (top middle),1,4–6 structures of known digallenes14 (Ar′ = 2,6-(Dipp)2C6H3; Ar* = 2,6-(Tripp)2C6H3) and digallynes.11,14,15 The structures of neutral digallanes (top right) and of the cationic compounds presented herein are also shown (R = alkyl, aryl, amido; L = CDPPh; Y = N donor groups). | ||
In this work, we show that Ga–Ga single and formal double bonds form within dications (Fig. 1). Ion pairing is avoided by using the weakly coordinating anion [pf]− = [Al{OC(CF3)3}4]−.13
Syntheses and molecular structures. The Ga+ source used in this work is [Ga(PhF)2][pf].16 Upon addition of a mesityl-substituted β-diketiminate ([NacNacMes]−) ligand, Ga+ disproportionates, giving the dicationic digallane [{Ga(NacNacMes)}2][pf]2·1.5oDFB ([1][pf]2·1.5oDFB) (eqn (1)) which is isostructural to neutral MgI dimers.17 Eqn (2) shows that the dicationic digallene [{Ga(CDPPh)}2][pf]2 ([2][pf]2) forms with the neutral ligand L = hexaphenylcarbodiphosphorane (CDPPh)18 as electron-rich, four-electron ligand, under conservation of the oxidation state of Ga. Obviously, [NacNacMes]− is more strongly coordinating than neutral CDPPh, inducing disproportionation of metastable Ga+19 (characterization in ESI†).‡
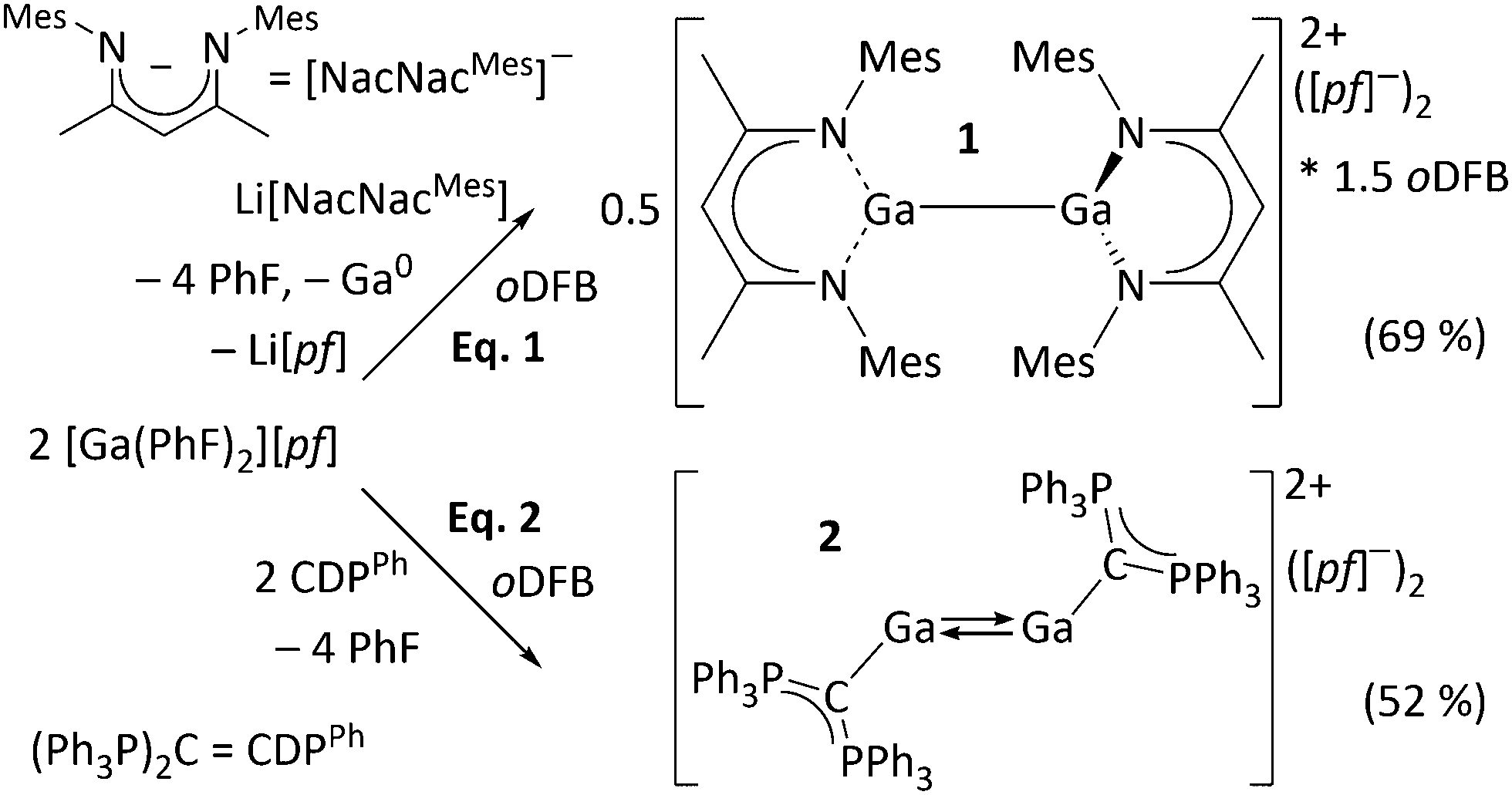
The molecular structure of the ecliptic dication 12+ is shown in Fig. 2. Despite several NacNacGa compounds being known,2012+ is the first featuring a Ga–Ga bond. The intact dimer 12+ exists in solution, as confirmed by DOSY NMR experiments (see ESI†). This is in line with quantum chemical calculations, which suggest that the dissociation is both endergonic in the gas phase and in oDFB (see below, eqn (5). Application of [In(PhF)2][pf] in the reaction leading to 22+ yielded [In(CDPPh)][pf] including a monomeric complex cation. However, the synthesis was not reproducible and the crystal structure is only discussed in the ESI.† In contrast to 12+, DOSY NMR experiments did not allow to clarify whether 22+ dissociates in solution (see ESI†). Its centrosymmetric molecular structure is displayed in Fig. 3. To the best of our knowledge, 22+ is the first isolated GaI–CDP complex. The structure contains a Ga2-dumbbell and a coplanar CCDP–Ga–Ga–CCDP moiety. Each Ga atom is coordinated by one CDPPh ligand in a trans-bent fashion that is typical for non-classical formal double bonds.3–6,9
 | ||
| Fig. 2 Molecular structure of 12+ in [1][pf]2·1.5oDFB. Only one of the two crystallographically independent 12+ units is shown. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at 50% probability level, the mesityl groups are shown as a wireframe model (black: C; blue: N; beige: Ga). | ||
 | ||
| Fig. 3 Molecular structure of 22+ in [2][pf]2. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at 50% probability level, the phenyl groups are shown as a wireframe model (black: C; purple: P; beige: Ga). | ||
Comparison with neutral digallanes and digallenes. The main structural parameters of the digallane 12+ and digallene 22+ are compared with related neutral species in Table 1. The correlation between Ga–Ga bond length and formal bond order is rather poor, underpinning the difficulty to assign bond orders in such compounds. The average Ga–Ga bond length in 12+ (238.22(9) pm) is remarkably short and only comparable to those in neutral five-membered GaII NHC dimer analogues, e.g. [GaII{[N(Dipp)C(Me)]2}]2 (236.34(9) pm)21 or [GaII{[N(tBu)CH]2}]2 (233.3(1) pm).22 The Ga–Ga bond in the singly bonded 12+ is only slightly longer than that in Robinson's formal digallyne22 and significantly shorter than in other digallanes(4). This is somewhat surprising, since Coulomb repulsion should lead to a bond lengthening in the dicationic dimer. Neutral dimers [{M(NacNac)}2] are also known for M = ZnI,23 and M = MgI.24 GaII is isoelectronic to ZnI and, ignoring the filled d orbitals, analogous to MgI. The Mg–Mg bond length in [{MgI(NacNacMes)}2], is almost 43 pm longer24 than the Ga–Ga bond in 12+. The positive charge residing on Ga, leading to contracted orbitals and, in addition, d-block contraction probably account for the shorter bonds. Accordingly, the corresponding ZnI dimer [{ZnI(NacNacMes)}2] has a similar M–M bond length (238.13(8) pm23) to the gallium dimer. However, the dihedral angle in the Ga dimer (84.65(8)°) is even higher than in the respective Zn (45.0(1)°) and Mg dimer (43.94(7)°).
| Entrya | Compound | d Ga–Ga [pm] | d E–Ga [pm] | E–Ga–Ga angle [°] | Ref. |
|---|---|---|---|---|---|
| a Includes as subscript the formally (!) assigned GaGa bond order of the respective compound. | |||||
1Ga![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Ga Ga |
Na2[Ar*GaGaAr*] | 231.9(3), 232.4(1) | 204(2) av. (E = C) | 131.0(4) av. (E = C) | 11 and 15 |
| 204.1(5) av. (E = C) | 131.07(17) av. (E = C) | ||||
| 2Ga–Ga | [HC(tBu)N]2GaGa[N(tBu)CH]2 | 233.3(1) | 183.8(6) av. (E = N) | 134.8(2) av. (E = N) | 22 |
| 3GaGa |
[(tBu2MeSi)(NHC)GaGa(NHC)(SiMetBu2)] | 234.1(3) | 209.7(4) (E = C) | 110.99(13) (E = C) | 25 |
| 125.88(17) (E = Si) | |||||
| 4GaGa |
Na2[Ar′GaGaAr′] | 234.7(1) | 205.9(5) (E = C) | 130.7(1) (E = C) | 14 |
| 5Ga–Ga | [(NacNacMes)GaGa(NacNacMes)]2+ (12+) | 238.22(9) | 184.68(18) av. (E = N) | 128.99(6) av. (E = N) | This work |
| 6Ga–Ga | (tmp)2GaGa(tmp)2 | 252.5(1) | 190.1(4) av. (E = N) | 120.7(1) av. (E = N) | 26 |
| 7Ga–Ga | [(Me3Si)2HC]2GaGa[CH(SiMe3)2]2 | 254.1(1) | 199.5(7) av. (E = C) | 122.0(2) av. (E = N) | 27 |
| 8GaGa |
[Ar′GaGaAr′] | 262.68(7) | 202.5(3) (E = C) | 123.16(7) (E = C) | 14 |
| 9GaGa |
[(CDPPh)GaGa(CDPPh)]2+ (22+) | 269.01(3) | 200.52(9) (E = C) | 118.18(3) (E = C) | This work |
| 10GaGa |
Ph2P(DippN)2GaGa(NDipp)2PPh2 | 278.73(12) | 207.9(2) av. (E = N) | 107.70(6) av. (E = N) | 28 |
The Ga–Ga distance in 22+ is longer than in a neutral digallene (entry 8 in Table 1), probably as a result of Coulomb repulsion between the positively charged [Ga(CDPPh)]+ fragments. The putative digallynes display shorter Ga–Ga bonds, due to the influence of the bridging Na atoms and the additional electrons in bonding orbitals (Fig. 1). In addition, the digallene in entry 3, stabilized by silyl- and NHC ligands,25 has a Ga–Ga distance similar to the Ga–Ga distance in putative digallynes, possibly due to the donation of electron density of the NHC-ligands into a Ga–Ga-bonding orbital.
The digallene dimer in entry 10 displays the longest Ga–Ga bond of the compounds presented in Table 1.28
Calculated thermodynamics. Eqn (3) and (4) include the formation energetics of 22+ and its (hypothetical) indium-analogue, which in our experiments remained monomeric in the solid state. The CDPPh complexation is favourable to the monocation (eqn (3)). Dimerization in eqn (4) is slightly favourable, but only in a polar oDFB solution (εr = 13.38;29 RI-BP86(D3BJ)/def2-TZVPP).
 | (3) |
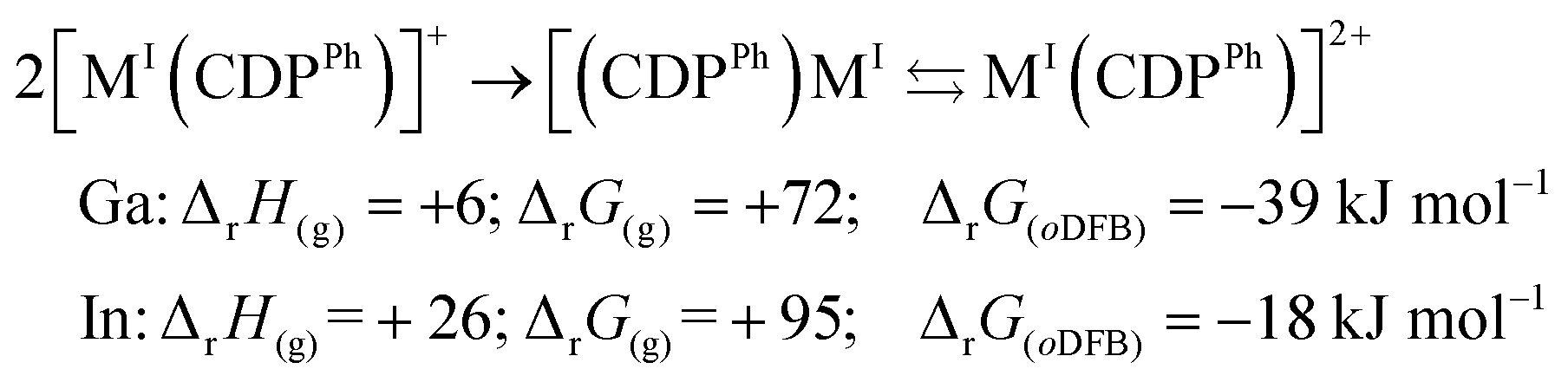 | (4) |
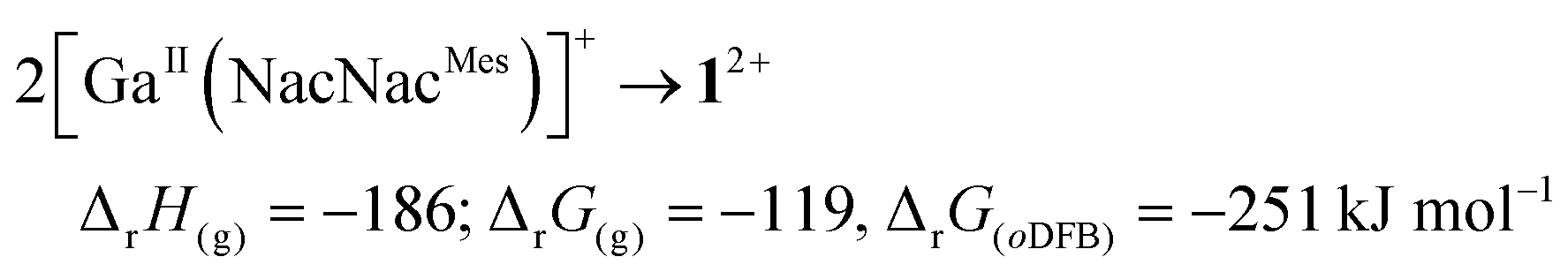 | (5) |
Bonding in the dicationic digallane and digallene. QTAIM charges δ of selected atoms in 12+ and 22+, bond path ellipticities (εBCP), electron densities on bond critical points (ρBCP) and the Ga–Ga Wiberg bond indices (WBI), are summarized in Table 2. Both, ρBCP(Ga–Ga) and WBI, are higher for 12+ compared to 22+. Additionally, the HOMO-LUMO gap is significantly higher in 12+ (2.6 vs. 2.1 eV; RI-BP86(D3BJ)/def2-TZVPP), again indicating a stronger Ga–Ga bond. The QTAIM charge of +1.09 on the formal GaII atoms suggests significant delocalization of the positive charge over the ligand in 12+. This, along with the contracted s and p orbitals in the dication, presumably accounts for the surprisingly short Ga–Ga bond. Ellipticity εBCP and WBI unequivocally classify the Ga–Ga bond as a single bond. By contrast, the bonding situation in the dicationic digallene is amazingly complex (resonance forms in Fig. 4, frontier orbitals in ESI†). The CCDP–Ga bonds can either be described as purely dative bonds (forms IV and VII in Fig. 4), leaving the positive charge on the Ga-centres, or as covalent bonds, formally shifting the positive charge to the ligand (forms I, V, VI).
| a For reasoning, see section 8.4 in ESI. |
|---|

|
 | ||
| Fig. 4 Limiting resonance forms I–III and arrow-based descriptions IV–VII of 22+. | ||
Invoking covalent double bonds between CCDP and Ga formally leaves a negative charge on the Ga atoms (forms II and III). Interestingly, the CCDP–Ga distance in 22+ is short at 200.6(1) pm and is even shorter than the covalent Cterphenyl–Ga bond in the first neutral digallene (202.5(3) pm).14 This indicates partial transfer of the positive charge to the CDP ligands as evident from entry 1 in Table 2 and is further supported by the nearly trigonal planar coordination mode around CCDP (∑bond![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) angles(CCDP) = 356.26(6)°). The simplified picture of a central sp2-hybridized CCDP atom and a delocalization of the positive charge along the P–CCDP–P axis as in form I would explain the observation. However, the calculated QTAIM charge on CCDP is negative at about −2 (Table 2 and forms IV–VII). This suggests that the CCDP–Ga bond also exhibits considerable electrostatic character, accounting for the surprisingly short CCDP–Ga bond. In fact, EDA-NOCV analysis confirms that the orbital-based and electrostatic contributions are equally important to describe the CCDP–Ga bond (see ESI†). The C–Ga bond in 22+ has a WBI less than unity, underlining the contribution of resonance forms IV and VII. Additionally, εBCP(CCDP–Ga) of ca. 0.11 (Table 2) and the EDA-NOCV analysis of [Ga(CDPPh)]+ indicate that both the σ- and the π-electron pairs of CCDP contribute to the formation of the CCDP–Ga bond (forms II, III and V–VII). Thus, the bonding situation in 22+ needs to be described by multiple resonance forms but is best visualized by resonance forms V–VII.
angles(CCDP) = 356.26(6)°). The simplified picture of a central sp2-hybridized CCDP atom and a delocalization of the positive charge along the P–CCDP–P axis as in form I would explain the observation. However, the calculated QTAIM charge on CCDP is negative at about −2 (Table 2 and forms IV–VII). This suggests that the CCDP–Ga bond also exhibits considerable electrostatic character, accounting for the surprisingly short CCDP–Ga bond. In fact, EDA-NOCV analysis confirms that the orbital-based and electrostatic contributions are equally important to describe the CCDP–Ga bond (see ESI†). The C–Ga bond in 22+ has a WBI less than unity, underlining the contribution of resonance forms IV and VII. Additionally, εBCP(CCDP–Ga) of ca. 0.11 (Table 2) and the EDA-NOCV analysis of [Ga(CDPPh)]+ indicate that both the σ- and the π-electron pairs of CCDP contribute to the formation of the CCDP–Ga bond (forms II, III and V–VII). Thus, the bonding situation in 22+ needs to be described by multiple resonance forms but is best visualized by resonance forms V–VII.
The non-classical double bond character of the GaGa bond in 22+ is clearly confirmed by an EDA-NOCV analysis: The HOMO of 22+ has pronounced Ga lone pair character (see ESI†) and accordingly EDA-NOCV analysis suggests that the most important interaction between the Ga atoms is the reciprocal donation of lone pair electron density into an empty 4p orbital. The resulting deformation densities Δρ(1)–(2) for the interaction between the [Ga(CDPPh)]+ fragments are shown in Fig. S-63 (ESI†).
In summary, we demonstrated that formation of single and formally double GaGa bonds is possible in dimeric, dicationic gallium compounds. Coulomb explosion can be prevented by employing electron rich ligands and the weakly coordinating [pf]− anion. 12+ is a dicationic digallane with very short GaGa bond, isostructural to a Jones Mg dimer. 22+ is the first dicationic digallane: Its bonding situation is complicated and has to be described by multiple resonance forms, best V–VII. It also represents the first univalent gallium-carbodiphosphorane-complex.
The authors acknowledge support by the Fonds of the Chemical Industry, the DFG in normal procedure, the state of Baden-Württemberg through bwHPC and the DFG through grant no. INST 40/467-1 and 575-1 FUGG (JUSTUS1 and 2 cluster).
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. P. Power, Chem. Rev., 1999, 99, 3463–3504 CrossRef CAS PubMed.
- (a) G. Trinquier, J. P. Malrieu and P. Riviere, J. Am. Chem. Soc., 1982, 104, 4529–4533 CrossRef CAS; (b) E. A. Carter and W. A. Goddard, J. Phys. Chem., 1986, 90, 998–1001 CrossRef CAS; (c) J. P. Malrieu and G. Trinquier, J. Am. Chem. Soc., 1989, 111, 5916–5921 CrossRef CAS.
- M. Driess and H. Grützmacher, Angew. Chem., Int. Ed. Engl., 1996, 35, 828–856 CrossRef CAS.
- G. Trinquier and J. P. Malrieu, J. Phys. Chem., 1990, 94, 6184–6196 CrossRef CAS.
- G. Trinquier and J. P. Malrieu, J. Am. Chem. Soc., 1987, 109, 5303–5315 CrossRef CAS.
- G. Treboux and J. C. Barthelat, J. Am. Chem. Soc., 1993, 115, 4870–4878 CrossRef CAS.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- H. Grützmacher and T. F. Fässler, Chem. – Eur. J., 2000, 6, 2317–2325 CrossRef.
- P. P. Power, Organometallics, 2020, 39, 4127–4138 CrossRef CAS.
- (a) M. Kaupp and P. v R. Schleyer, J. Am. Chem. Soc., 1993, 115, 1061–1073 CrossRef CAS; (b) K. W. Klinkhammer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2320–2322 CrossRef CAS; (c) Y. Xie, R. S. Grev, J. Gu, H. F. Schaefer, P. v R. Schleyer, J. Su, X.-W. Li and G. H. Robinson, J. Am. Chem. Soc., 1998, 120, 3773–3780 CrossRef CAS.
- J. Su, X.-W. Li, R. C. Crittendon and G. H. Robinson, J. Am. Chem. Soc., 1997, 119, 5471–5472 CrossRef CAS.
- (a) R. Dagani, Chem. Eng. News, 1998, 76, 31–35 CrossRef; (b) P. P. Power, J. Chem. Soc., Dalton Trans., 1998, 2939–2951 RSC; (c) G. H. Robinson, Acc. Chem. Res., 1999, 32, 773–782 CrossRef CAS; (d) A. Downs, Coord. Chem. Rev., 1999, 189, 59–100 CrossRef CAS; (e) G. H. Robinson, Chem. Commun., 2000, 2175–2181 RSC.
- (a) I. Krossing and A. Reisinger, Coord. Chem. Rev., 2006, 250, 2721–2744 CrossRef CAS; (b) T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789–899 RSC; (c) I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982–14024 CrossRef CAS PubMed.
- N. J. Hardman, R. J. Wright, A. D. Phillips and P. P. Power, Angew. Chem., Int. Ed., 2002, 41, 2842–2844 CrossRef CAS PubMed.
- B. Twamley and P. P. Power, Angew. Chem., Int. Ed., 2000, 39, 3500–3503 CrossRef CAS PubMed.
- (a) J. M. Slattery, A. Higelin, T. Bayer and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 3228–3231 CrossRef CAS PubMed; (b) A. Higelin, U. Sachs, S. Keller and I. Krossing, Chem. – Eur. J., 2012, 18, 10029–10034 CrossRef CAS PubMed; (c) K. Glootz, A. Barthélemy and I. Krossing, Angew. Chem., Int. Ed., 2021, 60, 208–211 CrossRef CAS PubMed; (d) A. Barthélemy, K. Glootz, H. Scherer, A. Hanske and I. Krossing, Chem. Sci., 2022, 13, 439–453 RSC.
- (a) A. Stasch and C. Jones, Dalton Trans., 2011, 40, 5659–5672 RSC; (b) C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- (a) W. C. Kaska, D. K. Mitchell and R. F. Reichelderfer, J. Organomet. Chem., 1973, 47, 391–402 CrossRef CAS; (b) R. Tonner, F. Oxler, B. Neumüller, W. Petz and G. Frenking, Angew. Chem., Int. Ed., 2006, 45, 8038–8042 CrossRef CAS PubMed; (c) R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Angew. Chem., Int. Ed., 2007, 46, 5263 CrossRef; (d) B. Inés, M. Patil, J. Carreras, R. Goddard, W. Thiel and M. Alcarazo, Angew. Chem., Int. Ed., 2011, 50, 8400–8403 CrossRef PubMed.
- P. Dabringhaus, A. Barthélemy and I. Krossing, Z. Anorg. Allg. Chem., 2021, 647, 1660–1673 CrossRef CAS.
- (a) S. Singh, J. Chai, A. Pal, V. Jancik, H. W. Roesky and R. Herbst-Irmer, Chem. Commun., 2007, 4934–4936 RSC; (b) A. Seifert, D. Scheid, G. Linti and T. Zessin, Chem. – Eur. J., 2009, 15, 12114–12120 CrossRef CAS PubMed; (c) D. W. N. Wilson, J. Feld and J. M. Goicoechea, Angew. Chem., Int. Ed., 2020, 59, 20914–20918 CrossRef CAS PubMed.
- R. J. Baker, C. Jones, D. P. Mills, G. A. Pierce and M. Waugh, Inorg. Chim. Acta, 2008, 361, 427–435 CrossRef CAS.
- D. S. Brown, A. Decken and A. H. Cowley, J. Am. Chem. Soc., 1995, 117, 5421–5422 CrossRef CAS.
- S. Schulz, D. Schuchmann, U. Westphal and M. Bolte, Organometallics, 2009, 28, 1590–1592 CrossRef CAS.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem. – Eur. J., 2010, 16, 938–955 CrossRef CAS PubMed.
- Z. Feng, Y. Fang, H. Ruan, Y. Zhao, G. Tan and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 6769–6774 CrossRef CAS PubMed.
- G. Linti, R. Frey and M. Schmidt, Z. Naturforsch. B, 1994, 49, 958–962 CrossRef CAS.
- W. Uhl, M. Layh and T. Hildenbrand, J. Organomet. Chem., 1989, 364, 289–300 CrossRef CAS.
- A. L. Hawley, C. A. Ohlin, L. Fohlmeister and A. Stasch, Chem. – Eur. J., 2017, 23, 447–455 CrossRef CAS PubMed.
- CRC handbook of chemistry and physics, ed. W. M. Haynes, D. R. Lide, T. J. Bruno and W. M. Haynes, CRC Press, Boca Raton, Florida, London, New York, 2017 Search PubMed.
- (a) Z. Zhu, R. C. Fischer, B. D. Ellis, E. Rivard, W. A. Merrill, M. M. Olmstead, P. P. Power, J. D. Guo, S. Nagase and L. Pu, Chem. – Eur. J., 2009, 15, 5263–5272 CrossRef CAS PubMed; (b) T. L. Allen, W. H. Fink and P. P. Power, J. Chem. Soc., Dalton Trans., 2000, 407–412 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2220000–2220002. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc06377h |
| ‡ In oDFB solutions of 22+, we reproducibly observed the formation of ca. 12% [H–CDPPh]+. The amount of protonated ligand is solvent-dependent. We suggest that the super basic ligand and Ga+ initiate intra-or intermolecular C–H bond activation of the ligand (see ESI†). |
| This journal is © The Royal Society of Chemistry 2023 |