
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOperando characterization of continuous flow CO2 electrolyzers: current status and future prospects
Dorottya
Hursán
 and
Csaba
Janáky
*
and
Csaba
Janáky
*
University of Szeged, Department of Physical Chemistry and Materials Science, Aradi sq. 1, Szeged, 6720, Hungary. E-mail: janaky@chem.u-szeged.hu
First published on 4th January 2023
Abstract
The performance of continuous-flow CO2 electrolyzers has substantially increased in recent years, achieving current density and selectivity (particularly for CO production) meeting the industrial targets. Further improvement is, however, necessary in terms of stability and energy efficiency, as well as in high-value multicarbon product formation. Accelerating this process requires deeper understanding of the complex interplay of chemical–physical processes taking place in CO2 electrolyzer cells. Operando characterization can provide these insights under working conditions, helping to identify the reasons for performance losses. Despite this fact, only relatively few studies have taken advantage of such methods up to now, applying operando techniques to characterize practically relevant CO2 electrolyzers. These studies include X-ray absorption- and Raman spectroscopy, fluorescent microscopy, scanning probe techniques, mass spectrometry, and radiography. Their objective was to characterize the catalyst structure, its microenviroment, membrane properties, etc., and relate them to the device performance (reaction rates and product distribution). Here we review the current state-of-the-art of operando methods, associated challenges, and also their future potential. We aim to motivate researchers to perform operando characterization in continuous-flow CO2 electrolyzers, to understand the reaction mechanism and device operation under practically relevant conditions, thereby advancing the field towards industrialization.
Introduction
Electrochemical CO2 reduction (CO2RR) is a rising technology to produce fuels and commodity chemicals in a possibly carbon neutral- or even negative way.1 Being an electrochemical process, it can be directly coupled to renewable electricity (e.g., solar, wind), assisting the storage of these intermittent energy sources in the form of chemical bonds,2 while reducing the dependence of our society on fossil fuel resources at the same time. Systematic investigation of CO2RR started in the 1990s when the main focus was on the exploration of selectivity trends for different metal electrodes.3 Over the years, materials engineering strategies, such as nanostructuring,4 alloying5,6 or novel materials design (e.g., non-metal electrodes),7,8 were explored to increase activity or to steer selectivity towards the formation of a given product. Additionally, the modification of the catalyst microenvironment, by electrode and electrolyte design, also turned out to be critically important to improve catalyst performance.9 In parallel, detailed mechanistic investigation of the CO2RR10 was also performed, which largely contributed to our current understanding of the reaction paths leading to certain products. In situ and operando spectroscopic investigations have become the most powerful tools for gaining mechanistic understanding,11 as only these methods are capable of tracking changes in the catalyst structure, its microenvironment, and reaction intermediates during operation. The improvement of spectroscopic techniques in terms of signal intensity (also in electrolytes), and spatial and temporal resolution has opened up new possibilities for operando characterization, and these techniques are rather commonly used in electrocatalysis research nowadays. Beyond online product detection methods, these include vibrational spectroscopy, X-ray based methods, local pH measurements and electron microscopy among others, thoroughly reviewed recently.10,12–15In the past 10–15 years, with the emergence of continuous-flow electrolyzer cells, the field has started to expand towards practical applications.16 As it became clear that the CO2RR has relevance for industrialization, electrolyzer development had also become a focus of research. Researchers concentrating on catalyst development tend to benchmark their catalysts in flow electrolyzers too17,18 to demonstrate the potential for practical application, already at the early stage of catalyst development. Several cell designs exist for low temperature CO2 electrolysis,16 which differ in the presence/absence of electrolytes and ion transport characteristics among others. Each of these cells has its specific advantages and limitations, but one feature is common: gas-phase CO2 is introduced to the cathode gas diffusion electrode (GDE), instead of being dissolved in the electrolyte as in the “traditional” H-type cells.19 The so-called triple-phase boundary, the gas/liquid/solid interface, results in improved mass transport for reactants and products. This leads to notably higher achievable CO2RR product partial current densities (exceeding 1 A cm−2),20,21 but also poses different challenges during system operation. For instance, carbonate precipitation and crossover is a major hurdle in the long-term and efficient operation of anion-exchange membrane (AEM)-containing CO2 electrolyzers.22,23 Furthermore, the different microenvironments in flow vs. batch electrolyzers,24 such as local pH and reactant/adsorbate concentrations, can open up new reaction pathways; therefore the mechanistic findings obtained in batch cells cannot explicitly be translated to flow conditions.
Despite the increasing number of flow cell studies to improve CO2RR performance, and operando investigations in batch-type cells dedicated to mechanistic understanding, only relatively few studies (less than 40) exist on the combination of the two. The main reason probably lies in the technical and practical challenges associated with the implementation of operando characterization in full electrolyzer devices (because of their complex and relatively fixed geometry). This would be, however, indeed necessary, as the interplay of several processes (e.g., cathode and anode reaction kinetics, ion transport, membrane properties) together determines the overall performance of these devices. Operando characterization of fuel cell and water electrolyzer devices is already at a more advanced stage.25,26 Considering their similarity to CO2 electrolyzers, these works can serve as examples and inspiration for the CO2RR community.
Previous works on CO2 electrolyzers have already nicely demonstrated the potential of operando methods towards the more realistic evaluation of catalyst/electrolyzer performance. Here, we review these studies with the aim to highlight how operando characterization can help to overcome the main challenges associated with CO2 (and also CO) reduction. We note that although high-temperature CO2 electrolyzers (i.e., solid oxide electrolyzers) offer high conversion efficiencies, because of the favorable thermodynamics, and fast reaction kinetics at elevated temperatures, they are out of the scope of this review, as they are very different from low temperature electrolyzers, which are our main focus here.
Continuous-flow CO2 reduction and status of the field
The main cell architectures for continuous-flow CO2 electrolysis have been thoroughly reviewed elsewhere; therefore here we give only a brief description of the different designs.16,23,27 The core component of continuous-flow CO2 electrolyzers is the gas diffusion electrode (GDE),28 allowing gas-phase CO2 to reach the catalyst, which results in enhanced mass transport. Here we note that we use the “flow cell” as a general denomination for all the CO2 electrolyzer devices employing gas-diffusion electrodes (GDE) with flowing gas-phase CO2 as the reactant, including both the zero-gap and the microfluidic architectures. The catalyst layer is deposited on one side of the gas diffusion layer facing the catholyte (if present) and the ion-exchange membrane (IEM). This latter separates the cathode and anode compartments. In a zero-gap configuration, the anode and cathode catalysts are in direct contact with the membrane separator, which together are called the membrane electrode assembly (MEA). The surface chemistry of the catalysts is largely determined by the membrane in such cells, and can be significantly different from the case when the catalysts are in contact with a bulk electrolyte (H-type cells or flow cells with electrolyte layers).29,30 The type of the separator determines the direction of the ion transport, which can be a cation-exchange (CEM), an anion-exchange (AEM), or a bipolar membrane (BPM; i.e., a CEM and an AEM stacked together). Importantly, none of these membranes is perfectly permselective,31 and even an AEM can let cations through (although in minor amounts), which turned out to be important in achieving high-performance AEM CO2 electrolysis.22The other major type of cell is the microfluidic device, employing a flowing electrolyte between the anode and cathode catalysts. In this configuration, it is possible that only one common electrolyte flows between the two electrodes,32 but membrane-separated (hybrid) cells, with separate anolyte and catholyte channels, are more commonly used for research purposes.33 In microfluidic electrolyzers a reference electrode can be placed in the catholyte and/or anolyte compartments, enabling the measurement of individual electrode potentials. This allows for easier decoupling of the processes happening in the cell. The microfluidic reactors are the most similar designs to the H-type cells, and thus are frequently used for catalyst testing at practically relevant current densities.
Remarkable progress has been made in recent years in terms of CO2RR selectivity and partial current density. This is particularly true for CO-production, which is very close to meet the industrial requirements.20,34 Our group was the first to reach 1 A cm−2 partial current density for CO formation, while maintaining high selectivity, conversion and low cell voltage in a zero-gap electrolyzer by applying a PiperIon membrane, which possesses a high carbonate ion conductance.20 Stable operation, exceeding 1000 h,34,35 has also been reported for CO2 to CO electroconversion; however there seems to be a trade-off between partial current density/selectivity and long-term operation.27 A recent roadmap on CO2RR highlighted that CO and ethanol could be the ideal products of electrochemical CO2 reduction, taking into account the market size, as well as the cost- and emission reduction when changing from current petrochemical production to electrosynthesis.36,37 CO can be upgraded to a wealth of products, such as transportation fuels through the Fischer–Tropsch process,38 and can also be electrochemically further reduced to C2+ products (CORR).38 The latter holds greater promise compared to the direct CO2 → C2+ conversion, considering energy efficiency and system design (carbonate-free operation). Interestingly, the highest current densities were reported for C2+ product formation21,27,39,40 in CO2RR. The high initial performance, however, was usually followed by quick deactivation, which may be attributed to catalyst restructuration/degradation or cell failure caused by either flooding or salt precipitation. For instance, a F- and K-modified Cu-catalyst showed a maximum of 53% FE for ethanol at a current density as high as 423 mA cm−2 recently; however, the longest reported performance test lasted only 12 hours with much lower (<20%) ethanol FE, which is still far from industrial relevance.41 We think that operando characterization will be a key tool to understand such performance loss mechanisms (Fig. 1).
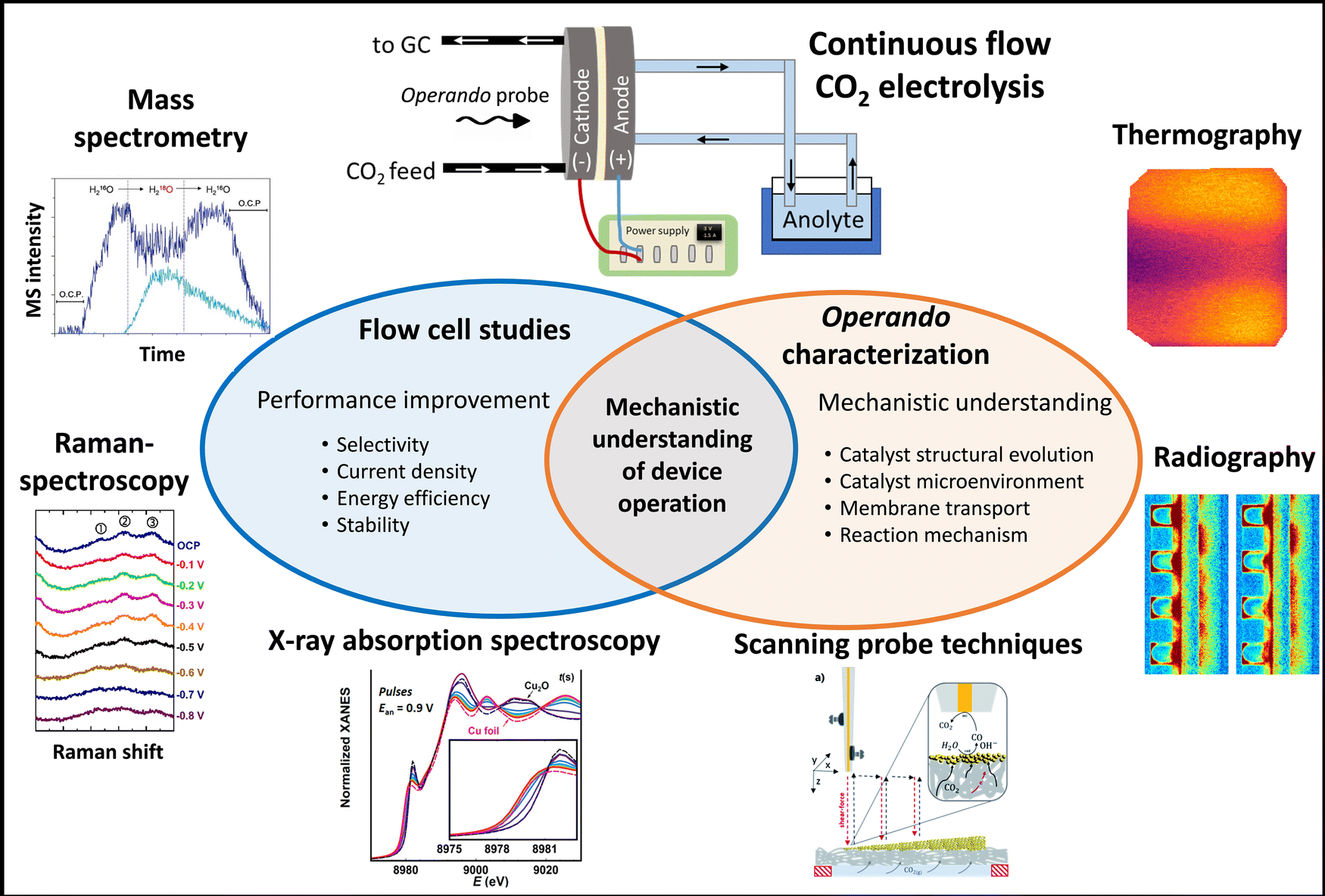 | ||
| Fig. 1 Combining continuous flow CO2 electrolysis with operando characterization. Reproduced from ref. 81, 100, 118, 125, 130 and 136 with permission from the copyright holders. Minor modifications have been made without altering the meaning of the original content. | ||
Batch versus flow electrolysis
In this section, we compare the conditions that prevail and the role of different cell components during CO2RR under low (H-type cells) and high (flow cells) current density conditions (Table 1). We aim to highlight how these different configurations, thus reaction environments, might influence reaction mechanisms and operando characterization tools under practically relevant conditions. In H-type cells, CO2 reaches the catalyst in the liquid-phase (i.e., dissolved in the electrolyte), which limits the maximum CO2RR current density (ca. to < 50 mA cm−2 for CO formation), because of the low solubility of CO2 in aqueous-electrolytes (ca. 30 mM).24 In contrast, in flow cells accommodating GDEs, gas-phase CO2 flow reaches the catalyst, significantly increasing the CO2 availability. Although the presence of a real three-phase boundary layer (gas–liquid–solid) has been debated,24,43 even if a thin electrolyte layer covers the active catalyst phase, the diffusion length for CO2 in GDE-based cells is at least one order of magnitude lower compared to that in batch cells.24 In H-type cells, bicarbonate electrolytes are almost exclusively used, as they can serve as a stock for CO2, increasing its effective concentration at the electrode, as demonstrated previously for both metal44 and carbon-based45 electrodes. As flow cells are not limited by CO2 solubility, they allow for the use of a much wider range of electrolytes.46 Hereby, the often-reported promotion of CO2RR by electrolyte components can be fully harnessed. Furthermore, the flow cell configuration can also assist mechanistic understanding, by allowing for a larger variability of experimental parameters,60 such as fluid flow rates or reactant compositions (gas mixtures), which can be better understood if combined with operando characterization.The use of alkaline electrolytes is widespread in flow CO2RR, because of the reduced CO2RR overpotentials, suppressed HER rates, and enhanced C2+ product formation in these environments.21,61 Performing CO2RR in strongly alkaline electrolytes in H-type cells, however, is not possible because the acid–base reaction between dissolved CO2 and OH− leads to significant pH decrease. As a consequence of the different electrolyte environments in flow and batch cells, the local pH may also differ in the two scenarios, influencing catalyst performance. A few studies reported local pH measurements during CO2RR by surface-enhanced infrared spectroscopy (ATR-SEIRAS),48,62 rotating ring disc (RRDE) voltammetry49 or scanning electrochemical microscopy (SECM)63 in batch configurations, and revealed notably (up to 5 units) higher pH near the electrode surface, compared to the bulk solution (Fig. 2(A) and (B)). This local pH variation can be attributed to the production of OH− (or consumption of H+) in both CO2RR and HER. It was found consistently that the surface pH (i.e., pH at the catalyst surface) is higher than the bulk pH of the electrolyte (i.e., pH at distances from the electrode surface larger than the diffusion layer thickness) during CO2RR when using bicarbonate electrolytes in both H-type and flow cells (Fig. 2(A) and (C)).48–51 During alkaline flow electrolysis, however, the neutralization reaction between CO2 and OH− can counterbalance the increase in the local pH (caused by the faradaic reactions), resulting in lower surface pH compared to the bulk electrolyte (Fig. 2(D)).52,53 This acid–base reaction can also be detrimental for electrolyzer operation if carbonate precipitates are formed.22,61,64 Additionally, the fluid streams in flow setups may also influence the local pH, either because of the continuous electrolyte refreshment or the change in the rate of CO2 + OH− reaction when the gas/electrolyte flow rates are varied.53
| Batch (H-type) cell | Flow cell | |
|---|---|---|
| a M: alkali metal cation. b CEM: cation exchange membrane, AEM: anion exchange membrane, BPM: bipolar membrane. | ||
| Achievable CO2RR current density20,21,24,27 | < 50 mA cm−2 | >1 A cm−2 |
| Reactant | Dissolved CO2/bicarbonate | Gas-phase/dissolved CO2 |
| Diffusion length of CO224,43,44,47 | ∼50 μm | ∼50 nm |
| Catholyte | MHCO3a | MOH, MHCO3, none |
| Local pH at the cathode48–53 | Near-neutral or alkaline | Alkaline |
| pH gradient48–53 | pHsurface > pHbulk (up to 5 pH unit difference) | Bicarbonate electrolyte: pHsurface > pHbulk, alkaline electrolyte: pHsurface < pHbulk |
| Catalyst support6,54–57 | Flat (glassy carbon, metal foil) or porous (carbon paper, metal foam) | Porous (carbon paper, metal foam, PTFE) |
| Ionomer16,29,58 | Not mandatory, mainly catalyst binder, but can change morphology, surface chemistry | Microfluidic: same role as for H-cell; zero-gap: ensures ionic conductance |
| Membrane59 | Type: CEM/AEM; role: product separation; does not limit cell performance | Type: CEM/AEM/BPMb/none; role: product separation, in zero-gap-cells it determines catalyst surface chemistry; can limit cell performance |
| Surface concentration of reactants/intermediates24 | Lower | Higher |
| Electrode configuration | 3-Electrode, cathode potential | 2 or 3-electrode, full cell voltage/cathode potential |
 | ||
| Fig. 2 Experimentally determined pH values at the catalyst surface in different cell configurations and electrolytes. (A) Local pH at the surface of a gold catalyst in a CO2-saturated KHCO3 electrolyte, determined by rotating ring disc voltammetry. Reproduced from ref. 49 with permission from John Wiley and Sons. (B) Surface pH of a copper electrode in contact with CO2-saturated phosphate buffers determined by surface-enhanced infrared spectroscopy. Reproduced from ref. 62. Copyright 2019 ACS. (C) Local pH measurements in a continuous-flow bicarbonate electrolyzer with a silver foam cathode by Raman spectroscopy. Reproduced from ref. 51. Copyright 2020 ACS. (D) Local pH measurements with Raman spectroscopy on the surface of a Cu GDE in contact with a flowing 1 M KOH electrolyte and gas-phase CO2. Reproduced from ref. 52. Copyright 2020 ACS. | ||
Electrolyte flow, in principle, can be advantageous for operando experiments, helping the removal of gas bubbles from the electrode surface which usually makes spectroscopic data collection challenging.12 Gas bubbles sticking to the electrode can also result in severe potential variations along the electrode surface,65 causing local activity changes. The porous, gas permeable structure of GDLs can mitigate this issue; however, their use also limits the implementation of certain operando techniques. For instance, FTIR spectroscopy (to detect surface adsorbates) cannot be performed in the more surface-sensitive ATR (internal reflection) configuration on GDEs, because it requires the deposition of a very thin catalyst layer on the surface of an ATR crystal.13,62,66 To mimic flow cell conditions, only external reflection (IRAS) can come into consideration, with an ultrathin electrolyte layer between the IR transparent window and the catalyst.13,67 This would come, however, at the expense of decreased surface sensitivity with additional technical challenges (reduced time resolution, solvent bands),13 which explains why operando IR spectroscopy has not been implemented in GDE-setups yet. On the other hand, carbon-based GDLs are particularly advantageous for operando X-ray absorption (XAS), as the relatively thin (few hundred μm) carbon papers are transparent for hard X-rays, as opposed to metal or thicker glassy carbon substrates commonly employed for H-cell studies.12
Apart from the catalyst microenvironment, it is also worth to compare the role of particular cell components in batch and flow electrolysis. To start with the core constituent, in principle the same or similar catalyst materials can be used in both setups. Nevertheless, the optimal reaction conditions can be substantially different in the two configurations; therefore catalysts have to be optimized for the two types of measurements separately. For instance, while HER dominated on Cu nanocatalysts in an H-type cell, when performing CO2RR using the same catalysts in an alkaline flow electrolyzer, FEH2 was largely suppressed with the concomitant increase in the FE for C2+ products (Fig. 3).17 Another example can be the case of nickel–nitrogen doped carbons, often outperforming metal catalysts in terms of CO production rate in H-cells and CO formation FE in flow cells, but their maximum reported stability was limited to a few tenth of hours in flow cells,68,69 because of rapid flooding at high current densities (note the similarity of the carbon skeleton to the carbon paper supports). One also has to consider which performance metric is in the focus of interest (e.g., selectivity, current density, energy- or CO2 conversion efficiency) as different system design rules might apply for the optimization of each.60
 | ||
| Fig. 3 Selectivity of the same Cu nanocatalysts in an H-cell using a CO2-saturated 0.1 M KHCO3 electrolyte (A) and in a gas-fed microfluidic flow cell with a flowing 1 M KOH electrolyte (B). Reproduced from ref. 17. Copyright 2020 ACS. | ||
Notably, flow cells bear higher experimental complexity, as several other processes other than the kinetics of cathode half reaction can limit their performance at high reaction rates, such as the paired anode process,27,70 the ionic conductivity of the membrane, or GDE flooding.20 Furthermore, the determination of the individual electrode potential is not always straightforward at high currents and particularly in zero-gap devices.71,72 Therefore, when we aim to correlate the catalyst structure/microenvironment in a flow electrolyzer to its performance using operando spectroscopy, we have to make sure that it is not limited by other factors.
Membranes are primarily used to separate the anode and cathode compartments of the cells, and therefore to inhibit product crossover. Moreover, they transport water and ions between the two half-cells and determine the type of ionic charge carriers (anions/cations) and the direction of their movement in the cell.59 In zero-gap devices, where the catalysts are in direct contact with the membrane, they control the microenvironment (e.g., pH and near-surface components) and thus the surface chemistry of the catalysts. Furthermore, they can also significantly influence the water management in the cell.73 Because of this, the catalyst layer should be preferably characterized together with the membrane; therefore the development of in situ/operando techniques for the characterization of full zero-gap devices or MEAs30 should be urgently promoted. The above effects are less pronounced in H-type cells and microfluidic flow cells, because of the presence of buffering electrolyte layers. The ionomer besides acting as a catalyst binder can also modify the catalyst microenvironment and morphology, moreover can also actively participate in the CO2RR/CORR by interacting with reactants or intermediates.29,74 In the MEA case, it also ensures ionic conduction between the catalyst particles as well as between the catalyst and the membrane.
Challenges in continuous flow CO2 electrolysis and how operando spectroscopy can overcome these
Before summarizing the existing reports on operando characterization of continuous-flow CO2 electrolyzers, we would like to give a brief overview of the main challenges associated with this rising technology. We also underscore how operando characterization can be a tool to help overcome these challenges (Table 2). Keeping the overall energy consumption of the cell as low as possible is one of the main goals from an economic point of view. The energy efficiency is directly related to the cell voltage, which is the sum of the anodic and cathodic half-cell voltages and ohmic losses originating from the membrane and electrolyte layers (if present). From a materials-science perspective, developing new catalysts with reduced overpotential towards the desired product is the most straightforward way to reduce the energy consumption.7,75,76 Finding alternative catalysts (e.g., bimetallics) or increasing the abundance of specific structural motifs or crystal facets17 selectively producing certain products are strategies to overcome the above-mentioned limitation. Operando spectroscopy, providing information on the catalyst bulk or surface structure (e.g., XAS, XPS, Raman), has largely contributed to catalyst improvement5,6,85 already, and will continue to do so hopefully under practically relevant conditions too. For a fixed anodic and cathodic reaction pair, however, we cannot go below the thermodynamic cell voltage, which is 1.33 V for CO production coupled with the anodic OER.77,86 Nevertheless, this value can be lowered to more than 1 V when using alternative anode reactions, for instance the increasingly studied glycerol oxidation.77,87| Challenges | Strategies to meet the challenge | Contribution of operando characterization |
|---|---|---|
| a XAS: X-ray absorption spectroscopy, XPS: X-ray photoelectron spectroscopy, EIS: electrochemical impedance spectroscopy, EC-MS: electrochemical mass spectrometry. | ||
| Improving energy efficiency5,6,59,77,78 | • Lowering anode and cathode potentials | • XASa, XPSa, Raman |
| ○ More active catalysts | ||
| ○ Alternative anode reactions | ||
| • Improving membrane conductivity | • Radiography, EISa | |
| Production of multi-carbon molecules4,79–81 | • Novel catalyst materials | • XAS, XPS, Raman |
| • Active site stabilization | ||
| • Optimization of local pH, CO2/CO concentration | • Local pH, Raman, IR | |
| • Mechanistic understanding of C–C coupling | • Raman, IR, EC-MSa | |
| • CO reduction | ||
| Stability improvement (>1000 h)22,59,82 | • Wet-proof GDEs | • Radiography |
| • Mitigating carbonate formation | • Local pH and ion concentration measurements | |
| • Improving membrane durability | • IR, Raman | |
| Efficient CO2 utilization23,83 | • Mitigation of CO2 and product crossover | • Radiography |
| • Local detection of CO2 and products | ||
| Scaling-up55,84 | • Scale-up or scale-out | • Local measurement of reactant/product concentrations, temperature, humidity (distribution) |
A significant portion of the energy input in flow electrolyzers can be lost at the membrane71,88 (Fig. 4(B)), because of its low ionic conductivity. As carbonate and bicarbonate ions were identified as main charge carriers in AEM CO2 electrolyzers20,89 (Fig. 4(A)), producing membranes that possess high conductivity for these ions is a prerequisite for further development. The water uptake of the membrane also significantly influences its ionic conductivity,31 which can be studied by neutron or X-ray radiography during electrolyzer operation.78,90,91 These techniques therefore have the potential to identify reasons for ohmic losses in CO2 electrolyzers.
 | ||
| Fig. 4 (A) Proposed carbon balance paths in an AEM electrolyzer with a flowing KHCO3 electrolyte. Reproduced from ref. 89. Copyright 2020 RSC. (B) Overpotentials during CO2RR at 200 mA cm−2 for each of the functional components in membrane/BPM (red), membrane/AEM (orange), and hybrid/AEM (navy) electrolyzers. Values were determined by using an analytical flow cell capable of resolving voltage drops across individual electrode and membrane components during CO2RR. Reproduced from ref. 71. Copyright 2019 ACS. (C) Alkali metal ion “activation” of an AEM CO2 electrolyzer operated with a pure water anolyte. Reproduced from ref. 22. Copyright 2021, the Author(s), under exclusive licence to Springer Nature Limited. | ||
The experimental conditions can have significant influence on the formation of multicarbon products. High local pH79 and intermediate surface CO concentration80 were found to be beneficial for C–C coupling, while suppressing the competitive HER and methane formation. To understand how these parameters affect reaction mechanisms, at high current densities, operando local pH measurements and Raman/IR spectroscopy (to detect surface CO2/CO concentrations) should be implemented in GDE-setups too. In addition, electrochemical mass spectrometry with isotopic labeling can also largely contribute to our understanding of mechanistic pathways towards certain products. Last but not least, reduction of CO has several benefits compared to CO2 reduction, such as the lower overpotential for C2+ products,92 higher near-surface CO concentration, and the absence of carbonate formation in alkaline electrolytes.93 As cell designs for CO and CO2 reduction are very alike, operando methods can be similarly implemented in CO electrolyzers as well giving additional insights into C–C coupling under high current density conditions.
Several degradation mechanisms can lead to unstable electrolyzer performance, such as GDE flooding, formation of carbonate precipitates, catalyst deactivation, and membrane degradation. The most commonly employed gas diffusion layers (GDLs) are carbon-based ones (i.e., carbon paper or cloth). One of the major obstacles when using these supports is their tendency to lose their hydrophobicity which consequently leads to the domination of the competing hydrogen evolution reaction (HER), resulting in flooding.94 In flow cells with electrolyte layers, flooding usually occurs even after a few hours (if not less) of operation,94 largely reducing the lifetime. To mitigate this issue, PTFE-based GDLs or metal foams are being investigated, but no breakthrough has been achieved so far. Nevertheless, the properties of the GDLs (thickness, porosity, PTFE content) largely influence the CO2RR performance, as demonstrated in others and our recent work too.42 The formation of (bi)carbonate precipitates under locally alkaline conditions (AEM electrolyzers) can also accelerate GDE flooding.95 Moreover, these precipitates block the transport of CO2 to the catalyst active sites too.22 This is a major hurdle particularly in zero-gap devices, because the thin electrolyte layer covering the catalyst can easily become saturated with these salts. Periodic rinsing of the cathode with water55 or pulsing the potential64 were two approaches suggested to mitigate this problem.
Operating zero-gap AEM CO2 electrolyzers with a pure water anolyte would be highly desired, as the absence of alkali cations in the system would inherently circumvent precipitate formation, in addition to the advantage of eliminating the use of expensive and dangerous alkali hydroxides. Our group recently showed that high performance water-based operation is only possible when small amounts of alkali cations (i.e., K+ or Cs+) are present near the cathode (Fig. 4(C)).22 Although the exact mechanism of their participation in the CO2 electrolysis is not clear yet, this coincides with the observed promotional effect of cations in CO2RR under batch electrolysis conditions.96–99
The aforementioned cell failure mechanisms are closely connected to the water management of the cell; thus their mitigation can be assisted by operando techniques that can give direct evidence on the relative humidity conditions or water content of the cell components. In this sense, radiographic analysis78,91,100 or the incorporation of relative humidity and temperature sensors in the electrolyzers73 are expected to contribute with novel insights. Additionally, methods that can locally measure the metal ion concentrations in the vicinity of the GDEs would also be highly desired.
Much less attention has been paid to possible membrane degradation mechanisms in CO2/CO electrolyzers.59 In general, CEMs have around one order of magnitude higher proven operational lifetimes compared to AEMs.101 One degradation mechanism of AEMs includes the nucleophilic attack of OH− on the cation functional groups and the polymeric backbone of the membrane, especially when they are not fully hydrated.102 Moreover, the formation of liquid products, such as ethanol, can also accelerate AEM degradation.59 In the future, more works should aim at both the post-mortem and in situ/operando characterization of ion exchange membranes,103,104 although the latter can be experimentally extremely challenging.
AEM CO2 electrolyzers suffer from significant loss of CO2 at the anode, because of the transport of carbonate and bicarbonate through the membrane, reducing carbon efficiency (Fig. 4(A)). CO2 electrolysis in an acidic cathode environment (e.g., using CEMs) could overcome this issue, but it generally results in low CO2RR selectivity, because of the favored HER. Recently, under certain experimental conditions, however, CO2 was reduced with high selectivity to CO and even to C2+ products in acidic electrolyzers. A common feature of these works was the abundance of weakly hydrated alkali metal cations (K+ or Cs+) in close proximity of the cathode.39,105–107 This finding may lay down a new research path for acidic CO2 utilization, which could largely benefit from operando characterization uncovering the local cathode microenvironment. Not only CO2, but also liquid products may cross the membrane, which is particularly significant for anionic species in AEM CO2 electrolyzers.108 This, on the one hand, inhibits precise FE calculations if not properly accounted for, and on the other hand results in extra downstream separation costs. Local and real-time detection of CO2RR products in the cathode vicinity is one possibility to mitigate the issue with performance assessment, but the challenges associated with detecting non-volatile liquid products using electrochemical mass spectrometry should be noted.109 A better and universal solution would be the development of AEM membranes with functional groups blocking the movement of all liquid products. Tuning the membrane water volume fraction was also found to be a useful strategy to reduce product crossover.110
Last, but not least, scaling up of CO2/CO electrolyzers is associated with several challenges (such as fluid- and heat management). One very promising scale-up strategy is the construction of large size and multi-cell stacks, as for fuel cells and water electrolyzers. Such cell stacks were already demonstrated for CO2 electrolyzers both with and without a liquid catholyte,55,111–113 including our pioneering work for a zero-gap architecture.55 As the size of the cell and the number of cell stacks increase, significant local variations in the different operational parameters within the device can be expected. For instance, CO2 concentration gradients can develop within large cells, particularly at high CO2 conversion rates. This can shift the selectivity towards HER; therefore measurement of reactant and product concentrations at different positions of the cell might be required. Similarly, mapping the temperature and relative humidity during operation can also become necessary by placing sensors inside the electrolyzer or using other methods, such as thermography. These data can additionally serve as inputs for multi-physics modeling which can help to identify optimal operational parameters and support technological development.
Operando characterization of CO2 electrolyzers
Table 3 summarizes the existing operando techniques in continuous-flow CO2 reduction with their main advantages and limitations. In the following subsections we give an overview of these focusing on what kind of information can be deduced from these measurements.| Operando technique | Information content/main advantages | Main limitations | Time resolution | Spatial resolution |
|---|---|---|---|---|
| a XAS: X-ray absorption spectroscopy, SECM: scanning electrochemical microscopy, AFM: atomic force microscopy, MS: mass spectrometry. b x and y are directions in the plane of the GDE, while z is perpendicular to that. | ||||
XASa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18,113,115,117,119–123 18,113,115,117,119–123 |
• Catalyst structure (oxidation state, coordination environment) | • Bulk sensitive | • Typically 20–30 minutes | N/A |
| • Only minor cell modification needed | • Synchrotron irradiation | • Subseconds for QXAFS | ||
| Raman spectroscopy51,52,124,125 | • Catalyst structure (oxidation state) | • SERS effect only on certain metal nanostructures | • Few tenths of seconds | • μm-scale (xb) |
| • Adsorbed intermediates/products | • Less suitable for zero-gap design | |||
| • Local pH with spatial resolution | ||||
| • Widely available at lab-scale | ||||
| Fluorescent microscopy50 | • Local pH measurement with spatial resolution in the x, y, z direction | • Fluorescent probe added to the electrolyte might change the local reaction environment | N/A | • μm- scale (x, y, zb) |
| • pH measurement within catalyst trenches | ||||
| UV-Vis spectroscopy30 | • pH measurement at the catalyst/membrane interface (MEA-design) | • Significantly different cell design compared to the real device (in situ) | N/A | • nm-scale (z) |
| SECMa | • Local reactivity and local pH | • Low achievable current densities | N/A | • μm-scale (x, y) |
| • Spatially resolved activity maps | ||||
| AFM126a |
• High resolution topography and mechanical property maps | • Small scan size | • Few minutes | • Few tenths/hundreds of nm |
| • Rapid flooding at high currents | ||||
| MS81,127a |
• Real-time detection of products/intermediates with low detection limit | • Deconvolution of signals (e.g., CO, CO2) can be challenging | • Seconds/tenths of seconds | N/A |
| • Mechanistic information | ||||
| • Isotopic information | ||||
| Neutron/X-ray radiography78,91,100,128,129 | • Water management | • Neutron/synchrotron facility | • Few seconds for X-ray few tenths of seconds for neutron | • μm – scale (x, y) |
| • Gas evolution | • Limited elemental information | |||
| • Precipitate formation | ||||
| • Operando cell failure diagnostics | ||||
| • Only minor cell modification needed | ||||
| Thermography130 | • Local activity mapping | • One-to-one correlation between temperature change and activity has to be proved | • Depends on catalyst substrate, layer thickness, etc. | |
| • Noninvasive probing to assess activity distribution | ||||
X-ray absorption spectroscopy
X-ray absorption spectroscopy is a core-level X-ray technique, measuring the X-ray absorption coefficient as a function of energy.114 It is element-specific, and suitable for the investigation of a wide range of materials including ordered, disordered, amorphous or even liquid-phase ones. Unlike X-ray photoelectron spectroscopy, it does not require high vacuum conditions; therefore it is excellently suited for operando measurements of electrocatalysts, reviewed recently.12 Nevertheless, to reach high signal to noise ratios, thus to allow for extraction of high quality data, usually synchrotron irradiation (high flux of high energy X-rays) is needed. The measured spectra can be divided into two regions.12 The XANES (X-ray absorption near edge structure) region is located in the vicinity of the absorption edge, and provides information on the chemical state of the absorbing atoms, local symmetry, and chemical bonding. The EXAFS (extended X-ray absorption fine structure) region, covering a range of up to a few thousand eV above the absorption edge, is sensitive to the arrangements of nearest neighbors around the absorbing atom, such as interatomic distances and coordination numbers. One limitation is its bulk-averaging nature, which must be kept in mind when studying electrocatalysts. This can be mitigated in part by measuring thin catalyst layers,115 provided that this enables sufficient signal intensity. Furthermore, recording an XAS spectrum typically takes 20–30 minutes, which impedes tracking fast processes. At some specialized beamlines, however, the quick-XAFS (QXAFS) method is available, allowing for time-resolution in the millisecond range.5,116–118X-ray absorption spectroscopy is currently the most widely applied operando spectroscopy to study the evolution of catalyst structure during CO2RR under high current density conditions.18,113,115,118–123 Because of its great compatibility with flow cell designs, the operando measurement requires only minor cell modifications (Fig. 5). The absence of electrolyte in the beam path when measuring in the back-illumination fluorescence mode eliminates complications associated with gas bubble formation in the electrolyte. Flooding or electrolyte perspiration through the pores of the GDE can, however, be detrimental for the measurement. To allow for the X-ray beam to pass through the cell body (usually made of metal or plastic), most frequently a polyimide window is cut into the cathode end plate and flow field. This, however, can disrupt the flow field (thus reactant transport), possibly resulting in performance variations compared to the unmodified cell. In the fuel cell field, the application of thinned graphite flow-fields, which are transparent to X-rays and have identical geometry as the flow-field in the original cell, has been reported.131–133 This design may be adopted in the future for CO2 electrolyzers as well, creating a more representative environment for operando tests.
 | ||
| Fig. 5 Schematic illustration and photograph of a microfluidic operando XAS cell allowing for fluorescence measurements. Reproduced from ref. 115. Copyright 2020 RSC. | ||
Different copper catalysts have been investigated by operando flow cell XAS in recent years.113,115,118–120 In one work, the effect of electrolyte, applied current density, and catalyst layer thickness was studied on a sputtered Cu cathode.115 No obvious changes were found for a 100 nm thick Cu catalyst layer when tested at different current densities (1 and 100 mA cm−2) in a KHCO3 electrolyte: in both cases only metallic Cu was present during CO2RR. Based on the determined Cu–Cu interatomic distances and Debye–Waller factors (measure of disorder in the crystal lattice), higher disorder was found for the 20 nm thick sample, compared to the 100 nm one under reaction conditions (100 mA cm−2), which seemed to be less favorable for C2+ product formation. In another work, a KOH-incorporated Cu catalyst (Cu–KOH) was tested by operando XAS in a zero-gap device.113 This catalyst, consisting of fully oxidized copper species (Cu2+) in the as-prepared state, showed improved performance for ethylene production (54.5% FE, 153 mA cm−2) compared to the reference Cu black, Cu2O and CuO samples. Operando XAS measurements revealed that although the material was reduced to a large extent to Cu0 during CO2RR (Ecell = 2.5–4 V), minor amounts of oxidized Cu species survived under the harsh reducing conditions. The Cu–KOH electrode contained a higher fraction of Cu(OH)2 compared to the reference Cu black, which was explained by its special (echinoid-shaped) morphology and defect-rich structure originating from the KOH incorporation. The high density of defect sites most probably induced a local pH increase, and resulted in the partial re-oxidation of the metallic Cu during CO2RR. The synergy between remaining Cu2O, Cu(OH)2 and metallic Cu was considered to be responsible for the improved ethylene selectivity. In contrast to the latter finding, full reduction of a Cu(OH)2 catalyst to metallic Cu was reported in a microfluidic operando XAS flow cell after 1 hour of reduction,120 possibly indicating that the different catalyst environments in microfluidic vs. zero-gap devices can affect the reducibility of Cu.
Pulsed CO2RR (p-CO2RR) was found to be an easy, yet versatile technique to steer the selectivity of copper catalysts, because of the unique changes induced in the catalyst structure, its microenvironment and the electric double layer when pulsing the potential.117 In this dynamic reaction environment, only operando techniques are able to provide mechanistic insights. In this regard, QXAFS with second/subsecond time resolution is a key tool to follow changes in the catalyst structure even during individual pulses117,118 (Fig. 6). Pulsed CO2RR was recently applied to a Cu nanocube catalyst in a microfluidic flow cell, and it was found that the C2/C1 selectivity can be tuned by adjusting anodic potential (Ea) while fixing the cathodic value (Ec).118 While at Ea = 0.9 V (vs. RHE), the selectivity towards C2+ products slightly increased compared to the potentiostatic reduction, higher Ea values (i.e., 1.2 V) favored CH4 formation over C2H4. Operando QXAFS revealed gradual reduction of the dominating Cu2O phase in the as-prepared catalyst to Cu0 during static reduction, as well as under pulsed conditions with lower Ea (Fig. 6(A)). Even though metallic Cu re-oxidized to Cu+ during the anodic pulse (Fig. 6(B)), its much faster reduction during the cathodic pulse resulted in a net reduction of the catalyst over time (Fig. 6(A)). With the higher anodic potential (Ea = 1.2 V), a small fraction of Cu2+ also appeared at the initial stage of CO2RR, but its concentration rapidly dropped to zero (conversion back to Cu+). In this case, a significant fraction of Cu+ remained intact (ca. 30%) for the whole duration of the experiment. These findings were complemented with the more surface sensitive SERS, which also confirmed the accumulation of Cu2O when pulsing with Ea = 1.2 V, originating from the reaction between metallic Cu and OH− ions during an anodic pulse.
 | ||
| Fig. 6 (A) Time-dependent XANES and FT-EXAFS spectra at the Cu K-edge during pulsed CO2RR on Cu nanocube catalysts. Results of the linear combination XANES fitting under potentiostatic and pulsed reduction. (B) Periodic oxidation and reduction of the pre-reduced catalysts followed by QXAFS during individual (60 s) pulses. Reproduced from ref. 118. Copyright 2021 the Authors, published by ACS. | ||
Operando QXAFS was also employed to study alloying processes in bimetallic catalysts during CO2RR, revealing active phases responsible for certain products. Zn-decorated Cu2O nanocubes with varying Zn-contents were investigated at current densities up to 500 mA cm−2 in a microfluidic flow cell.119 While the pure Cu2O exhibited high FE for C2 products (41% C2H4 and 12% EtOH at −500 mA cm−2), the increase in Zn-content resulted in a gradual enhancement of the CO selectivity (from 18 to 53%) and a decrease in C2+ products. The operando measurements revealed that most of the Cu+ (Cu2O) and Zn2+ (disordered ZnO) species in the as-prepared catalysts were reduced to the respective metallic states within 60 s. Linear combination analysis (LCA) of the XANES data suggested faster reduction of Zn2+ than Cu+, and revealed that the presence of Zn stabilizes Cu+ during CO2RR. The quick reduction of Cu2+ and Zn2+ was followed by a gradual and significantly slower transformation of the Cu and Zn phases into CuxZny alloys, which coincided with a time-dependent change in selectivity. Neural-network (NN) EXAFS analysis further uncovered that an increasingly Cu-rich (fcc-type) structure is formed at longer reaction times and that the contribution of a non-fcc type (Zn-rich) alloy is higher at higher initial Zn contents. QXAFS in combination with LCA-XANES and NN-EXAFS analysis was used to correlate redox peaks on the cyclic voltammograms of the bimetallic samples with the chemical evolution of Cu and Zn species, albeit at low current densities. This was complemented with operando SERS to deconvolute surface and sub-surface changes. These investigations revealed easier reduction of Cu+ located at the surface than in the bulk, and showed that the reduction peak of bulk Cu+ positively shifts in the presence of Zn (in a Cu–Zn alloy). The latter works nicely demonstrated the rich information content of operando XAS measurements when combined with advanced data analysis (machine learning) and complementary surface-sensitive techniques.118,119
Silver is the state-of-the-art catalyst for CO2 reduction to CO, but interestingly only a couple of studies aimed at its operando XAS investigation at high current densities.115,122 A sputtered Ag catalyst was studied in a microfluidic flow cell at current densities between 1 and 200 mA cm−2.115 An increase in crystallite size and a decrease in Ag–Ag distance during CO2RR were found, compared to the as-prepared (ex situ) sample. At higher current densities (≥100 mA cm−2), the presence of Ag–O bonds was also identified. Silver dendrites grown on a W seed were found to be efficient catalysts, showing 400 mA cm−2 CO partial current density in a zero-gap AEM electrolyzer.122 The structure of the catalyst was studied by operando XANES and depth resolved XPS measurements, which revealed residual Ag+ species (AgxCyOz) in the near-surface region under CO2RR conditions. The authors assumed that the large number of defects and undercoordinated Ag on the Ag-dendrite branches resulted in enhanced electric fields, which reduced the thermodynamic barrier for CO2RR and resulted in improved performance compared to the Ag/C benchmark.
With the aim to develop low-cost and earth-abundant catalysts for the conversion of CO2 into CO, Zn-based catalysts are being researched. A zinc pyrophosphate (Zn2P2O7) (pre)catalyst delivered up to 441 mA cm−2 current density for CO production at a low potential (−0.87 V vs. RHE), with this being among the best Zn-based CO2RR catalysts. The excellent performance was attributed to the stabilization of low-valent Znδ+ species during CO2RR as evidenced by time-dependent Raman and operando XAS measurements. Raman spectroscopy showed the presence of Zn–O bonds even after prolonged (300 min) electrolysis at 400 mA cm−2 current density. LCA of the XANES spectra revealed that 22% of the catalyst remained in an oxidized state after 35 minutes of CO2RR, but these measurements were performed only at 50 mA cm−2.123
The element-specificity and sensitivity to the local structure around the absorbing atom make XAS excellently suited for the investigation of metal–nitrogen doped carbon (M–N–C) catalysts, a promising family of single atom catalysts for CO2RR.7,54 The disordered nature and heterogeneity of these materials, and the sample-averaging nature of XAS, however, pose significant challenges in data analysis.12 Ni–N–C materials were found to be excellent catalysts for CO production, but their long-term stability is a major issue, particularly at high current densities.18,68,121 This might be overcome by understanding the evolution of the active metal sites during operation. A catalyst containing dinuclear Ni2 sites anchored on a N–C matrix exhibited 94.3% FE towards CO at 150 mA cm−2 current density in a microfluidic flow cell. Operando XAS measurements together with ex situ characterization revealed the presence of Ni–N4 moieties with D4h symmetry under open circuit conditions and a decrease in the Ni oxidation state with the application of reducing potentials. The Ni–N and Ni–Ni coordination numbers by EXAFS curve fitting in the as-prepared state were found to be 4.3 and 1.2, respectively. From this, the authors suggested the presence of a Ni2–N6 configuration, in which two Ni–N4 units are connected through bridge N-atoms. In the electrolyte (without polarization), these Ni2N6 motifs adsorbed oxygen containing species, and formed an O–Ni2–N6 configuration with decreased Ni–Ni distance having an oxygen bridge between the two Ni atoms. The application of a reducing potential resulted in further shortening of the Ni–Ni distance, indicating that the presence of dinuclear Ni sites is the key for the CO2RR activity.121 NiS nanoparticles were found to outperform a Ni–N–C catalyst in terms of FECO at current densities above 300 mA cm−2 in a zero-gap electrolyzer, recently.18 This was the first demonstration that a bulk Ni-compound exhibited high activity for CO production. To explain the origin of this intriguing selectivity, operando XAS analysis was performed. The high FECO for NiS was related to its similar local structure to that of Ni–N–C. According to the EXAFS analysis, the structure of the NiS electrode could be characterized by a distorted Oh symmetry with OxSy ligands. During CO2RR, this structure lost oxygen, and a distorted D4h symmetry was formed (Jahn-Teller effect), similar to the geometry of the Ni–N4 motifs in Ni–N–C. Prolonged electrolysis in alkaline media, however, led to the loss of S species and the formation of Ni-hydroxide (evidenced by XPS), and thus the mimicked electronic structure disappeared. This resulted in loss of CO2RR activity after 2 hours.18
Raman spectroscopy
Raman spectroscopy is an effective tool to study the chemical composition and structure of metal/metal oxide catalysts, as well as adsorbed intermediates and near-surface species during CO2RR.15,134 Surface-enhanced Raman spectroscopy (SERS) benefits from the enhancement of the Raman signal on certain metals (Ag, Au, Cu) and nanostructured electrodes, greatly improving the sensitivity and specificity of the technique.134 It has been widely applied to identify active sites and to detect changes in the oxidation state of catalysts during reactions.15 The possibility of detecting reaction intermediates, like *CO or *CHO, makes its use very important in mechanistic studies85 and in the investigation of the effect of reaction conditions (e.g., electrolyte effects). Furthermore, local pH measurements can also be performed, by detecting the near-surface ratio of buffer components, such as CO32−/HCO3− or H2PO42−/HPO42−/PO4314. One major advantage, compared to the complementary IR-spectroscopy, is the weak Raman scattering of water because of which measurements in aqueous electrolytes can be easily performed, which is very relevant for CO2RR studies. This allows for a larger flexibility of cell designs and makes it suitable for the investigation for gas diffusion electrodes too.51–53 Despite all these advantages, only a few works applied operando Raman spectroscopy under practically relevant conditions.51–53,124,125 The relatively fixed design of flow CO2 electrolyzers (e.g., anode and cathode strictly facing each other, membrane separation, carbon-based GDEs or porous frits as catalyst supports) limits the achievable cell designs for performing operando Raman spectroscopy on a full-cell device. To the best of our knowledge only two works attempted to use Raman-spectroscopy in full electrolyzers (Fig. 8), to measure the local pH.51,53 For the study of the catalyst surface structure and adsorbed intermediates, spectroelectrochemical flow cells, able to host GDEs (Fig. 7(A)), were reported, although in this configuration the operating conditions can be different from that of a practical device.52,124,125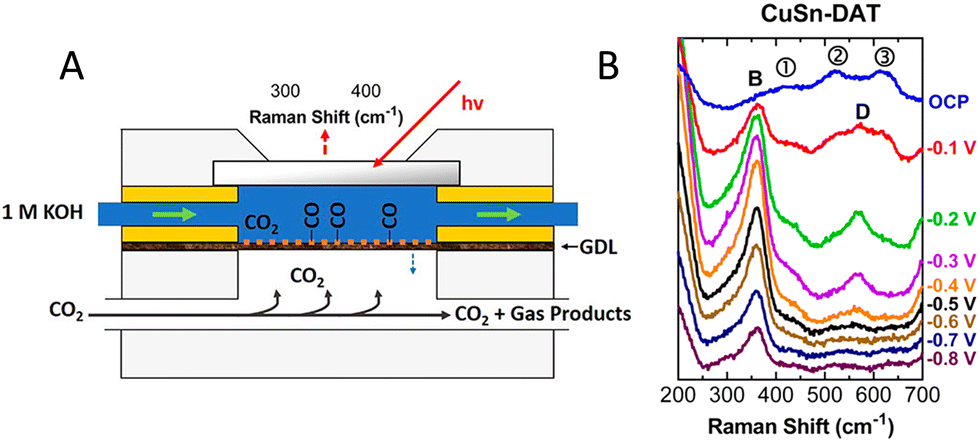 | ||
| Fig. 7 (A) Schematic and photograph of a Raman spectroelectrochemical flow cell hosting a gas diffusion electrode. The cell mimics the conditions of a microfluidic CO2 electrolyzer. (B) Operando SERS of a CuSn electrode during CO2RR measured in the cell. Peak B is assigned to the frustrated rotational mode of adsorbed CO on Cu. Peaks D and 1, 2, 3 are SnO2- and Cu2O-related bands. Reproduced from ref. 125. Copyright 2019 ACS. | ||
In one of these works, the potential-dependent structural evolution of the catalyst and the presence of surface adsorbed intermediates were studied on bare Cu, CuAg and CuSn alloys.125 Interestingly, the Cu2O-related peaks persisted during CO2RR at potentials down to −0.8 V vs. RHE (Fig. 7(B)), despite the thermodynamically favorable reduction to Cu under the given pH–potential conditions. The authors revealed that the intensity ratio of the peak related to the *CO stretching and the T2g mode of Cu2O showed very similar potential-dependence to the faradaic efficiency of CO. The highest ratio was found for the CuSn sample, correlating with its highest CO-selectivity. Overall, the results indicated that Cu2O was formed in the reaction of Cu with hydroxide ions (byproduct of CO2 reduction), but at higher concentration its presence blocked the active sites. Alloying Cu with Sn helped to reduce the passivating Cu2O layer to the zero-valent state, resulting in increased density of active sites. The higher CO availability on the CuSn catalyst surface also led to higher FE for C2+ products. The same group also studied polymer-incorporated Cu electrodes by operando Raman spectroscopy in a GDE-setup.124 The presence of polymers on the Cu surface altered the surface reactivity, and the degree of methylation of the polymers correlated with the CO2RR activity/selectivity. With an amine-rich polymer, 87% FE for ethylene was achieved in a highly alkaline medium (10 M KOH). On this electrode, operando Raman spectroscopy revealed the presence of an adsorbed CO (*CO) intermediate at a cathode potential of −0.47 V (vs. RHE), which could not be detected on the bare Cu and when a polymer with a high level of methylation was used. The latter catalysts showed significantly decreased ethylene (and overall CO2RR) selectivity. The presence of amino groups, thus, seemed to help the accumulation of *CO, thereby assisting C–C coupling. Peaks assigned to adsorbed carboxylate (*CO2−) were also clearly observed on the polyamine modified Cu, indicating enhanced stabilization of intermediates by the amine groups.
Beyond the characterization of catalyst structure and surface intermediates, the application of Raman spectroscopy to measure local pH is also becoming more and more common. The same spectroelectrochemical Raman cell as in ref. 125 was used to study the potential dependence of the local pH on a Cu GDE in contact with different electrolytes.52 The surface enhancement effect enabled to probe the local pH within 3 nm from the electrode surface (Fig. 2(D)). In a 1 M KOH electrolyte, the surface pH decreased from ca. 10 to 9.3 when the potential changed from −0.4 V to −1.3 V (vs. NHE). In parallel to this, peaks associated with basic copper carbonate (malachite) appeared, which showed similar potential dependence to the HCO3− peak. The authors explained this by the reaction of soluble Cu2+ species (originating from the initial reduction of CuO) with OH− and CO32− to form malachite. The consumption of the latter species resulted in a drop in the local pH (almost 5 pH unit difference compared to the bulk). At CO2RR potentials (<−1.2 V vs. NHE) or at longer timescales, the pH started to increase, because of the completion of the oxide reduction, thus depletion of soluble Cu2+, ceasing the malachite formation. The work highlighted that certain oxidized phases (i.e., malachite), which are thermodynamically not stable under the given potential–pH conditions, can still persist, presumably because of their sluggish reduction kinetics. The presence of these species, in turn, can steer the CO2RR selectivity.
Modified CO2 electrolyzer designs (full cells) were also reported that allow for local pH-measurements near the cathode (Fig. 8(A) and (B)). In one work, the pH gradient in the catholyte layer of a membrane-separated microfluidic cell (Fig. 8(A)) was measured in a distance of 40–120 μm from the cathode and extrapolated to the electrode surface.53 The modified flow cell was 3D-printed from a photosensitive resin and a plastic film cover was placed on the top of the cell to allow transparency for the beam. The Raman beam was parallel to the GDE and was moved in the perpendicular (x) direction towards the bulk of the electrolyte to measure the pH gradient. Without polarization in a 1 M KOH electrolyte, the pH at the electrode surface was 7.2, much lower than the bulk value, because of the neutralization reaction between OH− and CO2. Moving away from the electrode surface to the bulk electrolyte, the pH increased to above 11 at a distance of 120 μm. When applying 50 mA cm−2 current density to the cell (Fig. 8(C)), the pH increased at the electrode surface to ca. 9, because of the production of OH− during CO2RR. In this case, the region where HCO3− could be detected (i.e., pH < 11) was 40 μm narrower than at open circuit potential (OCP). With increasing current density, the HCO3− region shrank further and the cathode surface pH gradually increased (>12 at 150 mA cm−2). These results showed that the produced OH− during CO2RR cannot fully offset the OH− consumed by the reaction with CO2 and the usually observed nominal overpotential-reduction in alkaline electrolytes was attributed to the buildup of a Nernst potential at the cathode/electrolyte interface.53
 | ||
| Fig. 8 (A) Microfluidic flow cell designed for local pH measurements. Reproduced from ref. 53. Copyright 2020 ACS. (B) Bipolar membrane-bicarbonate electrolyzer for local pH measurements. Reproduced from ref. 51. Copyright 2020 ACS. (C) Measurement of local pH with Raman spectroscopy in the operando cell shown in panel C using a 1 M KOH electrolyte at 50 mA cm−2 current density. Measured HCO3− and CO32− concentrations as a function of distance from the electrode surface, fitted concentrations of dissolved species and the derived pH values. Reproduced from ref. 53. Copyright 2020 ACS. | ||
Local pH measurement in a zero-gap bicarbonate electrolyzer with a bipolar membrane (Fig. 8(B)) was also demonstrated.51 In this cell design the laser beam was perpendicular to the silver-foam cathode, which was illuminated through a sapphire window placed in the center of the cathodic flow plate. The use of a silver foam cathode was necessary to eliminate the carbon based GDLs which would obscure the electrocatalyst and the strong Raman signal of carbon would hinder the detection of HCO3− and CO32−. The cell modification, however, resulted in the deterioration of the performance (32% FE for CO at −1.3 V vs. Ag/AgCl). The surface pH increased during the reduction of bicarbonate, which became more prominent as the current density increased. When applying 200 mA cm−2 current density, the surface pH changed from 8.53 (OCP) to 10.31 after 5 minutes of electrolysis. A pH gradient established in this electrolyzer too: at 200 mA cm−2 the pH was around 1.3 units lower at a distance of 150 μm from the electrode, compared to the catalyst surface. Higher current densities led to thicker diffusion layers, explained by the production of OH− during bicarbonate electrolysis, which prevailed over the bubble-induced convection, transporting OH− away from the electrode surface. When increasing the temperature of the bicarbonate solution feed from 20 °C to 70 °C, the surface pH slightly increased (with 0.6 pH units) and the FE for H2 production dropped from 32% to 15% (at 50 mA cm−2 total current density). These results indicated that a higher surface pH diminishes HER in bicarbonate electrolyzers (in contrast to alkaline water electrolysis).
Ratiometric techniques for local pH measurements
Besides Raman spectroscopy, ratiometric techniques were also employed for local pH measurements. A pH-sensitive ratiometric probe (dye) has two or more absorption/emission bands that respond differently to the proton concentration.30 Within a certain pH-range, the ratio of the intensity of the absorption peaks at different wavelengths is directly correlated with the pH. Confocal fluorescent microscopy with micron-scale resolution in the x, y, z direction50 has been used to probe the pH within trenches of a Cu GDE. A two-color pH-sensitive fluorescent dye was added to the electrolyte (KHCO3). With increasing current density (up to 28 mA cm−2), the local pH increased both at the GDE surface and within the trenches (as expected), but spatial inhomogeneity was observed. As OH− ions are produced in both CO2RR and HER, the higher local pH was used as an indication for higher activity regions. When the average pH at the surface of the electrode was below 9.5, the electrode mostly produced C1 products and H2, while many C2 products were observed at pH values above 10. Randomly distributed trenches with an average width of (18 ± 8) μm were present in the microporous layer of the GDE. Interestingly, higher pH was observed within the narrow trenches indicating higher local activity in more confined regions of the GDE.A clever method to manipulate and measure the local pH in MEAs was recently demonstrated.30 The authors aimed to tune the local pH in a bipolar membrane electrolyzer at the catalyst/CEM interface to diminish acidity, and thus increase CO2RR selectivity. The local chemical environment of a BPM (consisting of a Nafion CEL and a graphene-oxide-based AEL) was modified by a weak-acidic polyelectrolyte consisting of poly(acrylic-acid) and poly(allylamine hydrochloride) (PAA–PAH) bilayers. Ratiometric dyes were covalently attached to the amine groups of PAH, allowing for in situ pH measurements at different distances from the cathode/CEL interface, depending on the number of PAH/PAA buffer layers between the dye and the Nafion. The local pH was quantified by UV-Vis spectroscopy. Without polarization, with the increasing number of PAA/PAH buffer layers, the pH gradually increased from ca. 3 to > 6. When applying a 4 mA cm−2 reverse bias, the pH in layers close to the Nafion decreased, because more protons were introduced into the CEL. In layers further away from the Nafion, the proton concentration, however, did not change significantly. This suggested that the pH changes occur within a much shorter distance in weak-acidic CEMs (50 nm), compared to typical boundary layer thicknesses at the electrified solid/liquid interface (20–200 μm).
Scanning probe techniques
Scanning probe techniques are powerful tools to investigate the surface topography and local activity of electrocatalysts. In scanning electrochemical microscopy (SECM) a nanometer-sized tip electrode is positioned in close proximity to the electrode surface functioning as a local probe to determine reaction rates or local concentrations. Its applicability to study GDEs was recently demonstrated for both ORR135 and CO2RR.136,137 A Pt microelectrode is suitable to detect OH− and H2O activities near the electrode, as the potential of PtO reduction depends on these parameters. With a shear-force based approach, a Pt microelectrode was positioned at 100 nm above a silver GDE.137 When applying increasingly negative potentials to the silver cathode, the PtO reduction potential shifted gradually towards more negative potentials. At the highest applied potential (−1.69 V vs. Ag/AgCl, ∼12 mA cm−2) the measured hydroxide/water activity ratio was beyond that was obtained for a 16 M KOH solution, indicating a highly alkaline environment near and inside the GDE.137 SECM was also used to investigate the effect of catalyst loading and CO2 back pressure on the activity (local CO product fluxes) of a gold GDE (Fig. 9(A)).136 Using a shear-force based approach, the CO formation rate was detected by measuring diffusion limited CO oxidation current on the Au nanoelectrode (tip) positioned at a sub-μm distance from the Au GDE. For a GDE with a shallow loading-gradient, the increase in catalyst loading led to a nearly linear increase in the amount of produced CO. Regions with increased activity (5–6 times higher currents) were also identified. At these hotspots, the CO-current was of similar magnitude or even higher at the low-loading areas, compared to those with high loading. This indicated that at low catalyst loading the formation of a three-phase boundary is more important to reach high activity, than the amount of catalyst. For the GDE with a steeper loading gradient, the activity decreased at the higher loading areas (Fig. 9(A)), because part of the catalyst nanoparticles were not accessible to the CO2. Recording the topography profile of the GDE showed that lower Z-positions (e.g., above a pore) coincided with higher CO-formation activity. A high-resolution activity map was also recorded within an area of similar loading. Surprisingly, large activity differences were found, indicating that inhomogeneous pore distribution significantly impacts local activity. The study also revealed that at lower CO2 back pressures, a large amount of catalyst remains unutilized, as the CO2 cannot reach the upper catalyst layers. Consequently, higher catalyst loading only leads to higher activity for CO2RR, when enough CO2 is supplied, and properties of the GDLs (hydrophobicity, pore structure, permeability) will have a significant impact on the optimal loading-CO2 back pressure combinations.136 | ||
| Fig. 9 Scanning probe methods applied to study CO2RR in GDE-configurations. (A) SECM experimental setup operated in a surface-generation tip-collection mode to measure local CO2RR activity on a Au-GDE with varying loadings (left). SECM array scans along the catalyst loading-gradient at different potentials applied to the GDE (right). The tip current is proportional to the CO production rate. Reproduced from ref. 136. Copyright 2019 the Authors, Published by RSC. (B) Schematic and photos of an EC-AFM cell (left). EC-AFM images of a Cu-sputtered GDE at 100 mA cm−2 (right). Reproduced from ref. 126. Copyright 2021 the Electrochemical Society. Reproduced by permission of IOP Publishing. All rights reserved. | ||
Electrochemical atomic force microscopy (EC-AFM) can provide operando topography and spatial maps of mechanical properties (e.g., adhesion, deformation), as well as insights into changes in catalyst surface faceting, agglomeration or degradation.126,138,139 While EC-AFM was previously limited to low current densities, because of bubble formation disturbing measurements, a suitable cell design hosting a GDE, and allowing for gas products to leave through the diffusion media, can enable high current density EC-AFM measurements.126 A commercial EC-AFM cell was modified to allow for gas flow below the GDE, with a stagnant electrolyte on the catalyst side (Fig. 9(B)), and its applicability was demonstrated at high current densities up to 100 mA cm−2. Technical challenges related to bubble formation, mechanical stability of the cell and trade-off between temporal resolution and scan size have been discussed. The reported cell allowed for the observation of morphological restructuring of catalysts, as well as carbonate precipitate formation. The investigated Cu films had very stable topography and mechanical property maps with no signs of restructuring or precipitate formation. Because of the small scan size, however, certain areas could remain hidden; therefore the technique needs to be further improved to increase the scan size. At higher current densities, electrolyte flooding also impeded long-term measurements (<3 min at 100 mA cm−2).
Mass spectrometry
Mass spectrometry offers real-time product detection, which is extremely important to capture rapid changes in the product distribution, for instance, during initial periods of catalysis and for dynamically changing systems (e.g., pulsed CO2RR). Furthermore, the detection of (unstable) reaction intermediates, present even in very small amounts close to the catalyst surface, can also largely contribute to our understanding of the reaction mechanism. In this regard, isotopic labeling is also of particular importance.47 Considering this fact, differential electrochemical mass spectrometry (DEMS) and online electrochemical mass spectrometry (OLEMS) became powerful tools for the real time detection of CO2/CO reduction products,109,140 despite some general limitations such as their inability to detect non-volatile species and the convolution of CO2 and CO signals.141,142 Notwithstanding the high mechanistic information content of electrochemical mass spectrometry (EC-MS) studies, interestingly, only few studies exist so far which took advantage of flow cells or GDE-configurations.81,127 A flow electrolyzer mass spectrometry (FEMS) technique combined with isotopic labeling was recently applied to study the origin of oxygen in CO/CO2RR intermediates (acetaldehyde) and products (ethanol, n-propanol).27 A membrane-less microfluidic cell with a flowing KOH electrolyte, and having a capillary probe for the MS placed in close proximity of the Cu GDE, was used for these studies (Fig. 10). The high current density (150 mA cm−2) obtained with the GDE setup allowed the detection of reactive species present in very low concentrations, which would be below the detection limit in an H-cell. By following m/z values relevant for n-propanol, acetaldehyde and ethanol, and using unlabeled and 18O-labeled CO and electrolyte (H218O) feeds, the authors found that while the O of acetaldehyde originated from CO, that of ethanol largely came from the electrolyte. This finding could be rationalized by a mechanism, in which the formed acetaldehyde goes through a rapid O-exchange with water, and then is further reduced to ethanol (Fig. 10(B)). The cross-coupling of acetaldehyde with CO, however, was found to be an unlikely route for the formation of C3 products. A gas accessible membrane electrode (GAME) system is composed of a cylindrical gas compartment with a porous polycarbonate-based GDE at the end, together placed in a stagnant electrolyte.140 In this configuration a GDE-interface is created, however, with a much thinner substrate (12 μm) compared to typical carbon-based GDEs (∼250 μm). This further enhances reactant and product transport, facilitating real-time detection of products with mass spectrometry. The electrocatalytic reaction sites and gas-sampling interface are collocated, and therefore the delay time for MS analysis is minimized. At the same time, an ultramicroelectrode (UME) can be positioned in proximity of the catalyst in the electrolyte for liquid product analysis or local pH measurements. With this setup the formation of H2, CH4 and C2H4 on a Cu–Au electrode was simultaneously detected with time resolution good enough to deconvolute the faradaic current during a cyclic voltammetry scan with 1 mV s−1 scan rate to the partial current of products. A Pt UME was also placed close (<100 μm) to the catalyst, and the voltammetric response of PtO reduction was used as a sensor for the local pH variation. | ||
| Fig. 10 (A) Schematic of the flow electrolyzer-mass spectrometry (FEMS) setup. (B) Proposed reaction mechanism for CO reduction to oxygenated products. Reproduced from ref. 81. Copyright Wiley-VCH GmbH. | ||
Besides detecting products and reaction intermediates, EC-MS methods could also be used to measure local CO2 concentrations near the catalyst surface. A DEMS method was reported to directly observe the local reaction environment during CO2RR in a custom-designed cell with dissolved CO2 as the reactant. The catalyst was directly deposited on the pervaporation membrane at the interface between the electrochemical cell and the mass spectrometer.141 By this approach the authors could experimentally observe the reaction between CO2 and the cathodically formed OH− and its influence on CO2RR. Such measurements would be extremely relevant for GDE-based continuous flow electrolyzers too, as the local CO2 concentration directly affects the CO2RR rate and the product distribution as well.
Neutron and X-ray imaging
Neutron- and X-ray radiography are non-destructive imaging techniques allowing in situ/operando characterization of processes taking place in energy devices, such as fuel cells, batteries or electrolyzers.143–145 The different interactions of neutrons and X-rays with matter make the two techniques largely complementary in materials science.144,146 Neutron radiography can give strong contrast for some elements located close to each other in the periodic table and is able to differentiate between isotopes as well (e.g., 1H and 2D).146 Neutrons penetrate, however, easily into materials composed of heavy elements (metals) which are typically used for the cell body of electrochemical devices. This makes neutron imaging excellently suited for in situ/operando investigations without the need for major cell modifications. In contrast, as X-rays interact with electrons of the atom, their attenuation increases with the increase in the atomic number of the element. Therefore, for X-ray imaging usually an X-ray window has to be placed in the cell to enable the beam to reach the core of the device. Synchrotron X-ray radiography has better spatial resolution (1 μm) than neutron imaging with short exposure times, however at the expense of reduced field of view.147 Both techniques have been widely used to characterize the water management in fuel cells or water electrolyzers.148–150 As usually gas-phase reactants and/or products are present in these devices, the observation of gas bubbles and their distribution within the cell is also crucial to study the two-phase transport phenomena,151 which can be performed with both neutron and X-ray radiography. Tomography (i.e., 3D spatially resolved imaging) is also feasible with both techniques, resulting in detailed characterization of the microstructure of the entire cell.150,152 The X-ray and neutron radiographic characterization of CO2 electrolyzers is still in its infancy, as we can find only a few reports on such studies. These are focused on the membrane hydration, water distribution across the MEA and gas bubble formation in the electrolyte layers.78,91,100,128,129To maintain a high ionic conductance, therefore fast ion transport in CO2 electrolyzers, it is necessary for the ion exchange membrane to be in a well-hydrated state.31 Recently neutron imaging was employed to directly quantify liquid water distribution across a MEA (Fig. 11(A)).91 Increasing the relative humidity (0–100%) of the reactant CO2 resulted in lower cell voltage, which stemmed from reduced ohmic losses, when a humidified reactant was used. To explain the origin of this observation, the through-plane (perpendicular to the flow-field) water-content of the MEA was characterized. The increase in the relative humidity resulted in a ca. 60% increase in the liquid water volume of the Nafion membrane at 575 mA cm−2 current density. The membrane, however, also experienced a gradual dehydration during operation when increasing the current density, which resulted in decreased ionic conductivity. The dehydration was most significant at the center, compared to the regions in contact with the anode and cathode catalysts (Fig. 11(A)). While this study gave interesting insights into the water transport by directly measuring the water uptake of the membrane, the cell significantly underperformed state-of-the art CO2 electrolyzers, possibly because of the modified cell design optimized for the neutron imaging experiment and the acidic local environment at the catalyst–CEM interface.
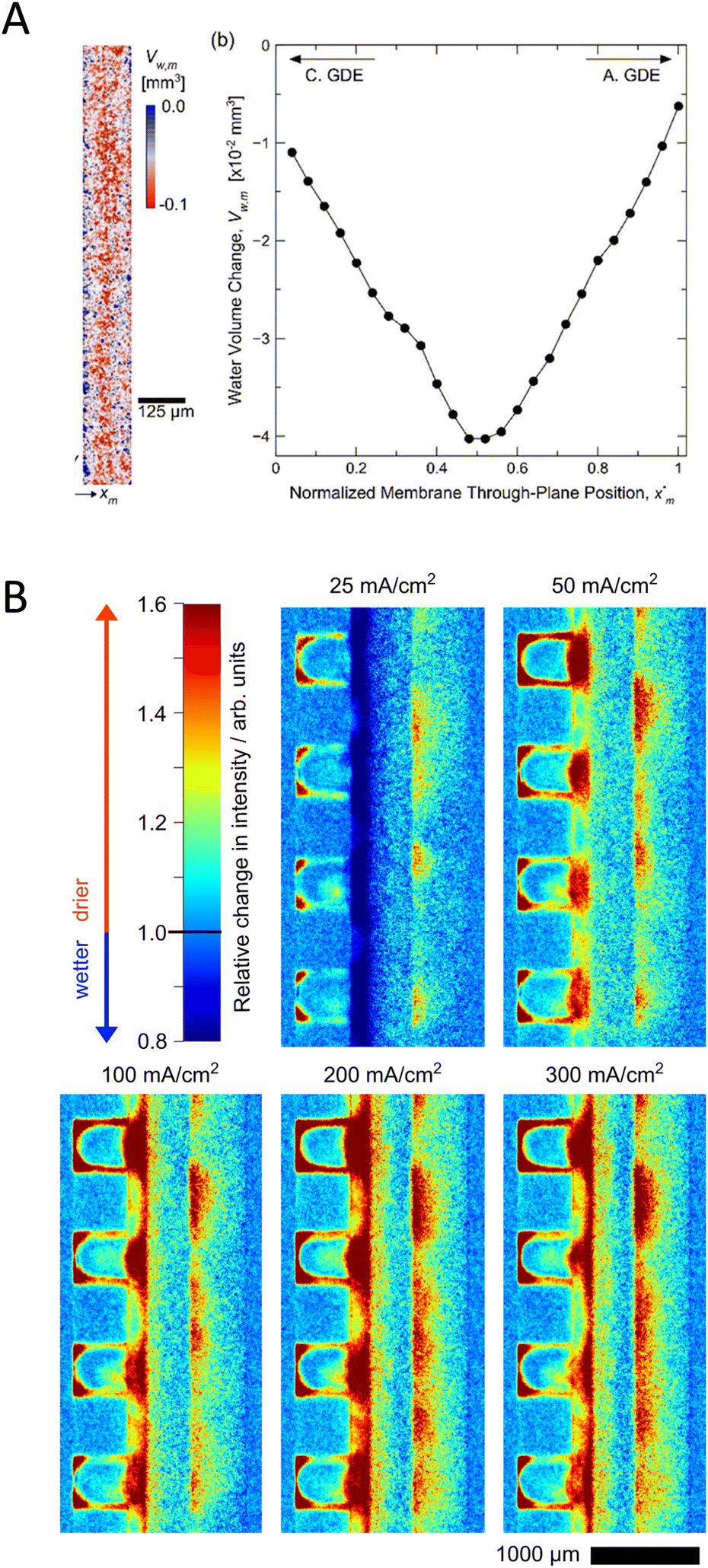 | ||
| Fig. 11 Operando visualisation and quantification of water management in CO2 electrolyzers. (A) Quantification of liquid water content in a MEA by neutron radiography. Processed image highlighting the water volume change in the CEM. Water-volume change across the membrane is plotted on the graph. Reproduced from ref. 91. Copyright 2020 ACS. (B) Relative change in the water-content of a zero-gap CO2 electrolyzer during operation. Reproduced from ref. 100. Copyright the Authors, published by Springer Nature. | ||
The same group also employed high spatial resolution (15 μm) neutron imaging to study the formation of gas bubbles in the catholyte layer of a CO2 electrolyzer, which they correlated to performance instability.128 The gas saturation of a 3 M KHCO3 electrolyte increased with increasing current density to 50 mA cm−2, then it reached a plateau. The dispersion in the cathode potential was correlated to the gas saturation of the electrolyte. In the lower current density region, a strong positive correlation was found between the two parameters (semi-stable operation), indicating that the unstable cathode potential was the result of gas accumulation in the electrolyte layer. At higher current densities, the number of gas removal pathways increased to maintain a constant electrolyte gas saturation. In this region the increased rate of transient gas accumulation (production) and its removal altered mass transport in the cell, and caused an increased cathode potential instability. The authors suggested that keeping the gas saturation below the plateau (oscillation) region would result in increased performance stability; therefore it is critical to improve gas management in CO2 electrolyzers. High resolution neutron imaging (6 μm) was also performed in a zero-gap AEM electrolyzer (with Ag catalyst) to study water distribution and to observe salt precipitation at the cathode.100 Up to 200 mA cm−2 the cathode became gradually dryer with increasing current density, because of the increased water consumption and electroosmotic drag from the cathode to the anode (Fig. 11(B)). At 300 mA cm−2, however, in some regions of the cathode the radiographic intensity even decreased. This counter-intuitive observation was explained by the accumulation of KHCO3 salts, which were preferably located in the channel vs. land area of the flow field. Although salt accumulation already started at 100 mA cm−2 current density, at this lower current gas transport was still sufficient to maintain high CO selectivity. A more pronounced bubble accumulation on the anode side in the region of the cathode land was also observed, suggesting higher local reaction rates in the cathode land region. This work nicely demonstrated the potential of neutron imaging to investigate water transport and salt precipitation in AEM-based CO2 electrolyzers, which might contribute to mitigate the carbonate problem in the future.
Synchrotron X-ray radiography was used to study the effect of operating temperature on the membrane hydration and gas/water content in a CEM zero-gap CO2 electrolyzer.78 The cell voltage and thus the overall power density reduced by ca. 35% when raising the temperature from 25 to 60 °C, as a result of the simultaneous reduction of activation, ohmic and mass transport losses. The radiographic images of the CEM at higher temperatures indicated higher water content, resulting in increased ionic conductivity and thus decreased ohmic losses. To study the anodic and cathodic mass transport limitations, the impact of temperature on gas saturation in the anode and cathode GDEs was also investigated. The authors found that mass transport losses increased with increasing current density. A modular cell for X-ray radiographic imaging of electrolyte distribution in strongly absorbing Ag GDEs was reported recently.129 Electrolyte intrusion into and distribution within the GDE were observed for ORR and CO2RR. For the CO2RR studies, a cell with AEM-separated anolyte and catholyte chambers was employed. To allow for the irradiation, an X-ray window was cut in the middle of the cell, and the cell was irradiated from the anode side. 300 mA cm−2 steady-state current density was reached at −1.0 V (vs. RHE) cathode potential; however, no selectivity data were reported. At lower potentials (−0.7 V), larger pore systems got filled up with the electrolyte, while at more negative potentials (−1.0 V vs. RHE) electrolyte droplets and gas bubbles also appeared on the gas and electrolyte side, respectively. On a more hydrophobic catalyst (higher PTFE-content) gas formation was already observed at −0.7 V, which became even more prominent at −1.0 V. Furthermore, electrolyte distribution was strongly retarded on this GDE, compared to the catalyst with lower PTFE-content. Formation of salt crystallites was observed at −0.7 V, which were washed away at −1.0 V, because of the intensified flooding at this potential.
As highlighted above, radiography has potential in the operando investigation of CO2 electrolyzers, in particular to identify the origin of overvoltages (e.g., membrane hydration) and cell failure mechanisms (e.g., precipitate formation, flooding, etc.). Neutron- and X-ray radiography are, however, expensive techniques as synchrotron or neutron facilities are required. Because of this, visual observation of the inner part of the cell by high-speed cameras is also very appealing to observe flooding or salt precipitation under laboratory conditions. In one work, the observation of precipitate formation was combined with the measurement of relative humidity (RH) and temperature by embedded sensors to characterize and model water transport within an AEM-separated zero-gap cell.73 The relative humidity was measured before and during CO2RR (at 100 mA cm−2) at different locations within the cell with dry (0% RH) and wet (70% RH) CO2 feeds. While the wet CO2 feed evenly humidified the cathode flow field, large spatial differences were observed when a dry feed was applied. It was shown that the operating parameters can be varied to adjust water transport across the membrane. The increased membrane transport may enhance cation crossover from the anolyte,22 resulting in detrimental precipitate formation at the cathode, but also promoting CO2RR. To visually observe this together with cathode flooding, a transparent window was incorporated into the cell. For the dry CO2 feed at 100 mA cm−2, liquid formation in the flow field could be observed after 20 minutes and salt precipitation after 120 minutes. The use of a wet CO2 feed, however, prolonged the salt and liquid accumulation to >150 mA cm−2, in accordance with the lower flux of water across the membrane.
Thermography
As with the increase of cell size or number of cell stacks, higher spatial variation in operating conditions is expected, probing local activities in a full electrolyzer device can assist scale-up efforts. Recently, location-specific catalytic activity was studied by infrared thermography,130 making use of the inefficient charge transfer and transport, resulting in heat generation. The authors postulated that any temperature change of the catalyst was primarily because of heat generation from the electrochemical reaction at the cathode. Therefore, observing the temperature change during operation can be a measure of electrochemical activity. For instance, a higher temperature increase was observed for Pt than Ag during HER at a fixed potential, in accordance with their known activity trend towards HER. Spatial mapping can also provide a means to examine transport phenomena or limiting chemical reactions, as uneven current distribution (thus temperature) can be the result of poor catalyst deposition, different aging of catalyst portions or spatially varying operating conditions. The influence of electrolyte flow on the reaction rate distribution was studied by changing the direction of the electrolyte flow from “bottom to top” to “side to side”. In the latter case, gas bubbles (O2) become trapped in the anode compartment, resulting in noisy LSV curves and rapidly changing temperature profiles of the cathode GDE. This was rationalized by shielding the Ni mesh anode by the buildup of gas, resulting in uneven current density distribution. As the reaction of CO2 with hydroxide ions is highly exothermic, carbonate formation can also be followed by thermography during CO2RR as demonstrated by a larger temperature increase when changing the gas feed from N2 to CO2.Conclusions and future prospects
As highlighted in this Feature Article, there is a tendency in the CO2RR research field to implement operando techniques in full electrolyzer devices or in GDE-setups to gain mechanistic understanding of the processes taking place under practically relevant conditions. The transition from low to high current density operando characterization, however, started only very recently, in the past 2–3 years. Consequently, there is still a large undiscovered research space, which has to be explored in the near future. Previous studies demonstrated the potential of operando characterization to serve as a tool to uncover the catalyst microenvironment, optimize working conditions, understand reaction mechanisms, or to identify reasons for device failure. Nevertheless, in several cases the operando techniques were applied to underperforming devices or without reporting activity/selectivity data. To make these measurements more representative of the real working environment, more intensive collaboration among electrochemists, spectroscopists and cell development engineers is needed to overcome technical challenges and maximize electrolyzer performance and spectroscopic data quality at the same time.Combination of different operando methods and their coupling to real-time product analysis would provide invaluable structure–performance correlations, helping to understand and deconvolute the multitude of processes taking place in a CO2 electrolyzer. As the CO2RR current densities have already reached industrially relevant values, the field should focus on improving energy-efficiency and durability of the electrolyzers. Operando methods can largely assist and accelerate the identification of the origin of voltage losses and cell deactivation mechanisms. In particular radiographic imaging will be an excellent tool for this when combined with elemental analysis. The selective production of high-value multicarbon products will largely benefit from the understanding of the C–C coupling mechanism at high current densities. Catalyst degradation studies, for instance by on-line ICP-MS,70,153 should also be performed in GDE-configurations in the future. Finally, more focus should be put on electrolyte-engineering, which was shown to be very efficient to tune selectivity and increase activity in H-type cells, but is only marginally researched in continuous-flow CO2RR.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding under the European Union's Horizon Europe research and innovation program from the European Research Council (ERC, Grant Agreement No. 101043617) and the FlowPhotoChem project (grant agreement ID: 862453). We also thank the Hungarian Renewable Energy National Laboratory for support, financed by the RRF-2.3.1-21-2022-00009 project.Notes and references
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- A. A. Samu, A. Kormányos, E. Kecsenovity, N. Szilágyi, B. Endrődi and C. Janáky, ACS Energy Lett., 2022, 7, 1859–1861 CrossRef CAS PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y.-W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- A. Herzog, A. Bergmann, H. S. Jeon, J. Timoshenko, S. Kühl, C. Rettenmaier, M. Lopez Luna, F. T. Haase and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2021, 60, 7426–7435 CrossRef CAS PubMed.
- A. M. Ismail, G. F. Samu, Á. Balog, E. Csapó and C. Janáky, ACS Energy Lett., 2019, 4, 48–53 CrossRef CAS.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- D. Hursán, A. A. Samu, L. Janovák, K. Artyushkova, T. Asset, P. Atanassov and C. Janáky, Joule, 2019, 3, 1719–1733 CrossRef PubMed.
- G. Marcandalli, M. C. O. Monteiro, A. Goyal and M. T. M. Koper, Acc. Chem. Res., 2022, 55, 1900–1911 CrossRef CAS PubMed.
- X. Li, S. Wang, L. Li, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2020, 142, 9567–9581 CAS.
- J. Li and J. Gong, Energy Environ. Sci., 2020, 13, 3748–3779 RSC.
- J. Timoshenko and B. Roldan Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed.
- R. Kas, O. Ayemoba, N. J. Firet, J. Middelkoop, W. A. Smith and A. Cuesta, ChemPhysChem, 2019, 20, 2904–2925 CrossRef CAS PubMed.
- M. C. O. Monteiro and M. T. M. Koper, Curr. Opin. Electrochem., 2021, 25, 100649 CrossRef CAS.
- H. Li, P. Wei, D. Gao and G. Wang, Curr. Opin. Green Sustainable Chem., 2022, 34, 100589 CrossRef CAS.
- B. Endrődi, G. Bencsik, F. Darvas, R. Jones, K. Rajeshwar and C. Janáky, Prog. Energy Combust. Sci., 2017, 62, 133 CrossRef.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- M. H. Han, D. Kim, S. Kim, S.-H. Yu, D. H. Won, B. K. Min, K. H. Chae, W. H. Lee and H.-S. Oh, Adv. Energy Mater., 2022, 12, 2201843 CrossRef CAS.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- B. Endrődi, E. Kecsenovity, A. Samu, T. Halmágyi, S. Rojas-Carbonell, L. Wang, Y. Yan and C. Janáky, Energy Environ. Sci., 2020, 13, 4098–4105 RSC.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- B. Endrődi, A. Samu, E. Kecsenovity, T. Halmágyi, D. Sebők and C. Janáky, Nat. Energy, 2021, 6, 439–448 CrossRef PubMed.
- D. Reinisch, B. Schmid, N. Martić, R. Krause, H. Landes, M. Hanebuth, K. J. J. Mayrhofer and G. Schmid, Zeitschrift für Physikalische Chemie, 2020, 234, 1115–1131 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- H. Liu, H. B. Tao and B. Liu, J. Phys. Chem. Lett., 2022, 13, 6520–6531 CrossRef CAS PubMed.
- Á. Vass, A. Kormányos, Z. Kószó, B. Endrődi and C. Janáky, ACS Catal., 2022, 12, 1037–1051 CrossRef PubMed.
- P. Kamat and P. Christopher, ACS Energy Lett., 2022, 7, 1469–1472 CrossRef CAS.
- C. Kim, J. C. Bui, X. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- Z. Yan, J. L. Hitt, Z. Zeng, M. A. Hickner and T. E. Mallouk, Nat. Chem., 2021, 13, 33–40 CrossRef CAS PubMed.
- G. M. Geise, M. A. Hickner and B. E. Logan, ACS Appl. Mater. Interfaces, 2013, 5, 10294–10301 CrossRef CAS PubMed.
- D. T. Whipple, E. C. Finke and P. J. A. Kenis, Electrochem. Solid-State Lett., 2010, 13, B109–B111 CrossRef CAS.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS.
- J. J. Kaczur, H. Yang, Z. Liu, S. D. Sajjad and R. I. Masel, Front. Chem., 2018, 6 Search PubMed.
- I. E. L. Stephens, K. Chan, A. Bagger, S. W. Boettcher, J. Bonin, E. Boutin, A. K. Buckley, R. Buonsanti, E. R. Cave, X. Chang, S. W. Chee, A. H. M. da Silva, P. de Luna, O. Einsle, B. Endrődi, M. Escudero-Escribano, J. V. Ferreira de Araujo, M. C. Figueiredo, C. Hahn, K. U. Hansen, S. Haussener, S. Hunegnaw, Z. Huo, Y. J. Hwang, C. Janáky, B. S. Jayathilake, F. Jiao, Z. P. Jovanov, P. Karimi, M. T. M. Koper, K. P. Kuhl, W. H. Lee, Z. Liang, X. Liu, S. Ma, M. Ma, H.-S. Oh, M. Robert, B. R. Cuenya, J. Rossmeisl, C. Roy, M. P. Ryan, E. H. Sargent, P. Sebastián-Pascual, B. Seger, L. Steier, P. Strasser, A. S. Varela, R. E. Vos, X. Wang, B. Xu, H. Yadegari and Y. Zhou, J. Phys. Energy, 2022, 4, 42003 CrossRef.
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536–1551 RSC.
- M. Jouny, G. S. Hutchings and F. Jiao, Nat. Catal., 2019, 2, 1062–1070 CrossRef CAS.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- C. Peng, S. Yang, G. Luo, S. Yan, M. Shakouri, J. Zhang, Y. Chen, W. Li, Z. Wang, T.-K. Sham and G. Zheng, Adv. Mater., 2022, 34, 2204476 CrossRef CAS PubMed.
- B. Kim, F. Hilman, M. Ariyoshi, S. Fujikawa and P. J. A. Kenis, J. Power Sources, 2016, 312, 192–198 CrossRef CAS.
- N. T. Nesbitt, T. Burdyny, H. Simonson, D. Salvatore, D. Bohra, R. Kas and W. A. Smith, ACS Catal., 2020, 10, 14093–14106 CrossRef CAS.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS PubMed.
- D. Hursán and C. Janáky, ACS Energy Lett., 2018, 3, 722–723 CrossRef PubMed.
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. A. Kenis, Phys. Chem. Chem. Phys., 2016, 18, 7075–7084 RSC.
- D. Hursán and C. Janáky, ACS Energy Lett., 2018, 3, 722 CrossRef PubMed.
- O. Ayemoba and A. Cuesta, ACS Appl. Mater. Interfaces, 2017, 9, 27377–27382 CrossRef CAS PubMed.
- F. Zhang and A. C. Co, Angew. Chem., Int. Ed., 2020, 59, 1674–1681 CrossRef CAS PubMed.
- A. J. Welch, A. Q. Fenwick, A. Böhme, H.-Y. Chen, I. Sullivan, X. Li, J. S. DuChene, C. Xiang and H. A. Atwater, J. Phys. Chem. C, 2021, 125, 20896–20904 CrossRef CAS.
- Z. Zhang, L. Melo, R. P. Jansonius, F. Habibzadeh, E. R. Grant and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 3101–3107 CrossRef CAS.
- D. A. Henckel, M. J. Counihan, H. E. Holmes, X. Chen, U. O. Nwabara, S. Verma, J. Rodríguez-López, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2021, 11, 255–263 CrossRef CAS.
- X. Lu, C. Zhu, Z. Wu, J. Xuan, J. S. Francisco and H. Wang, J. Am. Chem. Soc., 2020, 142, 15438–15444 CrossRef CAS PubMed.
- A. Roy, D. Hursán, K. Artyushkova, P. Atanassov, C. Janáky and A. Serov, Appl. Catal., B, 2018, 232, 512 CrossRef CAS.
- B. Endrődi, E. Kecsenovity, A. Samu, F. Darvas, R. V. Jones, V. Török, A. Danyi and C. Janáky, ACS Energy Lett., 2019, 4, 1770–1777 CrossRef PubMed.
- J. Zhang, W. Luo and A. Züttel, J. Mater. Chem. A, 2019, 7, 26285–26292 RSC.
- A. Ozden, F. Li, F. P. García de Arquer, A. Rosas-Hernández, A. Thevenon, Y. Wang, S.-F. Hung, X. Wang, B. Chen, J. Li, J. Wicks, M. Luo, Z. Wang, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, ACS Energy Lett., 2020, 5, 2811–2818 CrossRef CAS.
- T. Möller, T. Ngo Thanh, X. Wang, W. Ju, Z. Jovanov and P. Strasser, Energy Environ. Sci., 2021, 14, 5995–6006 RSC.
- S. Garg, C. A. Giron Rodriguez, T. E. Rufford, J. R. Varcoe and B. Seger, Energy Environ. Sci., 2022, 15, 4440–4469 RSC.
- S. S. Bhargava, F. Proietto, D. Azmoodeh, E. R. Cofell, D. A. Henckel, S. Verma, C. J. Brooks, A. A. Gewirth and P. J. A. Kenis, ChemElectroChem, 2020, 7, 2001–2011 CrossRef CAS.
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H.-R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, ACS Energy Lett., 2018, 3, 193–198 CrossRef CAS.
- K. Yang, R. Kas and W. A. Smith, J. Am. Chem. Soc., 2019, 141, 15891–15900 CrossRef CAS PubMed.
- M. C. O. Monteiro, A. Mirabal, L. Jacobse, K. Doblhoff-Dier, S. C. Barton and M. T. M. Koper, JACS Au, 2021, 1, 1915–1924 CrossRef CAS PubMed.
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O’Brien, J. Li, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS.
- C. Lee, B. Zhao, J. K. Lee, K. F. Fahy, K. Krause and A. Bazylak, iScience, 2020, 23, 101094 CrossRef CAS PubMed.
- C. M. Gunathunge, V. J. Ovalle and M. M. Waegele, Phys. Chem. Chem. Phys., 2017, 19, 30166–30172 RSC.
- C. W. Machan, M. D. Sampson, S. A. Chabolla, T. Dang and C. P. Kubiak, Organometallics, 2014, 33, 4550–4559 CrossRef CAS.
- T. Möller, W. Ju, A. Bagger, X. Wang, F. Luo, T. Ngo Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 RSC.
- X. Sheng, W. Ge, H. Jiang and C. Li, Adv. Mater., 2022, 34, 2201295 CrossRef CAS PubMed.
- Á. Vass, B. Endrődi, G. F. Samu, Á. Balog, A. Kormányos, S. Cherevko and C. Janáky, ACS Energy Lett., 2021, 6, 3801–3808 CrossRef PubMed.
- D. Salvatore and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 215–220 CrossRef CAS.
- Q. Xu, S. Z. Oener, G. Lindquist, H. Jiang, C. Li and S. W. Boettcher, ACS Energy Lett., 2021, 6, 305–312 CrossRef CAS.
- D. G. Wheeler, B. A. W. Mowbray, A. Reyes, F. Habibzadeh, J. He and C. P. Berlinguette, Energy Environ. Sci., 2020, 13, 5126–5134 RSC.
- D. M. Koshy, S. A. Akhade, A. Shugar, K. Abiose, J. Shi, S. Liang, J. S. Oakdale, S. E. Weitzner, J. B. Varley, E. B. Duoss, S. E. Baker, C. Hahn, Z. Bao and T. F. Jaramillo, J. Am. Chem. Soc., 2021, 143, 14712–14725 CrossRef CAS PubMed.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- J. Heyes, M. Dunwell and B. Xu, J. Phys. Chem. C, 2016, 120, 17334–17341 CrossRef CAS.
- Á. Vass, B. Endrődi and C. Janáky, Curr. Opin. Electrochem., 2021, 25, 100621 CrossRef.
- H. W. Shafaque, J. K. Lee, K. Krause, C. Lee, K. F. Fahy, P. Shrestha, M. Balakrishnan and A. Bazylak, Energy Convers. Manage., 2021, 243, 114302 CrossRef CAS.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, J. Electroanal. Chem., 2014, 716, 53–57 CrossRef CAS.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS.
- B. Hasa, M. Jouny, B. H. Ko, B. Xu and F. Jiao, Angew. Chem., Int. Ed., 2021, 60, 3277–3282 CrossRef CAS PubMed.
- M. Li, M. N. Idros, Y. Wu, T. Burdyny, S. Garg, X. S. Zhao, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2021, 9, 19369–19409 RSC.
- J. Zhang, W. Luo and A. Züttel, J. Catal., 2020, 385, 140–145 CrossRef CAS.
- R. Krause, D. Reinisch, C. Reller, H. Eckert, D. Hartmann, D. Taroata, K. Wiesner-Fleischer, A. Bulan, A. Lueken and G. Schmid, Chem. Ing. Tech., 2020, 92, 53–61 CrossRef CAS.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kühl, R. García-Muelas, N. López and B. R. Cuenya, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- K. U. Hansen, L. H. Cherniack and F. Jiao, ACS Energy Lett., 2022, 7, 4504–4511 CrossRef CAS.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- D. S. Hussey, D. Spernjak, A. Z. Weber, R. Mukundan, J. Fairweather, E. L. Brosha, J. Davey, J. S. Spendelow, D. L. Jacobson and R. L. Borup, J. Appl. Phys., 2012, 112, 104906 CrossRef PubMed.
- H. W. Shafaque, C. Lee, K. F. Fahy, J. K. Lee, J. M. LaManna, E. Baltic, D. S. Hussey, D. L. Jacobson and A. Bazylak, ACS Appl. Mater. Interfaces, 2020, 12, 54585–54595 CrossRef CAS PubMed.
- L. Wang, S. A. Nitopi, E. Bertheussen, M. Orazov, C. G. Morales-Guio, X. Liu, D. C. Higgins, K. Chan, J. K. Nørskov, C. Hahn and T. F. Jaramillo, ACS Catal., 2018, 8, 7445–7454 CrossRef CAS.
- A. Ozden, Y. Wang, F. Li, M. Luo, J. Sisler, A. Thevenon, A. Rosas-Hernández, T. Burdyny, Y. Lum, H. Yadegari, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, Joule, 2021, 5, 706–719 CrossRef CAS.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS.
- Y. Kong, H. Hu, M. Liu, Y. Hou, V. Kolivoška, S. Vesztergom and P. Broekmann, J. Catal., 2022, 408, 1–8 CrossRef CAS.
- A. Xu, N. Govindarajan, G. Kastlunger, S. Vijay and K. Chan, Acc. Chem. Res., 2022, 55, 495–503 CrossRef CAS PubMed.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- A. Murata and Y. Hori, Bull. Chem. Soc. Jpn., 1991, 64, 123–127 CrossRef CAS.
- J. Disch, L. Bohn, S. Koch, M. Schulz, Y. Han, A. Tengattini, L. Helfen, M. Breitwieser and S. Vierrath, Nat. Commun., 2022, 13, 6099 CrossRef CAS PubMed.
- N. Ul Hassan, M. Mandal, G. Huang, H. A. Firouzjaie, P. A. Kohl and W. E. Mustain, Adv. Energy Mater., 2020, 10, 2001986 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- I. Vincent, E.-C. Lee and H.-M. Kim, Sci. Rep., 2021, 11, 293 CrossRef CAS PubMed.
- S. Siracusano, S. Trocino, N. Briguglio, V. Baglio and A. S. Aricò, Materials, 2018, 11 Search PubMed.
- K. Yang, M. Li, S. Subramanian, M. A. Blommaert, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 4291–4298 CrossRef CAS PubMed.
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Nat. Catal., 2022, 5, 268–276 CrossRef CAS.
- M. C. O. Monteiro, M. F. Philips, K. J. P. Schouten and M. T. M. Koper, Nat. Commun., 2021, 12, 4943 CrossRef CAS PubMed.
- N. Wang, R. K. Miao, G. Lee, A. Vomiero, D. Sinton, A. H. Ip, H. Liang and E. H. Sargent, SmartMat, 2021, 2, 12–16 CrossRef CAS.
- E. L. Clark, M. R. Singh, Y. Kwon and A. T. Bell, Anal. Chem., 2015, 87, 8013–8020 CrossRef CAS PubMed.
- M. Krödel, B. M. Carter, D. Rall, J. Lohaus, M. Wessling and D. J. Miller, ACS Appl. Mater. Interfaces, 2020, 12, 12030–12042 CrossRef PubMed.
- N. Kato, S. Mizuno, M. Shiozawa, N. Nojiri, Y. Kawai, K. Fukumoto, T. Morikawa and Y. Takeda, Joule, 2021, 5, 687–705 CrossRef CAS.
- J. L. White, J. T. Herb, J. J. Kaczur, P. W. Majsztrik and A. B. Bocarsly, J. CO2 Util., 2014, 7, 1–5 CrossRef CAS.
- W. H. Lee, C. Lim, S. Y. Lee, K. H. Chae, C. H. Choi, U. Lee, B. K. Min, Y. J. Hwang and H.-S. Oh, Nano Energy, 2021, 84, 105859 CrossRef CAS.
- J. Yano and V. K. Yachandra, Photosynth. Res., 2009, 102, 241 CrossRef CAS PubMed.
- N. J. Firet, T. Burdyny, N. T. Nesbitt, S. Chandrashekar, A. Longo and W. A. Smith, Catal. Sci. Technol., 2020, 10, 5870–5885 RSC.
- O. Müller, M. Nachtegaal, J. Just, D. Lützenkirchen-Hecht and R. Frahm, J. Synchrotron Radiat., 2016, 23, 260–266 CrossRef PubMed.
- J. Timoshenko, A. Bergmann, C. Rettenmaier, A. Herzog, R. M. Arán-Ais, H. S. Jeon, F. T. Haase, U. Hejral, P. Grosse, S. Kühl, E. M. Davis, J. Tian, O. Magnussen and B. Roldan Cuenya, Nat. Catal., 2022, 5, 259–267 CrossRef CAS.
- H. S. Jeon, J. Timoshenko, C. Rettenmaier, A. Herzog, A. Yoon, S. W. Chee, S. Oener, U. Hejral, F. T. Haase and B. Roldan Cuenya, J. Am. Chem. Soc., 2021, 143, 7578–7587 CrossRef CAS PubMed.
- M. Rüscher, A. Herzog, J. Timoshenko, H. S. Jeon, W. Frandsen, S. Kühl and B. Roldan Cuenya, Catal. Sci. Technol., 2022, 12, 3028–3043 RSC.
- S.-F. Hung, F.-Y. Wu, Y.-H. Lu, T.-J. Lee, H.-J. Tsai, P.-H. Chen, Z.-Y. Lin, G.-L. Chen, W.-Y. Huang and W.-J. Zeng, Catal. Sci. Technol., 2022, 12, 2739–2743 RSC.
- T. Ding, X. Liu, Z. Tao, T. Liu, T. Chen, W. Zhang, X. Shen, D. Liu, S. Wang, B. Pang, D. Wu, L. Cao, L. Wang, T. Liu, Y. Li, H. Sheng, M. Zhu and T. Yao, J. Am. Chem. Soc., 2021, 143, 11317–11324 CrossRef CAS PubMed.
- W. H. Lee, C. Lim, E. Ban, S. Bae, J. Ko, H.-S. Lee, B. K. Min, K.-Y. Lee, J. S. Yu and H.-S. Oh, Appl. Catal., B, 2021, 297, 120427 CrossRef CAS.
- X. Y. Zhang, W. J. Li, J. Chen, X. F. Wu, Y. W. Liu, F. Mao, H. Y. Yuan, M. Zhu, S. Dai, H. F. Wang, P. Hu, C. Sun, P. F. Liu and H. G. Yang, Angew. Chem., Int. Ed., 2022, 61, e202202298 CAS.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CrossRef CAS.
- X. Chen, D. A. Henckel, U. O. Nwabara, Y. Li, A. I. Frenkel, T. T. Fister, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2020, 10, 672–682 CrossRef CAS.
- N. T. Nesbitt and W. A. Smith, J. Electrochem. Soc., 2021, 168, 44505 CrossRef CAS.
- G. Zhang, Y. Cui and A. Kucernak, ACS Catal., 2022, 12, 6180–6190 CrossRef CAS PubMed.
- K. Krause, J. K. Lee, C. Lee, H. W. Shafaque, P. J. Kim, K. F. Fahy, P. Shrestha, J. M. LaManna, E. Baltic, D. L. Jacobson, D. S. Hussey and A. Bazylak, J. Power Sources, 2022, 520, 230879 CrossRef CAS.
- H. Hoffmann, M. C. Paulisch, M. Gebhard, J. Osiewacz, M. Kutter, A. Hilger, T. Arlt, N. Kardjilov, B. Ellendorff, F. Beckmann, H. Markötter, M. Luik, T. Turek, I. Manke and C. Roth, J. Electrochem. Soc., 2022, 169, 044508 CrossRef.
- H.-P. Iglesias van Montfort and T. Burdyny, ACS Energy Lett., 2022, 7, 2410–2419 CrossRef CAS.
- A. S. Leach, J. Hack, M. Amboage, S. Diaz-Moreno, H. Huang, P. L. Cullen, M. Wilding, E. Magliocca, T. S. Miller, C. A. Howard, D. J. L. Brett, P. R. Shearing, P. F. McMillan, A. E. Russell and R. Jervis, J. Phys.: Condens. Matter, 2021, 33, 314002 CrossRef CAS PubMed.
- A. Siebel, Y. Gorlin, J. Durst, O. Proux, F. Hasché, M. Tromp and H. A. Gasteiger, ACS Catal., 2016, 6, 7326–7334 CrossRef CAS.
- R. J. K. Wiltshire, C. R. King, A. Rose, P. P. Wells, M. P. Hogarth, D. Thompsett and A. E. Russell, Electrochim. Acta, 2005, 50, 5208–5217 CrossRef CAS.
- A. J. Keeler, G. R. Salazar-Banda and A. E. Russell, Curr. Opin. Electrochem., 2019, 17, 90–96 CrossRef CAS.
- A. Botz, J. Clausmeyer, D. Öhl, T. Tarnev, D. Franzen, T. Turek and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 12285–12289 CrossRef CAS PubMed.
- M. C. O. Monteiro, S. Dieckhöfer, T. Bobrowski, T. Quast, D. Pavesi, M. T. M. Koper and W. Schuhmann, Chem. Sci., 2021, 12, 15682–15690 RSC.
- S. Dieckhöfer, D. Öhl, J. R. C. Junqueira, T. Quast, T. Turek and W. Schuhmann, Chem. – Eur. J., 2021, 27, 5906–5912 CrossRef PubMed.
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2018, 130, 6300–6305 CrossRef.
- I. Khalakhan, M. Vorokhta, P. Kúš, M. Dopita, M. Václavů, R. Fiala, N. Tsud, T. Skála and V. Matolín, Electrochim. Acta, 2017, 245, 760–769 CrossRef CAS.
- X. Wang, J. F. de Araújo, W. Ju, A. Bagger, H. Schmies, S. Kühl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS.
- E. L. Clark and A. T. Bell, J. Am. Chem. Soc., 2018, 140, 7012–7020 CrossRef CAS PubMed.
- C. J. Bondue and M. T. M. Koper, J. Electroanal. Chem., 2020, 875, 113842 CrossRef CAS.
- N. Kardjilov, I. Manke, R. Woracek, A. Hilger and J. Banhart, Mater. Today, 2018, 21, 652–672 CrossRef CAS.
- J. Banhart, A. Borbély, K. Dzieciol, F. Garcia-Moreno, I. Manke, N. Kardjilov, A. R. Kaysser-Pyzalla, M. Strobl and W. Treimer, Dedic. to Profr. Dr H.-P. Degischer Occas. his 65th Birthd., 2010, 101, 1069–1079.
- T. M. M. Heenan, C. Tan, J. Hack, D. J. L. Brett and P. R. Shearing, Mater. Today, 2019, 31, 69–85 CrossRef CAS.
- M. Strobl, I. Manke, N. Kardjilov, A. Hilger, M. Dawson and J. Banhart, J. Phys. D: Appl. Phys., 2009, 42, 243001 CrossRef.
- P. Boillat, E. H. Lehmann, P. Trtik and M. Cochet, Curr. Opin. Electrochem., 2017, 5, 3–10 CrossRef CAS.
- M. C. Paulisch, M. Gebhard, D. Franzen, A. Hilger, M. Osenberg, S. Marathe, C. Rau, B. Ellendorff, T. Turek, C. Roth and I. Manke, ACS Appl. Energy Mater., 2021, 4, 7497–7503 CrossRef CAS.
- Y. Wu, J. I. S. Cho, M. Whiteley, L. Rasha, T. P. Neville, R. Ziesche, R. Xu, R. Owen, N. Kulkarni, J. Hack, M. Maier, N. Kardjilov, H. Markötter, I. Manke, F. R. Wang, P. R. Shearing and D. J. L. Brett, Int. J. Hydrogen Energy, 2020, 45, 2195–2205 CrossRef CAS.
- R. F. Ziesche, J. Hack, L. Rasha, M. Maier, C. Tan, T. M. M. Heenan, H. Markötter, N. Kardjilov, I. Manke, W. Kockelmann, D. J. L. Brett and P. R. Shearing, Nat. Commun., 2022, 13, 1616 CrossRef CAS PubMed.
- W. Wang, S. Yu, K. Li, L. Ding, Z. Xie, Y. Li, G. Yang, D. A. Cullen, H. Yu, Z. Kang, J. A. Wrubel, Z. Ma, G. Bender, C. B. Capuano, A. Keane and F.-Y. Zhang, J. Power Sources, 2021, 516, 230641 CrossRef CAS.
- J. Hack, L. Rasha, P. L. Cullen, J. J. Bailey, T. P. Neville, P. R. Shearing, N. P. Brandon and D. J. L. Brett, Electrochim. Acta, 2020, 352, 136464 CrossRef CAS.
- F. D. Speck and S. Cherevko, Electrochem. Commun., 2020, 115, 106739 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |