
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Topological entrapment of macromolecules during the formation of metal–organic framework†
Nagi
Mizutani
a,
Nobuhiko
Hosono
 *b and
Takashi
Uemura
*b
*b and
Takashi
Uemura
*b
aDepartment of Advanced Materials Science, Graduate School of Frontier Sciences, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa, Chiba 277-8561, Japan
bDepartment of Applied Chemistry, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: nhosono@g.ecc.u-tokyo.ac.jp; uemurat@g.ecc.u-tokyo.ac.jp
First published on 10th January 2023
Abstract
Here we present our preliminary results on a novel approach to encapsulate large guest molecules in nanoporous materials, metal–organic frameworks (MOFs), via a newly discovered in situ crystal formation. This method has exciting prospects not only in the design of new organic/inorganic hybrids but also in capturing and separating molecules that are significantly larger than the actual pore size of the host MOF.
Molecular adsorption using porous materials, which is one of the essential means of capturing molecules in a solid phase, has been used in many applications such as gas separation and storage.1 However, this method inevitably imposes a geometric limitation on both the adsorbate (guest) and the adsorbent (host): the size of the guest molecules should be smaller than the pore size of the host. Therefore, the common notion is that large molecules with bulky and complicated structures/conformations cannot be captured in nanoporous materials with nanosized pore apertures. Recently, polymer adsorption within nanoporous materials has been reported;2 however, this phenomenon entails the diffusion of polymer chains into the constricted pores and imposes similar geometric limitations on the polymer shape. Furthermore, the polymer infiltration rate is generally slow due to large thermodynamic penalties.2 Herein, we propose a new strategy to entrap large molecules in nanoporous metal–organic frameworks (MOFs); it involves in situ MOF formation as a key process of molecular incarceration.
MOFs are emerging nanoporous materials with crystalline lattice structures formed through coordination bonding between organic ligands and metal ions.3 MOFs are of significant interest not only as adsorbents4 but also as materials applicable in advanced catalysts,5 sensing,6 and mass transportation.7 MOFs are conventionally synthesized via solvothermal methods using solvents that play an essential role in the homogeneous dissolution of the chemical reagents to facilitate crystal formation reactions.8 During the MOF crystal growth, solvent molecules are enfolded in the nanosized pores of the framework.9 In this study, we envisioned that if macromolecules are present during the MOF synthesis, they could be trapped and fixed in the MOF framework during its formation.
Nanoparticles,10 inorganic clusters,11 and some biomacromolecules12 have been introduced into MOFs by the in situ MOF formation method. In these cases, however, the guest species need to be smaller than the pore size of the MOF, otherwise lattice defects are generated to accommodate the oversized guests in the crystalline framework.12 Therefore, to date, the MOF crystallization process has rarely been considered as a rational means for capturing large polymeric molecules in the MOF nanopores.13 In this study, we found that successful guest encapsulation occurs when polymers are present during MOF crystallization. This enables guest entrapment in the MOF crystal, where the polymer chains are immobilized via physical entanglement with the MOF grid structure without inducing defects in the crystal structure. This unique mode of incarceration enables the permanent entrapment of cyclic macromolecules in the MOF grid. Whereas linear chains are prone to leach out of the MOF crystals, cyclic ones, which are topologically interlocked with the crystalline lattice, are retained intact even after rigorous washing procedures. The proposed in situ encapsulation method affords a new paradigm for capturing large molecules that cannot be introduced into already-formed porous materials via conventional adsorption.
In this study, we used [Zn2(ndc)2ted]n (hereafter termed as 1, ndc = 1,4-naphthalenedicarboxylate, ted = triethylenediamine, Fig. 1a) as the host MOF. 1 has a three-dimensional (3D) grid structure consisting of [Zn(ndc)]n layers. It has a porous square grid on the ab plane that is pillared by ted moieties along the c axis to form a 3D lattice. The primitive pore has two different apertures: the main aperture that directs to the c axis has size Ac of ca. 5.7 × 5.7 Å2 and the side aperture that directs to the a and b axes has size Aab of ca. 3.8 × 3.5 Å2 (Fig. 1a).14 Initially, we selected three common polymers, poly(methyl methacrylate) (PMMA, Mn = 7060 g mol−1), poly(4-vinylpyridine) (PVP, Mn = 6940 g mol−1), and polyethylene glycol (PEG, Mn = 1960 g mol−1) (Table S1, ESI†). The reactions conducted using each of these as an additive in the synthesis of 1 are designated as 1+PMMA, 1+PVP, and 1+PEG. In each reaction, the building components, Zn(NO3)2, H2ndc, and ted, were first mixed with an excess of the polymer in a glass vessel and then solubilized in a small quantity of N,N-dimethylformamide (DMF) (see ESI†). A high polymer concentration (50 mg mL−1) was used to ensure that any MOF nucleus encounters the polymer, leading to the enfolding of the polymer chains in the growing 3D grid of 1. After 24 h of reaction at 120 °C, white crystals were formed. They were carefully washed with solvents to remove the polymer adsorbed to the crystal surfaces (see ESI†).
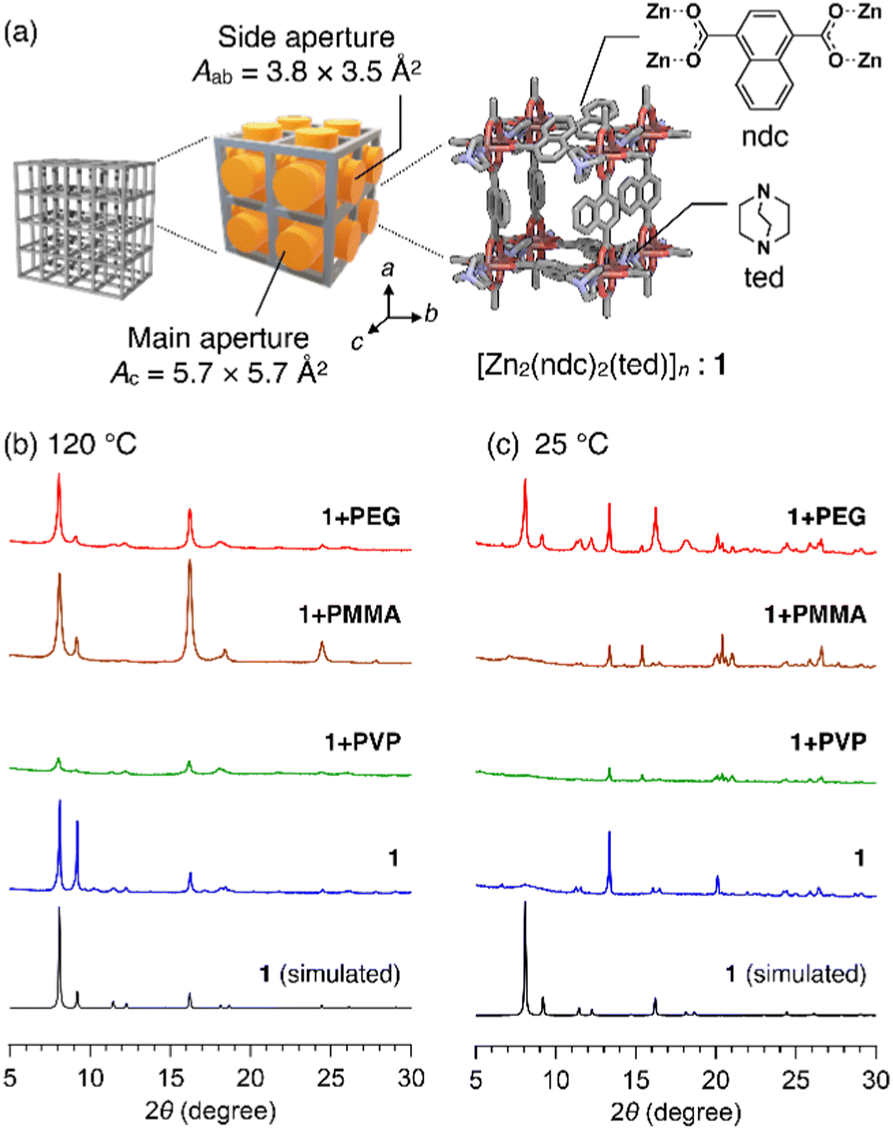 | ||
| Fig. 1 (a) Schematics of the crystal structure and pore apertures of 1. PXRD patterns of the products obtained by growing 1 at (b) 120 °C and (c) 25 °C in the presence of the respective polymers. Reaction type: (red) 1+PEG, (brown) 1+PMMA, (green) 1+PVP, and (blue) 1 without the polymer additive. Black line denotes the simulated PXRD pattern of the single crystal of 1. | ||
Interestingly, powder X-ray diffraction (PXRD) patterns of the products of 1+PMMA and 1+PEG indicated the successful formation of 1 (Fig. 1b). On the other hand, significantly weak diffraction peaks were obtained for 1+PVP, indicating the amorphous nature of the product. This could be due to the superior metal-coordination ability of the 4-vinylpyridine monomer units of PVP to that of the ndc carboxylate anions, which prevents the preferential formation of the Zn paddle-wheel clusters of 1. The crystals obtained from the 1+PMMA and 1+PEG reactions were further analyzed using 1H NMR spectroscopy after digesting them in an acidified deuterated solvent (DMSO-d6/DCl, 9/1, v/v) to investigate polymer inclusion in 1. Interestingly, the product of 1+PMMA showed no signals of PMMA, while that of 1+PEG clearly exhibited the signals of PEG (Fig. S1, ESI†). From the 1H NMR data, the amount of PEG encapsulated in 1 was determined to be 14 wt%.
The intriguing results obtained for the 1+PMMA and 1+PEG products prompted us to investigate the effect of polymer additives on the crystal formation process. We therefore conducted the same in situ encapsulation reactions at a significantly low temperature (25 °C). The synthesis of 1 at 25 °C without any polymer additive led to significantly deteriorated crystals of 1 (Fig. 1c). At 25 °C, the 1+PMMA and 1+PVP reactions also led to the same results, whereas the 1+PEG system provided high-quality crystals of 1. These observations indicate that PEG added to the reaction system significantly facilitates the crystal formation of 1, while the other two polymers do not.
Based on the low-temperature results, we infer that PMMA is not involved in the formation of 1 due to its relatively inert character and low compatibility with polar building components. On the other hand, PEG has potential metal-chelating ability,15 which is beneficial for stabilizing Zn2+ ions to promote the MOF nucleation process and thus enable the formation of 1 at a low temperature. Furthermore, this reasoning explains the mechanism of PEG encapsulation in 1. Owing to metal chelation at multiple sites on the chain, PEG can be always present in close proximity to Zn2+ ions. This leads to the aggressive involvement of PEG chains in the MOF lattice formation. In fact, the addition of ethylene glycol, a low-molecular-weight analogue (single monomer unit) of PEG, did not lead to crystals of 1 at 25 °C, because it could not induce the multidentate chelation effect owing to its short length (Fig. S2, ESI†).15
To better understand the in situ encapsulation mechanism, we varied the PEG concentration in the 1+PEG reaction at 120 °C. The relationship between the encapsulated and added PEG contents exhibited a non-linear trend (Fig. S3, ESI†). This result underpins the above-mentioned phenomenon of specific interaction between PEG and crystal nuclei of 1, leading to preferential rather than stochastic involvement of the PEG chains in the growing lattice. Remarkably, the reaction with a much higher PEG concentration (1.0 g mL−1), where PEG almost acts as a solvent, also resulted in the successful formation of 1 (Fig. S4, ESI†). In this case, the amount of encapsulated PEG was found to be 27 wt%, which agrees well with the previously reported maximum capacity of 1 (30 wt%).16 We further investigated the product with maximum PEG loading using differential scanning calorimetry (DSC) (Fig. S5, ESI†). The shift of the melting peak of PEG (∼54 °C) to a lower temperature suggests that the PEG chains are entirely encapsulated in the MOF phase and none of them are present outside of the MOF crystals.17 It should be noted that the enhanced crystal growth of some MOFs in poly(ionic liquid)s has been reported.18 The detailed and comprehensive investigations of the PEG-aided MOF formation mechanism will be pursued in our future study.
Solid-state HETCOR 2D NMR measurements on the product also supported the PEG impregnation of the pores of 1 (Fig. S6, ESI†). These observations indicate that the in situ crystallization method facilitates successful entrapment of PEG in 1. To represent this, the product of the 1+PEG reaction is hereafter denoted as 1⊃PEG. The crystal morphology of 1⊃PEG was investigated using scanning electron microscopy (SEM) (Fig. 2 and Fig. S7, ESI†). The crystal size of 1⊃PEG (28.1 ± 15.0 μm) was almost comparable to that of 1 prepared using DMF as the solvent instead of PEG (31.6 ± 22.1 μm), although the latter sample showed wider size distribution biased by the contamination of large particles of >100 μm (Fig. S8, ESI†).
 | ||
| Fig. 2 SEM images of (a) 1 prepared without any additive, (b) 1⊃PEG, and (c) 1⊃PEGins. 1⊃PEG was obtained by the in situ encapsulation of PEG during the formation of 1, while 1⊃PEGins was obtained by the post-incorporation of PEG in pre-synthesized 1. | ||
Next, we investigated the nanoscopic structure of the 1⊃PEG composite by FT-IR spectroscopy (Fig. S9, ESI†). The FT-IR profile in the range of 1550–1650 cm−1 provides information about the coordination bonding of the Zn paddle-wheel cluster in 1.19 The FT-IR spectrum of 1⊃PEG presented a dominant peak at 1637 cm−1, suggesting that the coordination bonding network in the crystal of 1 has limited defects (Fig. S9, ESI†). N2 adsorption measurements on the composite washed with tetrahydrofuran (THF) to remove the PEG guest (Fig. S10, ESI†) resulted in a Type I isotherm, which suggested no appreciable presence of mesopores. These observations suggest that PEG in the reaction mixture does not disrupt the growing MOF lattice while being incorporated, despite its sterically congested conformation. This is in stark contrast to the biomimetic mineralization of MOFs.12 The embedment of large biomacromolecules often generates lattice defects in the MOF crystal.
Furthermore, the effect of PEG entanglement in the 3D grid of 1 on PEG release was investigated by washing experiments to investigate if the topological restrictions impede PEG diffusion out of the MOF grid. For comparison, we prepared another PEG-loaded composite (denoted as 1⊃PEGins) using the post-synthetic insertion method.16 In brief, PEG (Mn = 1960 g mol−1) was mixed with crystals of 1 and heated at 120 °C for 18 h to facilitate the spontaneous infiltration of PEG chains into the nanopores of 1 (see ESI†). It should be noted that we used host 1 crystals with a narrow size distribution (29.2 ± 14.0 μm) for the 1⊃PEGins preparation to minimize any potential bias of the size differences in the results (see ESI,†Fig. 2 and Fig. S7 and S8). PEG infiltration is known to occur predominantly along the c-axis using the main aperture because of the diffusional preference.16,20 The PEG loading amount in the resultant 1⊃PEGins composite was determined to be 26 wt%. The FT-IR and PXRD data of 1⊃PEGins were comparable to that of 1⊃PEG, suggesting no appreciable difference between the coordination structures of the 1⊃PEG and 1⊃PEGins composites (Fig. S9, ESI†).
For washing, the composites were soaked in an excess of THF and ultrasonicated for 10 min at 25 °C. Then, the amount of PEG in 1 was determined using 1H NMR spectral analysis of acid-digested samples (see ESI†). The washing process was repeated thrice to monitor the progressive decrease in the residual PEG amount in 1 (Fig. 3a). The initial PEG loading amounts of both the 1⊃PEG and 1⊃PEGins composites were comparable. However, the residual PEG amounts differed appreciably after the washing process. 1⊃PEG preserved 20% of the initially loaded PEG after three washing cycles, while 1⊃PEGins retained only 11%. The comparable particle sizes of both composites rule out the size effect (Fig. S8, ESI†). Thus, 1⊃PEGins permitted easier PEG elution than 1⊃PEG, indicating that the method of PEG introduction affects the PEG chain configuration in the porous lattice of 1. That is, PEG chains in 1⊃PEG and 1⊃PEGins may have different impregnation characteristics (Fig. 3b and c). 1⊃PEG adopts an entangled form of PEG, with the chains penetrating the pores in all three directions because the MOF formation and PEG enfolding occur simultaneously in a random manner (Fig. 3b). On the other hand, 1⊃PEGins has an aligned form of PEG chains because PEG diffusion occurs predominantly along the c-axis of 1, resulting in a less-entangled configuration of PEG in the framework (Fig. 3c). This geometric feature of the aligned PEG aids facile diffusion, resulting in the rapid release of PEG molecules from 1 compared to that of the entangled form. We also found that the molecular weight of PEG affects the guest encapsulation efficiency and retention behavior (see ESI†). PEGs with larger MW showed stronger retention in 1 after the washing cycles, while extremely larger MW (>200 kg mol−1) resulted in low encapsulation efficiency due to the potential volume exclusion effect (Fig. S11 and S12, ESI†).
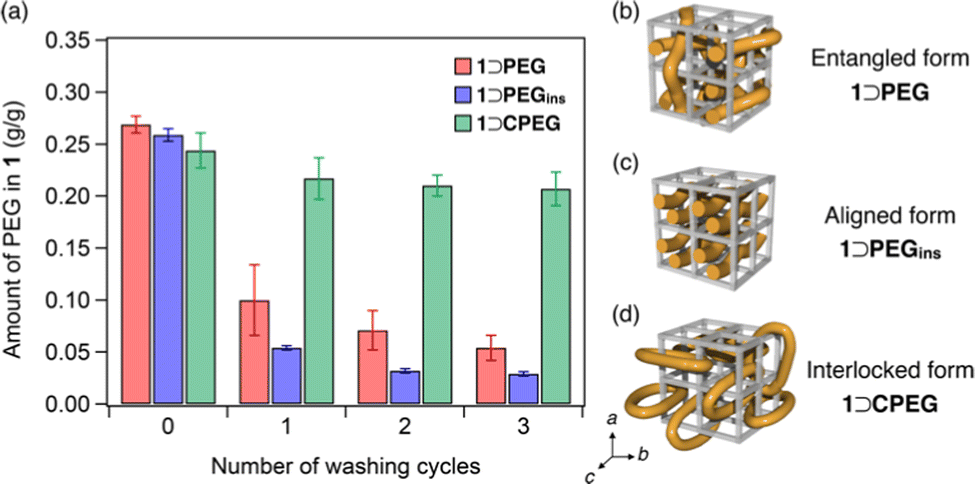 | ||
| Fig. 3 (a) Amount of residual PEG in 1 for the (red) 1⊃PEG, (blue) 1⊃PEGins, and (green) 1⊃CPEG composites after repeated washing. Error bars indicate standard errors (N = 3). Schematics of the plausible mode of polymer encapsulation for (b) 1⊃PEG, (c) 1⊃PEGins, and (d) 1⊃CPEG. | ||
The new guest-entrapping mode in the MOF prompted us to examine the encapsulation of cyclic macromolecules. Although it has been demonstrated that cyclic PEG cannot be introduced into 1 by the direct insertion method owing to size limitation and diffusional issues,21 we envisioned that it could be achieved using the present in situ encapsulation method. Furthermore, this would realize another class of MOF/polymer hybrids, in which cyclic chains are topologically interlocked with the 3D crystalline lattice (Fig. 3d). We synthesized a cyclic PEG sample (CPEG) via the end-to-end cyclization reaction of linear PEG (Mn = 1960) as reported earlier (Fig. S13, ESI†).21 The in situ formation of 1 using CPEG as the additive was performed under conditions identical to those of 1⊃PEG formation (see ESI†). The reaction successfully afforded the 1⊃CPEG composite, as confirmed by PXRD, DSC, solid-state HETCOR 2D NMR, SEM, particle size distribution, and FT-IR analyses (Fig. S4–S9, ESI†). The amount of CPEG loaded in 1 was determined to be 24 wt%, comparable to those of the 1⊃PEG and 1⊃PEGins composites. Notably, washing experiments revealed a remarkably higher retention ability of 1⊃CPEG than those of 1⊃PEG and 1⊃PEGins, although they are chemically identical species (Fig. 3a). 1⊃CPEG retained 85% of the originally loaded PEG even after three washing cycles, whereas the other two lost most of it. These results confirm the interlocked nature of cyclic PEG in 1 (Fig. 3d), resulting in the permanent immobilization of the PEG chain in the MOF crystal through mechanical bonding.22
Finally, considering the principle of the MOF-based topological guest entrapment, we anticipated the discrimination of cyclic and linear chains (Fig. S14, ESI†). In general, the separation of cyclic and linear polymers is considered highly difficult and often entails arduous procedures because they have almost identical physical attributes.23 To demonstrate the separation of the cyclic polymer, a crude product of the conventional PEG cyclization reaction, which contains a complex mixture of the cyclic product, linear precursor, and extended linear by-products (see ESI,† and Fig. S15), was used in the synthesis of 1. When the crystalline product formed was rigorously washed through repeated sonication/decantation cycles, the linear chains were completely washed off, whereas the cyclic products remained entrapped in the MOF lattice. Indeed, only cyclic PEGs were detected in the MOF phase in the comprehensive analyses of the digested sample using MALDI TOF-MS, 13C NMR, and size-exclusion chromatography (Fig. S15–S17, ESI†), highlighting the potential of this method in hitherto difficult molecular separation.
In summary, we demonstrated a new method to encapsulate large molecular species in MOFs by leveraging the in situ crystal formation process. This method allows the successful incorporation of macromolecules that cannot be directly introduced into the nanopores of MOFs via conventional adsorption. Owing to this feature, cyclic polymers are topologically trapped in the grid of the MOF lattice, resulting in a newly defined interlocked form of a polymer/MOF hybrid, in which the polymer is permanently immobilized. The proposed method is expected to expand the horizon in the design of organic–inorganic hybrids, which often face the fundamental issue of the easy release of the guest molecules from the host during practical solvent treatment and washing processes.
This work was supported by JSPS KAKENHI Grant No. 21J21641 (N. M.), 21H01981 (N. H.), 21H00385 (N. H.), and 21H04687 (T. U.). N. H. acknowledges the Katsu Research Encouragement Award of the University of Tokyo and the UTEC-UTokyo FSI Research Grant for financial support. N. M. is grateful to the Program for Leading Graduate School (MERIT-WINGS).
Conflicts of interest
There are no conflicts to declare.References
- P. A. Wright, Microporous Framework Solids, The Royal Society of Chemistry, Cambridge, UK, 2007 Search PubMed.
- (a) P. Duan, J. C. Moreton, S. R. Tavares, R. Semino, G. Maurin, S. M. Cohen and K. Schmidt-Rohr, J. Am. Chem. Soc., 2019, 141, 7589–7595 CrossRef CAS PubMed; (b) N. Oe, N. Hosono and T. Uemura, Chem. Sci., 2021, 12, 12576–12586 RSC.
- (a) O. Yaghi, M. O’keeffe, N. Ockwig, H. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed; (b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- (a) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed; (b) Y. Su, K. Otake, J.-J. Zheng, S. Horike, S. Kitagawa and C. Gu, Nature, 2022, 611, 289–294 CrossRef CAS PubMed.
- (a) Z. Hu and D. Zhao, CrystEngComm, 2017, 19, 4066–4081 RSC; (b) Q. Wang, Q. Gao, A. M. Al-Enizi, A. Nafady and S. Ma, Inorg. Chem. Front., 2019, 7, 300–339 RSC.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- M. D. Allendorf, V. Stavila, M. Witman, C. K. Brozek and C. H. Hendon, J. Am. Chem. Soc., 2021, 143, 6705–6723 CrossRef CAS PubMed.
- S. Han, S. C. Warren, S. M. Yoon, C. D. Malliakas, X. Hou, Y. Wei, M. G. Kanatzidis and B. A. Grzybowski, J. Am. Chem. Soc., 2015, 137, 8169–8175 CrossRef CAS PubMed.
- Z.-M. Zhang, T. Zhang, C. Wang, Z. Lin, L.-S. Long and W. Lin, J. Am. Chem. Soc., 2015, 137, 3197–3200 CrossRef CAS PubMed.
- (a) K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed; (b) P.-H. Tong, L. Zhu, Y. Zang, J. Li, X.-P. He and T. D. James, Chem. Commun., 2021, 57, 12098–12110 RSC.
- S. Takamaizawa, M. Furihata, S. Takeda, K. Yamaguchi and W. Mori, Macromolecules, 2000, 33, 6222–6227 CrossRef CAS.
- (a) H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem. – Eur. J., 2005, 11, 3521–3529 CrossRef CAS PubMed; (b) T. Uemura, Y. Ono, K. Kitagawa and S. Kitagawa, Macromolecules, 2008, 41, 87–94 CrossRef CAS.
- H. Noma, T. Koga, C. Hirakawa, K. Nonaka, K. Shobu, T. Kaibuki and S. Moriyama, Electrochem. Soc. Trans, 2014, 58, 77–88 Search PubMed.
- (a) B. Le Ouay, C. Watanabe, S. Mochizuki, M. Takayanagi, M. Nagaoka, T. Kitao and T. Uemura, Nat. Commun., 2018, 9, 3635 CrossRef PubMed; (b) N. Mizutani, N. Hosono, B. Le Ouay, T. Kitao, R. Matsuura, T. Kubo and T. Uemura, J. Am. Chem. Soc., 2020, 142, 3701–3705 CrossRef CAS PubMed.
- T. Uemura, N. Yanai, S. Watanabe, H. Tanaka, R. Numaguchi, M. T. Miyahara, Y. Ohta, M. Nagaoka and S. Kitagawa, Nat. Commun., 2010, 1, 83 CrossRef PubMed.
- J.-K. Sun, Y. I. Sobolev, W. Zhang, Q. Zhuang and B. A. Grzybowski, Nature, 2020, 579, 73–79 CrossRef CAS PubMed.
- (a) V. Zeleňák, Z. Vargová and K. Györyová, Spectrochim. Acta, Part A, 2007, 66, 262–272 CrossRef PubMed; (b) K. Tan, N. Nijem, P. Canepa, Q. Gong, J. Li, T. Thonhauser and Y. J. Chabal, Chem. Mater., 2012, 24, 3153–3167 CrossRef CAS; (c) Z. Su, Y.-R. Miao, G. Zhang, J. T. Miller and K. S. Suslick, Chem. Sci., 2017, 8, 8004–8011 RSC.
- T. Uemura, G. Washino, S. Kitagawa, H. Takahashi, A. Yoshida, K. Takeyasu, M. Takayanagi and M. Nagaoka, J. Phys. Chem. C, 2015, 119, 21504–21514 CrossRef CAS.
- T. Sawayama, Y. Wang, T. Watanabe, M. Takayanagi, T. Yamamoto, N. Hosono and T. Uemura, Angew. Chem., Int. Ed., 2021, 60, 11830–11834 CrossRef CAS PubMed.
- B. H. Wilson and S. J. Loeb, Chem, 2020, 6, 1604–1612 CAS.
- (a) Z. Jia and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2085–2097 CrossRef CAS; (b) F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 12, 433–444 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, PXRD, NMR, FT-IR, DSC, SEM, particle size distribution, gas adsorption isotherm, SEC, and MALDI TOF-MS data. See DOI: https://doi.org/10.1039/d2cc06330a |
| This journal is © The Royal Society of Chemistry 2023 |