
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cationic dialanes with fluxional π-bridged cyclopentadienyl ligands†
Philipp
Dabringhaus
 ,
Silja
Zedlitz
and
Ingo
Krossing
*
,
Silja
Zedlitz
and
Ingo
Krossing
*
Albert-Ludwigs University Freiburg, Albertstr. 21, Freiburg 79104, Germany. E-mail: krossing@uni-freiburg.de
First published on 21st November 2022
Abstract
Unique π-cyclopentadienyl bridged dialanes are synthesized as complex salts with aluminate anions by comproportionation of aluminocenium cations [AlIII(Cp)(Cp*)]+/[AlIIICp2]+ with [(AlICp*)4]. Very short Al–Al bond lengths occur in positively charged Al24+ fragments. Intriguingly, the prepared asymmetric dialane shows a unique fluxional coordination of the cyclopentadienyl ligands.
The first unsupported dialane was reported already in 1988 by Uhl.1 Since then, a large variety of dialanes of type R2Al–AlR2 and their adducts have been prepared, mostly via the reduction of the respective aluminium halides with alkali metals.2 Only recently, anionic ethane-analogous examples [R3Al–AlR3]2− were described.3 In the course of the intense search for and study of aluminylenes and dialumenes, dialanes bridged by alkenes, alkynes, or aromatic hydrocarbons have been prepared.4,5 This portfolio of bridged dialanes was extended recently by an oxo-bridged anionic dialane with a highly strained Al–Al σ-bond6 as well as bis-phosphinidene-bridged dialanes.7 The reversible insertion of aluminylenes into Al–H bonds is a novel pathway to dialanes8 and allows for the synthesis of the only known cationic dialanes.9
Surprisingly, although cyclopentadienyl ligands have been fundamentally important for the development of Al(I) complexes like in the first Al(I) compound [(AlCp*)4] (Cp* = C5Me5)10 or in a first Al4+ cluster salt,11 only very few examples of AlII–AlII bound structures stabilized with Cp ligands are known. Complexes of type [(Al(X)Cp*)2] (X = Br, I) were reported as intermediates during the reduction of Cp*AlX2 complexes to [(AlCp*)4].12 Analogues with the bulkier Cp3t-ligand (Cp3t = 1,3,5-tBu-C5H2) are also known.13 Recently, a bis(aluminocenophane) I with a short Al–Al bond was reported (Chart 1).14 In all of these compounds, either more ionic η5-coordinated or more covalent η1-bonded cyclopentadienyl ligands were observed. π-Cp ligands bridging over two metal centres were hitherto reported for very few group 10 metal examples.15 In the main group, the only known examples comprise π-Cp bridged MII–MII units characterised for M = Ga16 (II) and M = In17 (III).
 | ||
| Chart 1 Recently reported aluminocenophane as well as a Cp-bridged digallane and a diindane. TMEDA = tetramethylethylenediamine, TMS = trimethylsilyl. | ||
Here, we report on the syntheses of cationic cyclopentadienyl bridged dialanes via comproportionation of AlICp* and aluminocenium cations, which were generated in situ by the reduction of Sn(II)/Ge(II) cations or synthesized separately. The starting point for the synthesis of novel cationic dialanes was the reactivity study of Schnöckels [(AlCp*)4] towards cyclopentadienylgermanium18 and -tin19 cations [MCp][F{Al(ORF)3}2] (M = Ge, Sn, RF = C(CF3)3), sparked by our discovery of mixed low-valent group 13 complex salts.20 Reaction of [MCp]+ salts with 0.25 [(AlCp*)4] yielded a mixture of various compounds from which single crystals of the asymmetric aluminocenium cation [Al(Cp)(Cp*)][F{Al(ORF)3}2] grew (ESI†). Interestingly, addition of 0.5 equivalents of [(AlCp*)4] yielded an elusive π-cyclopentadienyl bridged dialane [Cp(AlCp*)2][F{Al(ORF)3}2] 1A as a colourless crystal (eqn (1), ESI†). Reduction of [MCp]+ by AlCp* was accompanied by Cp abstraction to intermittently form the mixed aluminocenium ion [AlIII(Cp)(Cp*)]+, which probably did undergo comproportionation to 1A with another AlICp* molecule. This reactivity is surprising, since AlICp* typically forms mixed valent donor–acceptor complexes of type Cp*Al → AlR3 (R = t-Bu,21 C6F522) upon reaction with AlIII-Lewis acids. Hence, the formation of divalent 1A would require the presence of the mixed valent Lewis acid–base adduct [Cp*AlI → AlIII(Cp*)(Cp)]+2. Notably, a closely related compound has been isolated with [CpGaI → GaIIICp2I].23

To study this interplay between a mixed-valent AlI → AlIII adduct as in 2 and AlII–AlII dialane 1, we developed a straightforward synthesis of these complexes starting from accessible Al(III) and Al(I) species. We chose the hitherto elusive parent aluminocenium salt [AlIIICp2][Al(ORF)4] 3 as the starting material. Although the synthesis of [AlCp2]+ was reported in 1996 as an ion pair with a [MeB(C6F5)3]− counterion24 and low-temperature NMR analysis clearly confirmed its successful preparation by the characteristic signal at δ27Al = −126.4, no scXRD suitable crystals could be grown. Its structural characterization was achieved in 2009 by switching to a less coordinating anion, i.e. [Al(ORF)4]− in 3, prepared by addition of [H(OEt2)2][Al(ORF)4] to AlCp3.25 Yet, formation of the ether-adduct [AlCp2(OEt2)2]+ and the instability of solutions of 3 in DCM at temperatures above −20 °C impeded a clean isolation and full characterisation of 3. Therefore, we developed an easily scalable synthesis of 3 (ESI†). A solution of AlCp3, in situ prepared from AlCl3 and MgCp2 in toluene26 (Scheme 1a), was reacted with [Ph3C][Al(ORF)4] at room temperature to yield clean 3 as a colourless precipitate in high yield (89%, Scheme 1b).25 The purity of 3 was verified by NMR spectroscopy in fluorobenzene at room temperature, giving sharp lines at δ1H = 6.45 and at δ27Al = −130.9. We note that the reduced ion pairing of the cation with the anion [Al(ORF)4]−vs. [MeB(C6F5)3]− leads to a pronounced high-field shift by −4.5 ppm. Aluminocenium cation 3 is a potent and accessible Lewis acid (FIAgas = 746, FIADCM = 241, HIAgas = 716, and HIADCM = 133)27 and its reactivity in Lewis acid catalysis or as a cationic Al-source will likely spark future research.
 | ||
| Scheme 1 Synthesis of Cp-bridged dialanes 1B and 4via the parent aluminocenium cation 3. 1,2-DFB = 1,2-difluorobenzene; RF = C(CF3)3. | ||
To study the redox reaction between trivalent 3 and AlICp*, 0.25 equivalents of [(AlCp*)4] were added to a 1,2-DFB solution of 3 at −40 °C (Scheme 1d). Here, the asymmetric Cp-bridged dialane [Cp(AlCp*)(AlCp)][Al(ORF)4] 4 was isolated instead of a Lewis acid–base adduct as formulated for 2 (ESI†). Increased addition of 0.5 equivalents [(AlCp*)4] to 3 results in the formation of the [Al(ORF)4]− salt of the symmetric dialane [Cp(AlCp*)2]+1B, which is accompanied by the formation of metallic aluminium and hints to the formation of highly labile [(AlCp)4] (ESI†).28 Moreover, the reaction is accompanied by the formation of the known28 and rt-stable [CpAl(AlCp*)3] (δAlCp* = −76.3, δAlCp = −108.1). If the parent aluminocenium cation 3 is reacted with 1.25 equiv. [(AlCp*)4], a clean reaction to 1B and [CpAl(AlCp*)3] is observed and both compounds could be isolated (Scheme 1c, ESI†). Intriguingly, 1B can also be generated cleanly from 4 by addition of our previously reported low-valent Al4-cluster cation,11 accompanied by Al(0) precipitation (ESI,†Scheme 1e).
The molecular structures of the symmetric dialanes similarly display short Al–Al bond lengths of 2.518(1) Å in 1A and 2.508(1) Å in 1B (Fig. 1), which are only slightly longer compared to the Al–Al bonds in aluminocenophane I14 or highly strained cyclic dialanes.6,29 Moreover, a similar orientation of the bridging Cp-unit on the Al–Al bond is observed in 1A and 1B with distances of 2.297(1) Å and 2.287(1) Å between the Cp-centroids and the midpoint of the Al–Al bond respectively. C–C bond lengths in the bridging Cp ligand differ insignificantly with the shortest bond representing the C2–C3 bond (1.384(4) Å in 1A, 1.393(3) Å in 1B) and the longest representing the C1–C2/C3–C4 bonds (1.417(4) Å in 1AB). The molecular structure of 4 includes four different cations in the asymmetric unit with three different Cp orientations relative to the Al–Al bond. With an average Al–Al bond length of 2.487 Å in 4 (shortest: 2.481(1) Å, dAl–Al, range = 2.481(1)–2.492(1) Å), they are even shorter than in the symmetric dialanes 1A and 1B. Globally, the only shorter Al–Al bond known to date resides in 1,2-dialuminacyclobutene (2.467(2) Å).5 Moreover, no considerable changes in C–C bond lengths in the bridging Cp ligands are observed. The flexible coordination of the bridging Cp ligand in 4 is already indicated in the multiple structures detected by scXRD. This flexibility is further confirmed using the 1H NMR spectrum of 4, where only one singlet is observed at 6.22 ppm for all protons of the inequivalent Cp ligands (ESI†). Hence, not only a rotation of the bridging Cp, but also a rapid exchange between the bridging and the terminal Cp ligands occurs. Moreover, only one broad singlet at δ27Al = −47 is observed for 4 (ESI†). A static structure has computed shifts of the Cp- and Cp*-coordinated aluminium atoms at −63 and −38 ppm respectively. Hence, a complete exchange of the Cp-type ligands in the asymmetric dialane is quick via structure 2 rendering all Al atoms and the Cp ligands equivalent on the timescale of NMR. No freezing of the ligand exchange could be observed upon cooling the probe to −40 °C (ESI†). The symmetric dialane displays a broad singlet at δ27Al = −40 (δcalc = −41). These observations are supported by the computed thermodynamics, which reveal an insignificant activation barrier of 0.6/3.2 kJ mol−1 for the rotation of the Cp ring in 4/1 respectively (Fig. 2). The exchange of the bridging and terminal Cp units in 4 was computed to be a concerted mechanism with a very small activation energy of only 23.2 kJ mol−1. A higher barrier of +44 kJ mol−1 was computed for Cp–Cp* exchange (Fig. 2). The transition states of these ligand exchange TSI and TSII resemble the structures expected for classic donor–acceptor AlI → AlIII complexes of type 2 (vide infra). This fluxional coordination reflects the versatile electronic bonding situation in 4.
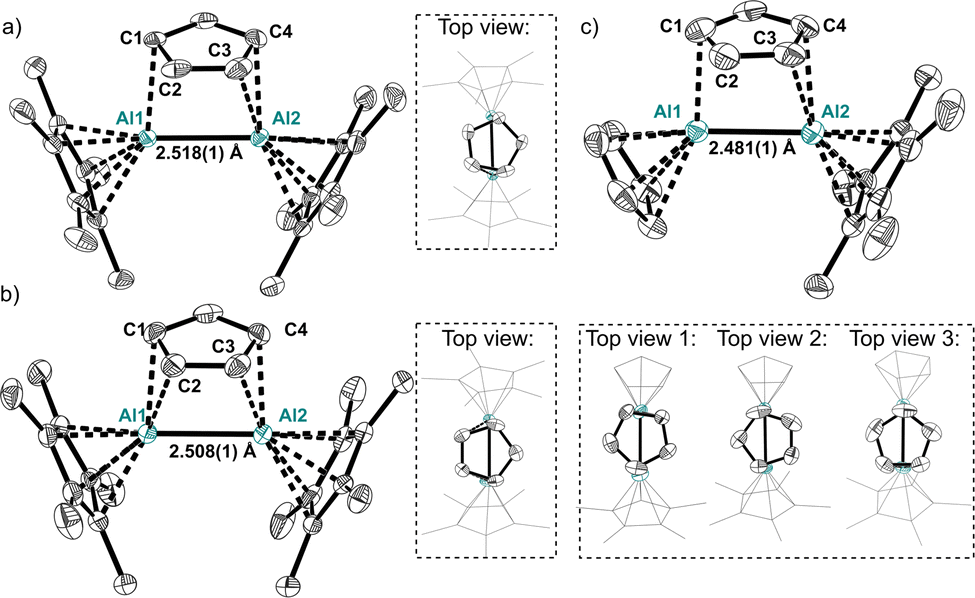 | ||
| Fig. 1 Molecular structures of cations 1A (a), 1B (b) and 4 (c) along with views of different orientations of the bridging Cp ligand. H-atoms and anions are omitted for clarity. Thermal displacement of the ellipsoids is set at 50% probability. | ||
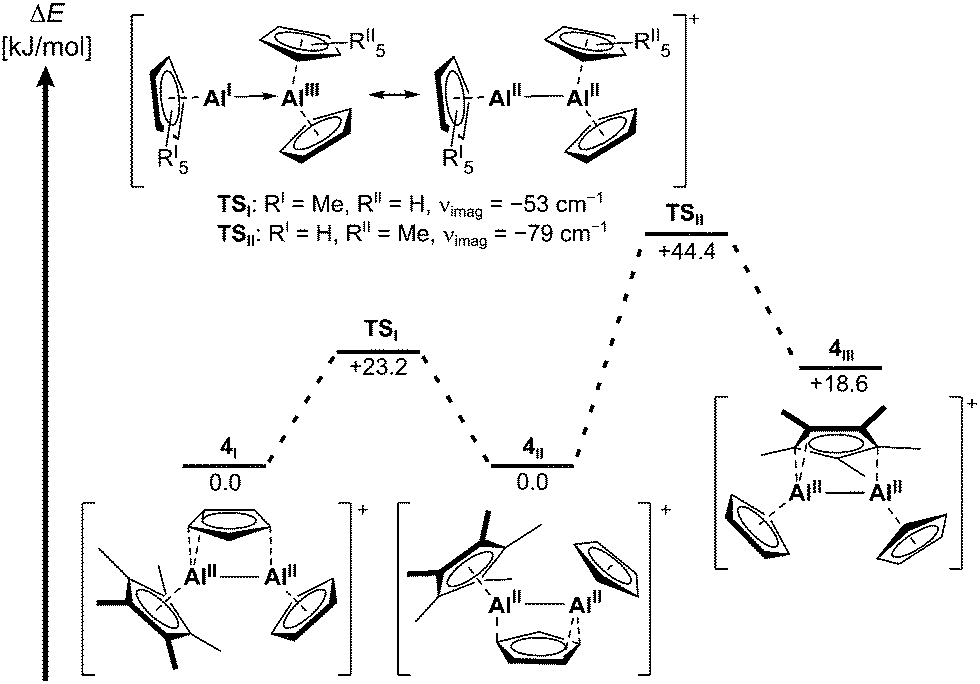 | ||
Fig. 2 Reaction profile for exchange of Cp* and Cp ligands in 4 (bp86-d3bj/def2-svp level of DFT). TSI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) andII are not intermediates, but include one imaginary frequency. andII are not intermediates, but include one imaginary frequency. | ||
The bonding in dialanes 1 and 4 was further analysed by DFT. Natural bond orbital (NBO) analyses on the Al–Al bonds reveal a σ-bonding interaction with large s character of the Al atoms (>60%) and Wiberg bond orders of 0.99 and 0.96 for the symmetric and asymmetric dialanes respectively (ESI†). The QTAIM analysis (quantum theory of atoms in molecules) yields highly positive charges on the central Al2 fragment in 4 (Fig. 3a, for 1 see the ESI†). Moreover, a non-nuclear attractor (NNA) resides between the Al atoms in the cations. Similarly to isoelectronic Na2 and [Mg–Mg]2+ fragments,30,31 NNAs in the formal [Al–Al]4+ dimers 1 and 4 originate from significant build-up of electron density upon bonding of Al atoms with high s character30 and were also calculated for the recently reported aluminocenophane I.14 Due to the NNAs, QTAIM charges of the individual Al atoms cannot be discussed (ESI†). In the EDA-NOCV analysis of 4, the interaction of the bridged Cp is best described by coordination of a Cp− anion to the [CpAl–AlCp*]2+ dimer (ESI†). Here, the attractive bonding is dominated by the electrostatic interaction (ΔEelstat. = −275 kcal mol−1 (65%), ΔEOrb = −131 kcal mol−1 (31%)). For the alternative fragmentation into Cp˙ and [CpAl–AlCp*]+˙, the ΔEOrb value is −142 kcal mol−1 only slightly higher. Since the ionic fragmentation overestimates ΔEelstat, while the fragmentation into radicals underestimates ΔEelstat, the reality most likely lies between the two extremes. In both fragmentations, the interaction between the σ* orbital of the Al–Al bond and the Cp ligand represents the major orbital interaction (Fig. 3b).
 | ||
| Fig. 3 (a) and (c) Distribution of the Laplacian Δρr in the Al–RCP(Cp)–Al planes of 4 (a) and TSI (c) with BCP (dark blue), RCP (ring critical point, red) and NNA (green) along with computed QTAIM charges. (b) and (d) Plots of the deformation density Δρ (isovalue 0.001) associated with the major orbital interaction ΔEOrb (values in kcal mol−1) for fragmentation of 4 in [CpAlAlCp*]2+ (singlet) and Cp− (singlet) (b) and TSI in [AlCp2]˙(doublet) and [AlCp*]+˙ (doublet) (d) and the eigenvalues ν of Δρ. Charge flows from yellow to purple. (e) Gas phase and solution thermodynamics (in 1,2-DFB) for the observed dialane formation as well as their disproportionation into Al(I) and Al(III) species. All values are in kJ mol−1. Computed at the pbe0-d3bj/def2-tzvpp/bp86-d3bj/def2-svp level of DFT, solvation energies with cosmo-rs theory at the bp86-d3bj/def2-svp level. | ||
Intrigued by the fluxionality of the ligands in 4, the computed transition states TSI and TSII were analysed by means of EDA-NOCV and QTAIM (Fig. 3c and d, for TSII see the ESI†). Similar charges of the central Al2 fragment are computed for the cations. Contour plots of the Laplacians of TSI and TSII indicate an asymmetry in bonding interaction when compared to 4 (Fig. 3c, ESI†). In the EDA-NOCV analysis, Al–Al bonds in TSI and TSII are best described as covalent bonds by fragmentation into an [Al(Cp)(Cp/Cp*)]˙ and an [Al(Cp/Cp*)]+˙ fragment (Fig. 3d, ESI†). Surprisingly, the small eigenvalues of the Δρ plots reveal minor charge accumulation at the η5-Cp coordinated Al atom. Hence, in contrast to the expected donor–acceptor character in the transition states, TSI and TSII are best described as covalently bound and polarized dialanes.
Finally, the overall thermodynamics for the presented synthetic procedure were computed (Fig. 3e). Exergonic reaction enthalpies are obtained for all observed reaction steps, both in the gas phase and in solution. Moreover, the symmetric dialane 1 is computed to be stable against disproportionation, which is in-line with the experiment. Intriguingly, the disproportionation of the asymmetric dialane 4 into AlICp and [AlCpCp*]+ is more favourable compared to the reformation of [AlCp2]+ and AlICp*. Hence, in principle dialane 4 could function as a stable transfer source of the elusive AlICp to a suitable acceptor. Note that solutions of ‘AlICp’ decompose above −60 °C.
In summary, we report cyclopentadienyl bridged, cationic dialanes formed via comproportionation between aluminocenium cations and [(AlCp*)4]. The asymmetric dialane shows a unique fluxional, ionic coordination of the Cp ligands at the central [Al–Al]4+ units. The reactivity of the reported dialanes, combining the electrophilicity of cationic Al species with the reducing ability of dialanes, will be studied in further research.
This work was supported by the Fonds of the Chemical Industry and the DFG (grant KR2046/35-1). Support came from the state of Baden-Württemberg through bwHPC and the DFG through grant no INST 40/467-1 and 575-1 FUGG (JUSTUS1/2 cluster).
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. Uhl, Z. Naturforsch. B, 1988, 43, 1113 CrossRef CAS.
- (a) Y. Zhao, Y. Liu, L. Yang, J.-G. Yu, S. Li, B. Wu and X.-J. Yang, Chem. – Eur. J., 2012, 18, 6022 CrossRef CAS; (b) K. Koshino and R. Kinjo, J. Am. Chem. Soc., 2020, 142, 9057 CrossRef CAS; (c) S. Kurumada, S. Takamori and M. Yamashita, Nat. Chem., 2020, 12, 36 CrossRef CAS PubMed.
- (a) C. J. Snyder, P. Zavalij, K. Bowen, H. Schnöckel and B. Eichhorn, Dalton Trans., 2015, 44, 2956 RSC; (b) S. J. Bonyhady, N. Holzmann, G. Frenking, A. Stasch and C. Jones, Angew. Chem., Int. Ed., 2017, 56, 8527 CrossRef CAS.
- (a) P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, J. Am. Chem. Soc., 2017, 139, 14384 CrossRef CAS PubMed; (b) C. Weetman, A. Porzelt, P. Bag, F. Hanusch and S. Inoue, Chem. Sci., 2020, 11, 4817 RSC; (c) R. L. Falconer, K. M. Byrne, G. S. Nichol, T. Krämer and M. J. Cowley, Angew. Chem., Int. Ed., 2021, 60, 24702 CrossRef CAS.
- T. Agou, K. Nagata and N. Tokitoh, Angew. Chem., Int. Ed., 2013, 52, 10818 CrossRef CAS.
- K. Koshino and R. Kinjo, J. Am. Chem. Soc., 2021, 143, 18172 CrossRef CAS PubMed.
- S. Nees, F. Fantuzzi, T. Wellnitz, M. Fischer, J.-E. Siewert, J. T. Goettel, A. Hofmann, M. Härterich, H. Braunschweig and C. Hering-Junghans, Angew. Chem., Int. Ed., 2021, 60, 24318 CrossRef CAS.
- (a) T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195 CrossRef CAS; (b) R. L. Falconer, G. S. Nichol, I. V. Smolyar, S. L. Cockroft and M. J. Cowley, Angew. Chem., Int. Ed., 2021, 60, 2047 CrossRef CAS; (c) C. Bakewell, K. Hobson and C. J. Carmalt, Angew. Chem., Int. Ed., 2022, 61, e202205901 CrossRef CAS.
- (a) L. J. Morris, A. Carpentier, L. Maron and J. Okuda, Chem. Commun., 2021, 57, 9454 RSC; (b) J. Kretsch, A. Kreyenschmidt, T. Schillmöller, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2020, 59, 13690 CrossRef CAS.
- C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1991, 30, 564 CrossRef.
- P. Dabringhaus, J. Willrett and I. Krossing, Nat. Chem., 2022, 14, 1151 CrossRef CAS.
- S. G. Minasian and J. Arnold, Chem. Commun., 2008, 4043 RSC.
- A. Hofmann, T. Tröster, T. Kupfer and H. Braunschweig, Chem. Sci., 2019, 10, 3421 RSC.
- W. Haider, D. M. Andrada, I.-A. Bischoff, V. Huch and A. Schäfer, Dalton Trans., 2019, 48, 14953 RSC.
- (a) R. Beck and S. A. Johnson, Organometallics, 2013, 32, 2944 CrossRef CAS; (b) H. Werner, A. Kühn, D. J. Tune, C. Krüger, D. J. Brauer, J. C. Sekutowski and Y.-H. Tsay, Chem. Ber., 1977, 110, 1763 CrossRef CAS; (c) G. Hierlmeier, A. Hinz, R. Wolf and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 431 CrossRef CAS PubMed.
- R. J. Baker, C. Jones, M. Kloth and J. A. Platts, Organometallics, 2004, 23, 4811 CrossRef CAS.
- Z. D. Brown, Z. Zhu, B. D. Ellis and P. P. Power, Main Group Chem., 2010, 9, 111 CAS.
- M. Schorpp and I. Krossing, Chem. – Eur. J., 2020, 26, 14109 CrossRef CAS PubMed.
- M. Schleep, C. Hettich, J. Velázquez Rojas, D. Kratzert, T. Ludwig, K. Lieberth and I. Krossing, Angew. Chem., Int. Ed., 2017, 56, 2880 CrossRef CAS.
- P. Dabringhaus and I. Krossing, Chem. Sci., 2022, 13, 12078 RSC.
- S. Schulz, A. Kuczkowski, D. Schuchmann, U. Flörke and M. Nieger, Organometallics, 2006, 25, 5487 CrossRef CAS.
- J. D. Gorden, C. L. B. Macdonald and A. H. Cowley, Chem. Commun., 2001, 75 RSC.
- C. Schenk, R. Köppe, H. Schnöckel and A. Schnepf, Eur. J. Inorg. Chem., 2011, 3681 CrossRef CAS.
- M. Bochmann and D. M. Dawson, Angew. Chem., Int. Ed. Engl., 1996, 35, 2226 CrossRef CAS.
- M. Huber, A. Kurek, I. Krossing, R. Mülhaupt and H. Schnöckel, Z. Anorg. Allg. Chem., 2009, 635, 1787 CrossRef CAS.
- J. D. Fisher, P. H. M. Budzelaar, P. J. Shapiro, R. J. Staples, G. P. A. Yap and A. L. Rheingold, Organometallics, 1997, 16, 871 CrossRef CAS.
- (a) P. Erdmann and L. Greb, ChemPhysChem, 2021, 22, 935 CrossRef CAS PubMed; (b) Computed on DLPNO-CCSD(T)/aug-cc-pVQZ//PBEh-3c/def2-mSVP level of DFT with CPCM; (c) P. Erdmann, J. Leitner, J. Schwarz and L. Greb, Chem. Phys. Chem., 2020, 21, 987 CrossRef CAS PubMed.
- H. Sitzmann, M. F. Lappert, C. Dohmeier, C. Üffing and H. Schnöckel, J. Organomet. Chem., 1998, 561, 203 CrossRef CAS.
- C. Cui, X. Li, C. Wang, J. Zhang, J. Cheng and X. Zhu, Angew. Chem., Int. Ed., 2006, 45, 2245 CrossRef CAS.
- J. A. Platts, J. Overgaard, C. Jones, B. B. Iversen and A. Stasch, J. Phys. Chem. A, 2011, 115, 194 CrossRef CAS.
- (a) L.-C. Wu, C. Jones, A. Stasch, J. A. Platts and J. Overgaard, Eur. J. Inorg. Chem., 2014, 5536 CrossRef CAS; (b) S. Besnainou, M. Roux and R. Daude, C. R. Hebd. Seances Acad. Sci., 1955, 311 CAS; (c) J. Cioslowski, J. Phys. Chem., 1990, 94, 5496 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2210630–2210633, and 2214274. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc05786g |
| This journal is © The Royal Society of Chemistry 2023 |