
Photo/Ni dual-catalyzed radical defluorinative sulfonylation to synthesize gem-difluoro allylsulfones†
Yiran
Xu
,
Shengchun
Wang
,
Zhao
Liu
,
Mian
Guo
* and
Aiwen
Lei
 *
*
College of Chemistry and Molecular Sciences, The Institute for Advanced Studies (IAS), Wuhan University, Wuhan 430072, P. R. China. E-mail: aiwenlei@whu.edu.cn; whguomian@whu.edu.cn
First published on 2nd March 2023
Abstract
Radical defluorinative functionalization of α-trifluoromethyl styrenes represents an effective way toward gem-difluoroalkenes. There are general interests in developing novel synthetic protocols for defluorinative functionalization with various types of radicals. However, reports on the preparation of gem-difluoro allylsulfones via an S-centered radical pathway are limited. Herein, we developed a photo/nickel dual-catalyzed defluorinative sulfonylation that rapidly and reliably synthesizes gem-difluoro allylsulfones. The merit of this protocol is exhibited by its mild conditions and wide scope, thus providing a novel strategy for the sulfonyl radical participating in radical defluorinative coupling.
As a type of carbonyl isostere, gem-difluoroalkenes are usually of unique metabolic stability, bioactivity, and target specificity, thus providing more opportunities for drug discovery (Scheme 1).1 Therefore, the synthesis of gem-difluoroalkenes has recently been an emerging goal in organic and medicinal chemistry. Until now, several strategies have been developed for the preparation of gem-difluoroalkenes. Classic methods, such as Wittig-type and Reformatsky decarboxylation reactions, usually involve highly reactive species and/or harsh conditions, and result in a limited compatibility of functional groups. As a convergent approach, SN2-type reactions, in which fluoride is lost by nucleophilic attack on CF3, require strong nucleophiles and may limit their substrate scope.2 Distinctive in mechanisms, the revival of radical chemistry has provided new opportunities to prepare gem-difluoroalkenes, in which the defluorination of CF3 is achieved by a Ni/Cr-promoted β-F elimination3 (Scheme 2A) or a photo-/electro-induced radical/polar cross-cover4 (Scheme 2B). However, most studies of radical defluorinative coupling are focused on C-centered radicals or B-centered radicals (Scheme 2B). The exploration of other types of radicals to synthesize diversified gem-difluoroalkenes is in crucial demand, yet challenging.
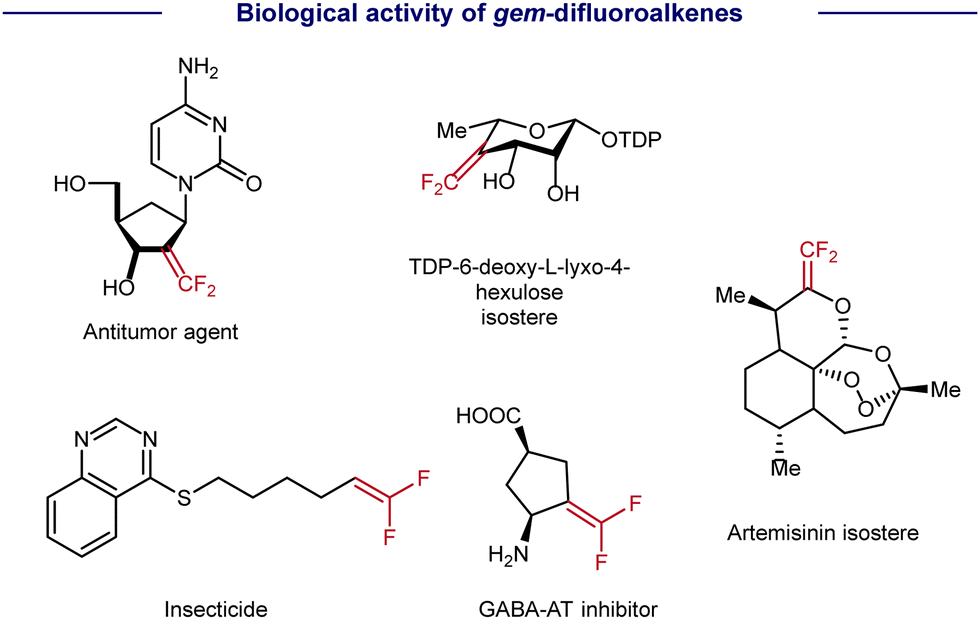 | ||
| Scheme 1 Representative gem-difluoroalkenes with biological activity. | ||
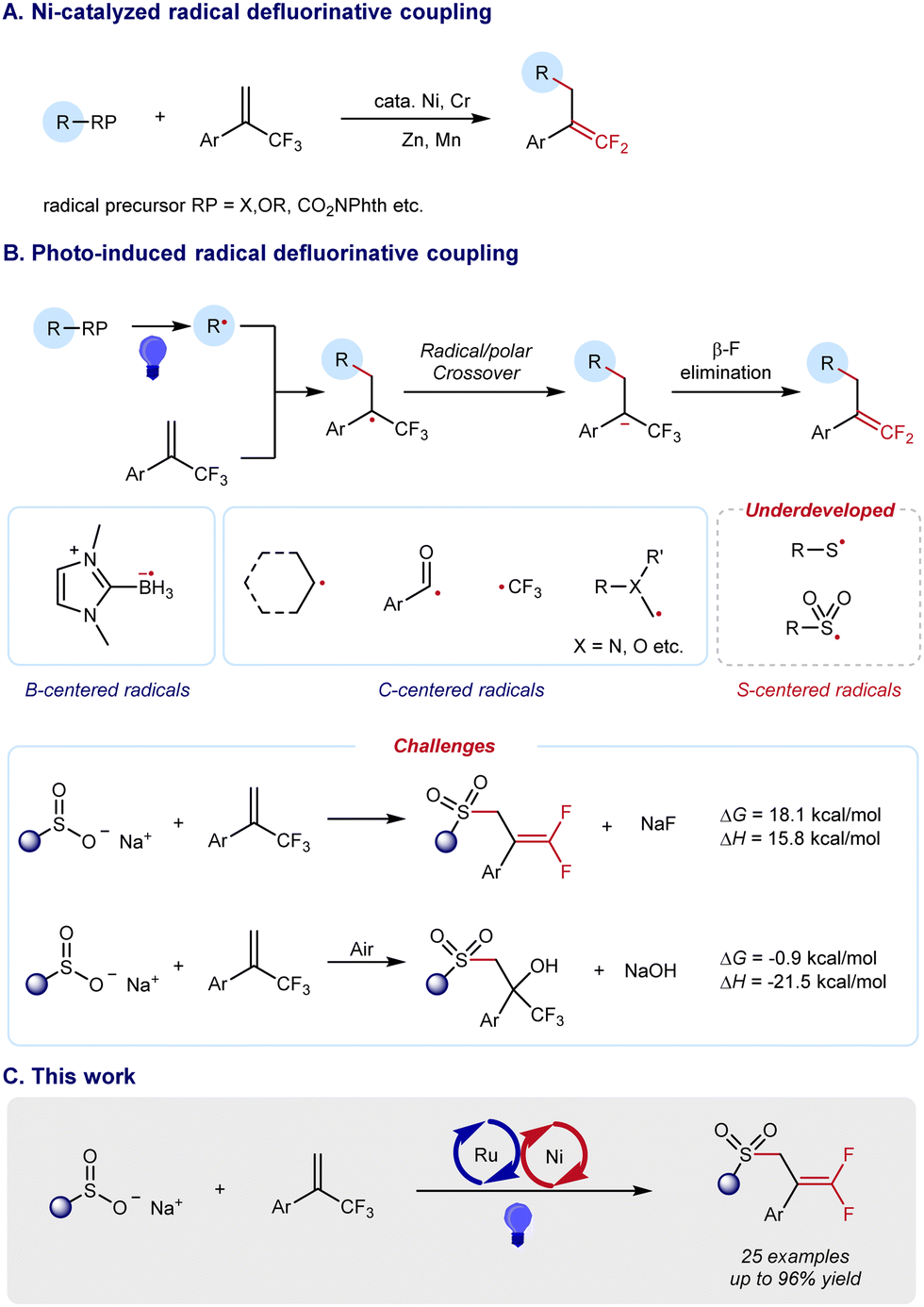 | ||
| Scheme 2 Recent advances in radical defluorinative functionalization to synthesize gem-difluoroalkenes. (A) Ni catalyzed radical defluorinative coupling. (B) Photo-induced radical defluorinative coupling. (C) Outline of this work. | ||
Due to the unique properties of the C–S bond, the construction of S-containing compounds has drawn much attention from synthetic chemists over recent decades.5 Among these organic sulfur compounds, allylsulfones serve as versatile synthetic blocks and can be effectively transformed to other value-added chemicals.6 Thus, we became interested in the synthesis of gem-difluoro allylsulfones which have the potential to integrate the nature of both gem-difluoro alkenes and allylsulfones. However, the synthesis of gem-difluoro allylsulfones via an S-centered radical pathway remains elusive. Challenges still exist in such desired transformations. Defluorinative sulfonylation to synthesize gem-difluoro allylsulfones is endergonic by 17.9 kcal mol−1, which is thermodynamically unfeasible. In addition, an aerobic difunctionalization to produce functionalized CF3-substituted tertiary alcohol is exergonic by −1.2 kcal mol−1, which is thermodynamically spontaneous (see the detailed DFT calculation in Fig. S1, ESI†).
To address such a thermodynamic challenge, we focused our attention to the photocatalytic organic reaction that utilizes visible light as energy input, providing a green and sustainable synthetic protocol. Merging photocatalysis and nickel catalysis, we considered that a radical defluorinative coupling could be achieved based on the addition of a sulfonyl radical to α-(trifluoromethyl)-styrenes, where the subsequent β-F elimination could be effectively promoted by a nickel catalyst (Scheme 2C). This dual-catalyzed process would offer a solution to avoid a fast side reaction that leads to α-trifluoromethyl-β-sulfonyl tertiary alcohols with trace air. Herein, we report the successful execution of this design plan.
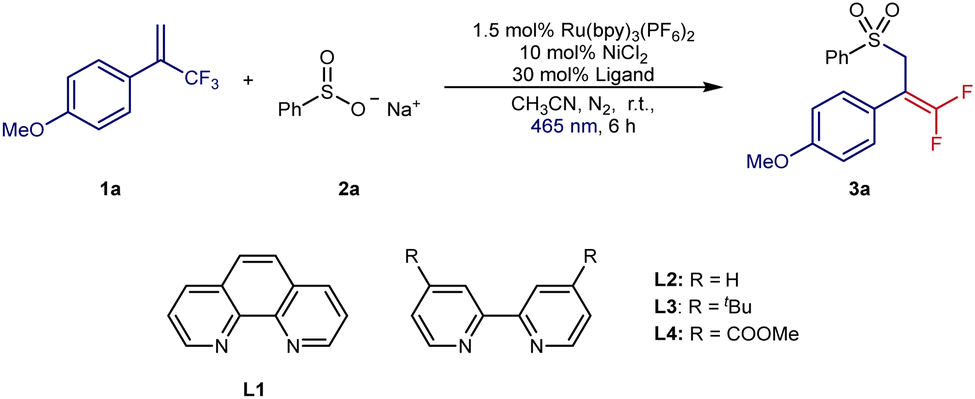
At the outset of our investigation, we chose 1-methoxy-4-(3,3,3-trifluoroprop-1-en-2-yl)benzene 1a as the radical acceptor and inexpensive sodium benzenesulfinate 2a as the sulfonyl radical precursor (Table 1). The initial examination was focused on the ligands with Ru(bpy)3(PF6)2 as the photocatalyst and NiCl2 as the metal catalyst (entries 1–4). To our delight, the desired transformation was successfully realized in 93% isolated yield with 30 mol% L2 as ligand. By merging Ru(bpy)3(PF6)2 and Ni(bpy)3Cl2, the best results were found in MeCN under blue LED irradiation at room temperature to obtain 3a in 96% GC yield and 90% isolated yield. Control experiments (entries 6–9) proved that Ru(bpy)3(PF6)2, Ni(bpy)3Cl2 and irradiation were essential for this organic transformation. Without the nickel catalyst, only a trace amount of product could obtain and the yield was lowered to 39% without the bipyridine ligand. When using eosin Y or an iridium complex as photocatalysts instead of Ru(bpy)3(PF6)2, lower yields of 3a were found (entries 10 and 11). This transformation exhibited a much lower yield with DCE as the solvent and failed with THF as the solvent (entries 12 and 13). Similar to other radical defluorinative couplings, the synthesis of gem-difluoro allylsulfones failed in air (entry 14).
|
|
||
|---|---|---|
| Entry | Variation from the standard conditionsa | Yieldb [%] |
| a Conditions: 1a (0.15 mmol, 1.0 equiv.), 2a (0.20 mmol, 1.33 equiv.), Ru(bpy)3(PF6)2 (1.5 mol%), NiCl2 (10 mol%), ligand (30 mol%) in MeCN (2.0 mL) under N2 atmosphere and irradiation with blue LED (465 nm) for 6 h. b Yields were determined by GC-FID with decane as the internal standard; isolated yield is shown in parentheses. | ||
| 1 | L1 was used | 21 |
| 2 | L2 was used | 93 |
| 3 | L3 was used | 65 |
| 4 | L4 was used | 36 |
| 5 | 10 mol% Ni(bpy)3Cl2 was used | 96 (90) |
| 6 | Without ligand | 39 |
| 7 | Without Ru(bpy)3(PF6)2 | n.d. |
| 8 | Without Ni(bpy)3Cl2 | Trace |
| 9 | Without irradiation | n.d. |
| 10 | Eosin Y as photocatalyst | n.d. |
| 11 | [Ir(dFCF3ppy)2((4,4′-dCF3bpy))](PF6) as photocatalyst | 12 |
| 12 | DCE as solvent | 21 |
| 13 | THF as solvent | n.d. |
| 14 | Air instead of N2 | n.d. |
With the optimal conditions in hand, we turned our attention to exploring the generality of our photo/Ni dual-catalyzed radical defluorinative coupling of α-trifluoromethyl styrene. As shown in Scheme 3, the scope was largely insensitive to electronic changes at the para and meta positions of trifluoromethylated alkenes (3a–3k). However, this defluorinative coupling failed with otho-substituted trifluoromethylated alkenes (see the ESI†). Interestingly, other cyclic motifs of CF3-substituted alkenes were also suitable radical acceptors for this organic transformation, including naphthalene (3l), benzodioxole (3m), benzodioxan (3n), and N-Boc pyrrole (3o).
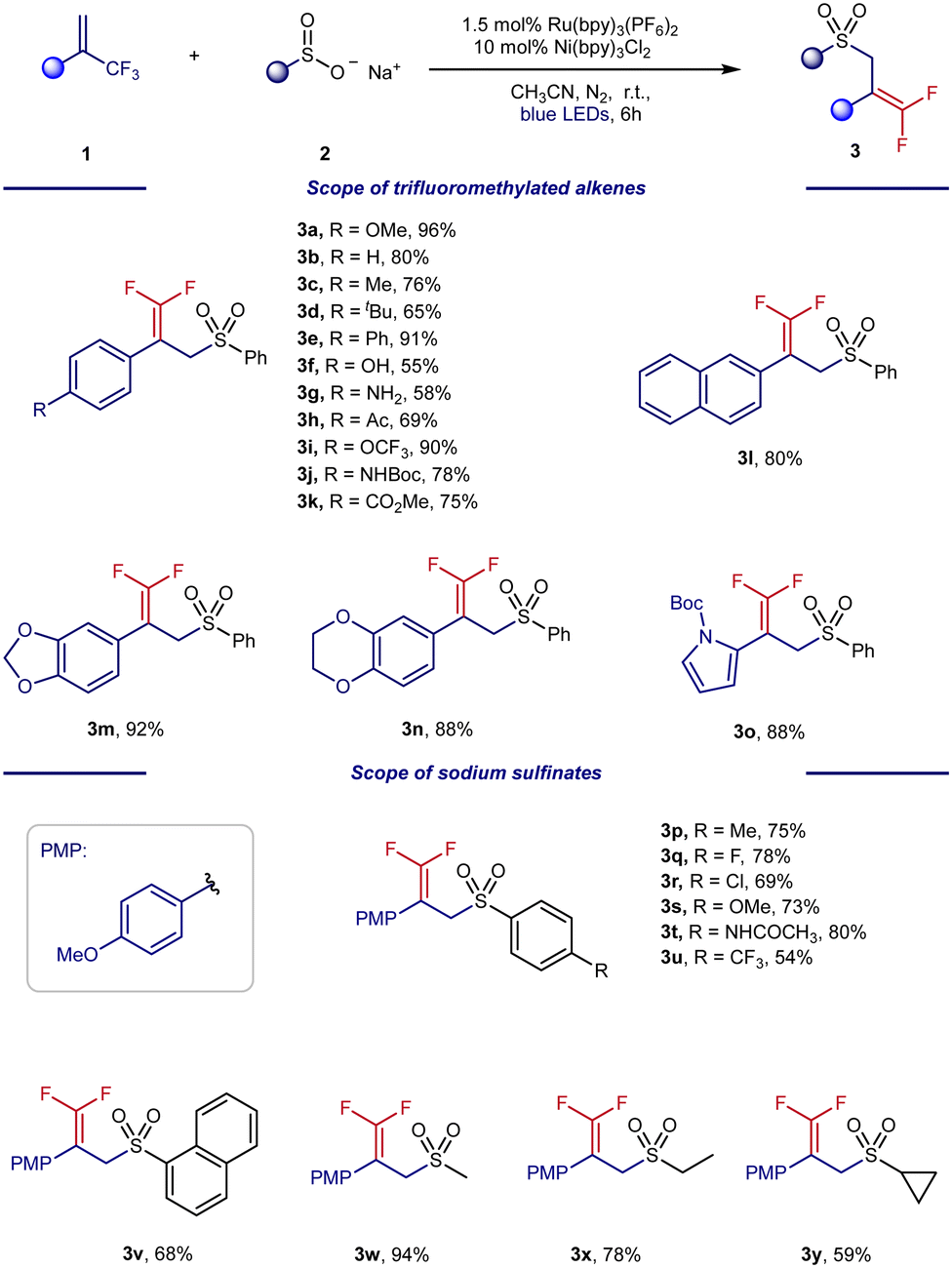 | ||
| Scheme 3 Scope of substrates. Reaction conditions: 1 (0.15 mmol, 1.0 equiv.), 2 (0.2 mmol, 1.33 equiv.), Ru(bpy)3(PF6)2 1.5 mol%, Ni(bpy)3Cl2 10 mol%, MeCN (2.0 mL), room temperature, N2, 6 h. Yields of isolated products are shown. | ||
Next, the scope of the sodium sulfinates was examined. A series of para-substituted sodium benzenesulfinates, including halides (3q and 3r), amides (3t), and trifluoromethyl (3u), were all well tolerated, forming the desired products in 54–80% yield. In addition, 2-naphthylsulfinic acid sodium (3v) was also a suitable sulfonyl radical precursor for this transformation, providing 64% yield. Pleasingly, sodium alkylsulfinates (3w–3y) were also well-tolerated under the reaction conditions and provided yields of 59–94%. To further explore the potential applications of this synthetic method, we carried out a gram-scale experiment (Scheme 4). The gram-scale reaction between 1a and 2a afforded the corresponding gem-difluoroalkenes 3a in 96% isolated yield.
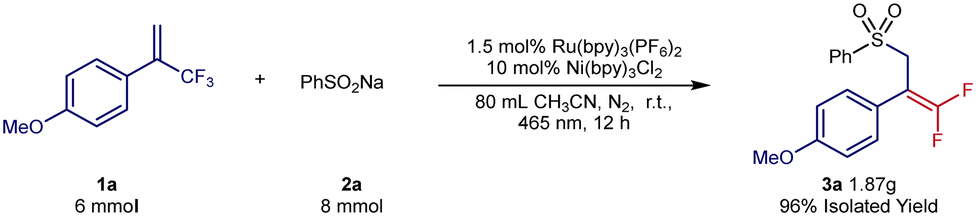 | ||
| Scheme 4 Gram-scale experiment. 1a (6 mmol, 1.0 equiv.), 2a (8 mmol, 1.33 equiv.), Ru(bpy)3(PF6)2 (0.09 mmol, 1.5 mol%), Ni(bpy)3Cl2 (0.6 mmol, 10 mol%), 80 mL CH3CN, r.t., 465 nm, 12 h. | ||
Based on previous reports,4h,7 a plausible mechanism of this radical defluorinative sulfonylation is shown in Scheme 4. Firstly, sodium benzenesulfonate is preferentially oxidized by photo-excited RuII(bpy)3 to generate the corresponding sulfonyl radical and RuI complex. Then, radical addition occurs between the sulfonyl radical and 2a to form a benzyl carbon radical I, which can further react with NiII to form alkyl-NiIII species II. Next, the final product 3a is obtained via a β-F elimination from II, and at the same time NiIII-F is generated. Finally, RuII(bpy)3 and NiII are regenerated via a single electron transfer between the RuI complex and NiIII-F and the catalytic cycle is completed (Scheme 5).
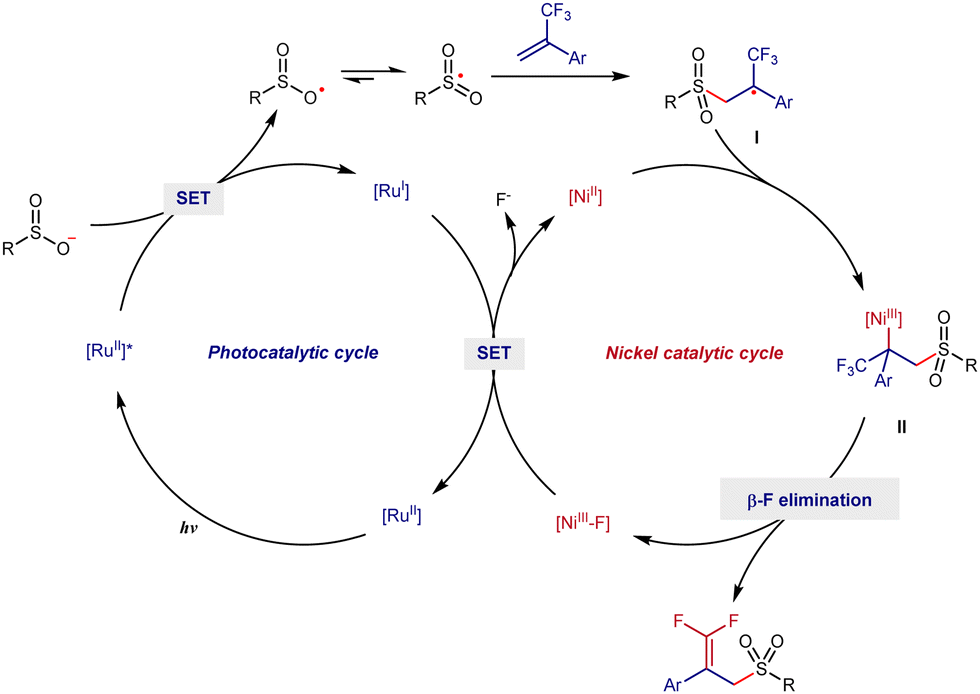 | ||
| Scheme 5 Proposed mechanism. | ||
We have demonstrated that a radical defluorinative sulfonyla-tion, consisting of the addition of a sulfonyl radical to alkenes and a nickel-promoted β-F elimination, leads to a challenging coupling of sodium sulfinates and CF3-substituted alkenes to synthesize a series of gem-difluoro allylsulfones. This protocol features mild conditions, a facile synthesis, and a wide scope of substrates. We believe that this method not only provides a rare example of a sulfonyl radical participating in the synthesis of gem-difluoro allylsulfones, but also represents a new strategy of photo/nickel dual-catalyzed defluorinative functionalization.
This work was supported by the National Key R&D Program of China (No. 2021YFA1500104), the National Natural Science Foundation of China (No. 22031008 and No. 22171216), the Science Foundation of Wuhan (No. 2020010601012192), and the Postdoctoral Foundation of Hubei Province (No. 211000025). We thank Prof. Xiaotian Qi for his efforts in DFT calculations. The numerical calculations in this paper were performed on the supercomputing system at the Supercomputing Center of Wuhan University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Bobek, I. Kavai and E. De Clercq, J. Med. Chem., 1987, 30, 1494 CrossRef CAS PubMed; (b) C. Leriche, X. He, C. W. Chang and H. W. Liu, J. Am. Chem. Soc., 2003, 125, 6348 CrossRef CAS PubMed; (c) K. Fujii, Y. Nakamoto, K. Hatan and Y. Kanetsuki, JP2006016331 A, 2006; (d) Y. Pan, J. Qiu and R. B. Silverman, J. Med. Chem., 2003, 46, 5292 CrossRef CAS PubMed; (e) G. Magueur, B. Crousse, M. Ourévitch, D. Bonnet-Delpon and J.-P. Bégué, J. Fluorine Chem., 2006, 127, 637 CrossRef CAS.
- (a) G. Chelucci, Chem. Rev., 2012, 112, 1344 CrossRef PubMed; (b) X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375 CrossRef CAS; (c) M. Hu, Z. He, B. Gao, L. Li, C. Ni and J. Hu, J. Am. Chem. Soc., 2013, 135, 17302 CrossRef CAS PubMed; (d) M. Wang, X. Pu, Y. Zhao, P. Wang, Z. Li, C. Zhu and Z. Shi, J. Am. Chem. Soc., 2018, 140, 9061 CrossRef CAS PubMed; (e) P. H. S. Paioti, J. Del Pozo, M. S. Mikus, J. Lee, M. J. Koh, F. Romiti, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2019, 141, 19917 CrossRef CAS PubMed.
- (a) Y. Lan, F. Yang and C. Wang, ACS Catal., 2018, 8, 9245 CrossRef CAS; (b) Z. Lin, Y. Lan and C. Wang, ACS Catal., 2018, 9, 775 CrossRef; (c) X. Lu, X. X. Wang, T. J. Gong, J. J. Pi, S. J. He and Y. Fu, Chem. Sci., 2019, 10, 809 RSC; (d) C. Zhu, Z. Y. Liu, L. Tang, H. Zhang, Y. F. Zhang, P. J. Walsh and C. Feng, Nat. Commun., 2020, 11, 4860 CrossRef CAS PubMed; (e) C. Zhang, Z. Lin, Y. Zhu and C. Wang, J. Am. Chem. Soc., 2021, 143, 11602 CrossRef CAS PubMed; (f) Y. Ping, Q. Pan, Y. Guo, Y. Liu, X. Li, M. Wang and W. Kong, J. Am. Chem. Soc., 2022, 144, 11626 CrossRef CAS PubMed.
- (a) T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390 CrossRef CAS PubMed; (b) F.-L. Qing, X.-Y. Liu, J.-A. Ma, Q. Shen, Q. Song and P. Tang, CCS Chem., 2022, 4, 2518 CrossRef CAS; (c) F. Tian, G. Yan and J. Yu, Chem. Commun., 2019, 55, 13486 RSC; (d) W. Xu, H. Jiang, J. Leng, H. W. Ong and J. Wu, Angew. Chem., Int. Ed., 2020, 59, 4009 CrossRef CAS PubMed; (e) T. Xiao, L. Li and L. Zhou, J. Org. Chem., 2016, 81, 7908 CrossRef CAS PubMed; (f) S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073 CrossRef CAS PubMed; (g) J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 3723 CrossRef CAS PubMed; (h) L.-H. Wu, J.-K. Cheng, L. Shen, Z.-L. Shen and T.-P. Loh, Adv. Synth. Catal., 2018, 360, 3894 CrossRef CAS; (i) W. J. Yue, C. S. Day and R. Martin, J. Am. Chem. Soc., 2021, 143, 6395 CrossRef CAS PubMed; (j) W. Chen, S. Ni, Y. Wang and Y. Pan, Org. Lett., 2022, 24, 3647 CrossRef CAS PubMed; (k) X. Yan, S. Wang, Z. Liu, Y. Luo, P. Wang, W. Shi, X. Qi, Z. Huang and A. Lei, Sci. China: Chem., 2022, 65, 762 CrossRef CAS; (l) C. Zhu, M. M. Sun, K. Chen, H. Liu and C. Feng, Angew. Chem., Int. Ed., 2021, 60, 20237 CrossRef CAS PubMed; (m) Z. Cai, R. Gu, W. Si, Y. Xiang, J. Sun, Y. Jiao and X. Zhang, Green Chem., 2022, 24, 6830 RSC; (n) F. Li, C. Pei and R. M. Koenigs, Angew. Chem., Int. Ed., 2022, 61, e202111892 CAS; (o) B. Wang, C. T. Wang, X. S. Li, X. Y. Liu and Y. M. Liang, Org. Lett., 2022, 24, 6566 CrossRef CAS PubMed; (p) X. Wang, C. Wang and C. Bolm, Org. Lett., 2022, 24, 7461 CrossRef CAS PubMed.
- (a) P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807 CrossRef CAS PubMed; (b) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC; (c) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016 CrossRef CAS PubMed; (d) P. Renzi, E. Azzi, A. Lanfranco, R. Moro and A. Deagostino, Synthesis, 2021, 3440 CrossRef.
- (a) A. El-Awa, M. N. Noshi, X. M. du Jourdin and P. L. Fuchs, Chem. Rev., 2009, 109, 2315 CrossRef CAS PubMed; (b) X. Chen, S. Hussain, S. Parveen, S. Zhang, Y. Yang and C. Zhu, Curr. Med. Chem., 2012, 19, 3578 CrossRef CAS PubMed.
- (a) Q. Liu and L.-Z. Wu, Natl. Sci. Rev., 2017, 4, 359 CrossRef CAS; (b) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485 CrossRef CAS PubMed; (c) Z. Li, C. Li, Y. Ding and H. Huo, Coord. Chem. Rev., 2022, 460, 214479 CrossRef CAS; (d) T. Ichitsuka, T. Fujita and J. Ichikawa, ACS Catal., 2015, 5, 5947 CrossRef CAS; (e) F. Chen, X. Xu, Y. He, G. Huang and S. Zhu, Angew. Chem., Int. Ed., 2020, 59, 5398 CrossRef CAS PubMed; (f) C. Yao, S. Wang, J. Norton and M. Hammond, J. Am. Chem. Soc., 2020, 142, 4793 CrossRef CAS PubMed; (g) J. S. Lin, X. Y. Dong, T. T. Li, N. C. Jiang, B. Tan and X. Y. Liu, J. Am. Chem. Soc., 2016, 138, 9357 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc05934g |
| This journal is © The Royal Society of Chemistry 2023 |