
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid-state NMR – a complementary technique for protein framework characterization†
Linda
Cerofolini
 ab,
Kiefer O.
Ramberg
c,
Luis C.
Padilla
ad,
Paweł
Antonik
c,
Enrico
Ravera
abd,
Claudio
Luchinat
abd,
Marco
Fragai
*abd and
Peter B.
Crowley
*c
ab,
Kiefer O.
Ramberg
c,
Luis C.
Padilla
ad,
Paweł
Antonik
c,
Enrico
Ravera
abd,
Claudio
Luchinat
abd,
Marco
Fragai
*abd and
Peter B.
Crowley
*c
aMagnetic Resonance Center (CERM), University of Florence, Via L. Sacconi 6, Sesto Fiorentino 50019, Italy. E-mail: fragai@cerm.unifi.it
bConsorzio Interuniversitario Risonanze Magnetiche di Metalloproteine (CIRMMP), Via L. Sacconi 6, Sesto Fiorentino 50019, Italy
cSchool of Biological and Chemical Sciences, University of Galway, University Road, Galway H91 TK33, Ireland. E-mail: peter.crowley@nuigalway.ie
dDepartment of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 3, Sesto Fiorentino 50019, Italy
First published on 15th December 2022
Abstract
Protein frameworks are an emerging class of biomaterial with medical and technological applications. Frameworks are studied mainly by X-ray diffraction or scattering techniques. Complementary strategies are required. Here, we report solid-state NMR analyses of a microcrystalline protein–macrocycle framework and the rehydrated freeze-dried protein. This methodology may aid the characterization of low-crystallinity frameworks.
Protein crystals, which for decades have enabled advances in biomedical research, are currently in development as reaction vessels and templating devices with potential therapeutic applications.1,2 Such materials are attractive as they are sustainable, biocompatible, programmable (from primary structure to crystalline assembly), and possess highly selective recognition and catalytic activities. Considerable effort is being invested in porous protein crystals, which possess large well-defined pores that permit the uptake and release of substrate/product cargo, enabling, for example, controlled drug delivery. Such “frameworks”, with solvent contents >50%, can be achieved by using naturally porous cage proteins such as ferritin3,4 and viral capsids,5,6 or by engineered protein assembly with inducers that direct the formation of porous structures.7–12 Among the assembly inducing strategies, metal ions/complexes3,9 and organic ligands7,8,10,11 are being used to noncovalently crosslink proteins. For example, Chen and co-workers reported an example of ligand-induced assembly based on rhodamine-sugar conjugates that enabled frameworks of concanavalin A.7 Commercially-available macrocycles such as cucurbit[7]uril and sulfonato-calix[8]arene (sclx8) have been shown to yield frameworks of different proteins including Ralstonia solanacearum lectin (RSL).10–12
To date, the structural characterization of protein frameworks has relied mainly on X-ray diffraction or scattering techniques.4,6,10,11 However, alternative strategies are required to cater for frameworks that have reduced crystallinity or that lose crystallinity upon guest uptake.4,10,12 Spectroscopic methods could be useful for framework characterization, providing additional information on the residues involved in ligand complexation. One such method is solid-state NMR (ssNMR), which thanks to on-going experimental advances yields spectra of microcrystalline, sedimented or freeze-dried proteins comparable in quality to solution-state spectra used for structural studies.13–28 The sample conditions, in particular the protein hydration state (e.g. rehydration of freeze-dried samples), are crucial for achieving good quality spectra.13,14,19
Recently, we showed that solid-state spectral quality is sufficient to ensure resonance assignment, epitope mapping, and the calculation of structural models.22,27 Here, we present a ssNMR characterization of microcrystalline RSL – sclx8 precipitates,11 which form spontaneously at low pH and low salt. Complete solution-state NMR assignments of RSL in the sugar-free (BMRB 25952), D-mannose-(BMRB 25950) or L-fucose-bound (BMRB 25951) forms made this protein an ideal candidate.29 For comparison, a freeze-dried sample of RSL devoid of sclx8 was characterized also. Such comparative analysis of the same protein in two distinct conditions provides information on the protein–macrocycle framework11 that are additive with respect to the crystallographic data. Thus, we demonstrate the potential of ssNMR for the characterization of ligand–induced protein frameworks. We also put forward calixarene-mediated protein precipitation as a means of rapidly generating ordered protein solids suitable for ssNMR analysis.
RSL is a thermostable trimer with a 6-bladed β-propeller fold (C3-symmetry) and micromolar affinity for L-fucose and related sugars.29,30 The ∼40% sequence identity between the N- and C-terminal blades of the RSL monomer results in pseudo C6 symmetry.10 Frameworks of RSL and sclx8 have been characterized by X-ray crystallography and evidence for protein–calixarene binding in solution was obtained by NMR spectroscopy.11 RSL co-crystallizes with sclx8 in at least three space groups depending on the precipitant and pH conditions (Table 1). Crystal forms II and III are frameworks (solvent content >50%) in which the crystal packing is dictated by the calixarene and there are no protein–protein contacts. In contrast, the freeze-dried RSL (devoid of sclx8) requires protein–protein contacts. Crystal form III is particularly interesting as it occurs in the absence of a precipitant such as ammonium sulfate or polyethylene glycol. Millimetre-scale crystals, suitable for X-ray diffraction, grow within hours at pH 4 and 4 °C. Microcrystalline RSL – sclx8 precipitates are obtained within seconds at pH 3.4 and room temperature.11
Samples of microcrystalline RSL – sclx8 (Fig. S1, ESI†) or freeze-dried RSL were prepared with uniformly 13C/15N-labelled protein in 20 mM potassium phosphate, 50 mM NaCl, 5 mM D-fructose, plus 0 or 10 mM sclx8 and pH adjusted to 3.4 (by adding HCl). Microcrystalline (9.0 mg) or freeze-dried (13.4 mg) samples were packed into 3.2 mm zirconia rotors (Bruker). All spectra were acquired on a Bruker Avance III spectrometer operating at 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) MHz (19T, 201.2MHz 13C Larmor frequency) equipped with a Bruker 3.2mm Efree NCH probe-head. Spectra were recorded at 14 or 20 kHz magic-angle spinning (MAS) frequency and the sample temperature was maintained at ∼280K. Complete details on sample preparation and data collection (Table S1, ESI†) are given in the ESI.†
MHz (19T, 201.2MHz 13C Larmor frequency) equipped with a Bruker 3.2mm Efree NCH probe-head. Spectra were recorded at 14 or 20 kHz magic-angle spinning (MAS) frequency and the sample temperature was maintained at ∼280K. Complete details on sample preparation and data collection (Table S1, ESI†) are given in the ESI.†
The ssNMR spectra of microcrystalline RSL – sclx8 exhibit sharp, well-resolved signals and the 2D 15N–13C NCA spectrum was assigned by comparison with the solution state assignments for L-fucose-bound RSL (BMRB 25951).29 The assignment was confirmed by analysing the 3D ssNMR spectra (Fig. S2, ESI†) and is reported in Fig. 1 and Table S2 (ESI†). As noted previously for the solution state assignments, homologous residues in the N- and C-terminal blades of the RSL monomer had similar chemical shifts in the solid-state spectrum (Fig. 1, e.g. D32/D77; G33/G78; G35/G80; W36/W81; G39/G84). All of the 2D 15N–13C NCA resonances, except for the N-terminus (S1-Q4), K34 and the C-terminus (T89-N90) were assigned. The N- and C-termini of RSL are mobile, as evidenced by high temperature factors in X-ray structures, which may explain the lack of cross-peaks.11,30 K34 is a key residue in the sclx8 binding site and the occurrence of multiple conformations may explain the absence of a well-resolved cross-peak. The K34 side chain is well-defined in RSL – sclx8 co-crystal structures at pH ∼ 4 but tends to be disordered at high pH.11 While the microcrystalline sample used for ssNMR was obtained under conditions similar to the P3 crystal form (Table 1), there are differences in the samples that may give rise to multiple K34 conformations and resonance broadening.
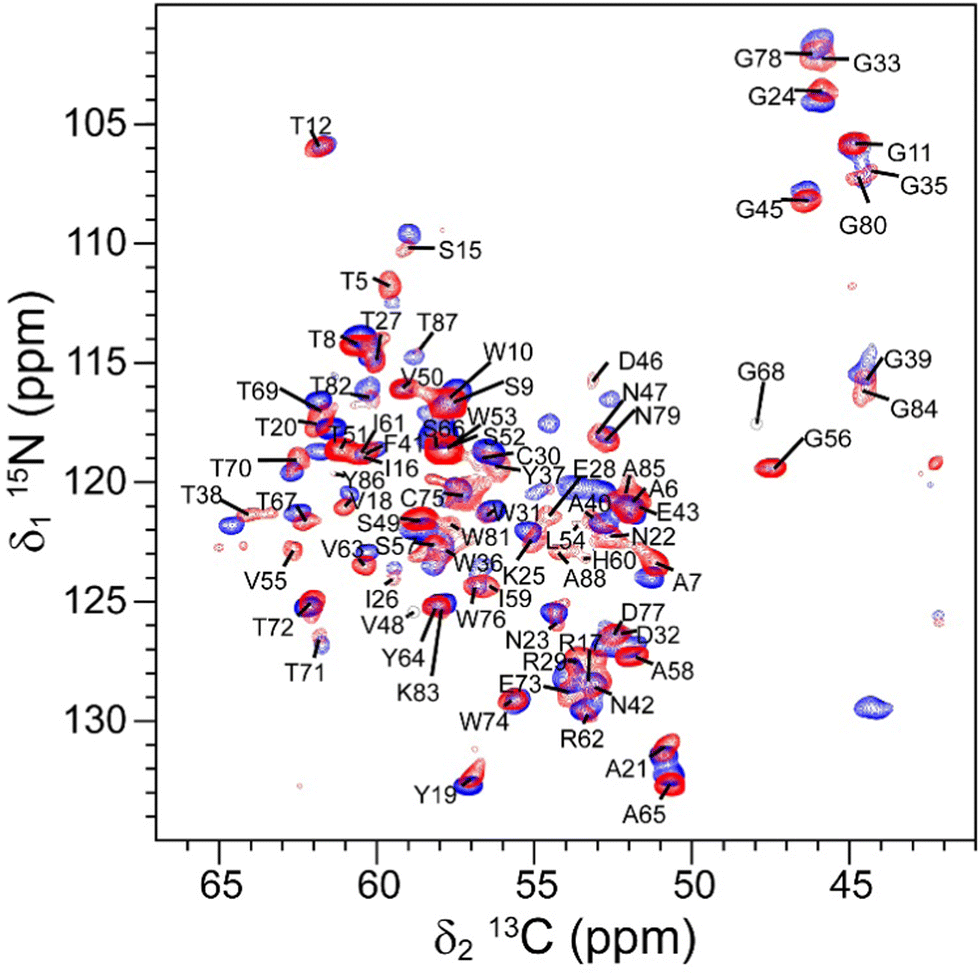 | ||
| Fig. 1 The assigned 2D 15N–13C NCA solid-state spectrum of microcrystalline RSL – sclx8 (red) overlaid on the spectrum of rehydrated freeze-dried RSL (blue). | ||
The RSL side chains were assigned by analysing the 3D 15N–13C NCACX (Fig. S2, ESI†) and 2D 13C–13C dipolar assisted rotational resonance (DARR) solid-state spectra (Table S2, ESI†). A visual inspection of the DARR spectrum (Fig. 2) revealed that ∼40% of the signals were lower intensity than the rest. The lower intensity signals belong to residues V13-R17, I26-E28, C30-A40, D46-V48, H60-R62, G80-N90 spread throughout the protein but involved or close to the sugar and sclx8 binding sites. Structural heterogeneity29 may be responsible for this signal broadening. The occurrence of cross-peaks in the 2D 13C–13C DARR spectra was consistent with preservation of the trimeric structure of RSL, as expected for this highly stable protein. Cross-peaks between T5 and N47, A6 and S49, and S9 and T51, were assigned in the 13C–13C DARR spectrum with a 100 ms mixing time. These residues are located on adjacent β-strands at the inter-monomer interface and line the central channel of the β-propeller fold. Signal overlap prevented the identification of other inter-monomer cross-peaks.
 | ||
| Fig. 2 Overlaid 2D 13C–13C DARR and 15N–13C NCO spectra of microcrystalline RSL – sclx8 (red) and rehydrated freeze-dried RSL (blue). | ||
For comparison, freeze-dried RSL was characterized by ssNMR. The freeze-dried sample was rehydrated until the maximum resolution was achieved in the 1D {1H}13C cross-polarization (CP) spectrum (ωH = 78 kHz; ωC = 50 kHz, Fig. S3, ESI†).13,14,19 This optimisation process required ∼24 hours, in stark contrast to the microcrystalline sample that required no optimisation. A comparison of the 1D {1H}13C CP spectra of microcrystalline RSL – sclx8 and rehydrated freeze-dried RSL revealed slightly broader signals in the latter (Fig. S4, ESI†). 1D projections along the 13C dimension of representative signals showed 5–10% broader peaks in rehydrated freeze-dried RSL compared to microcrystalline RSL – sclx8 (Fig. S5, ESI†). The signal-to-noise ratio was ∼1.1-fold better for the rehydrated freeze-dried sample (230) compared to microcrystalline RSL – sclx8 (203), which can be attributed to the higher amount of protein in the former and to the porous nature (60% solvent) of the latter.19 The 2D 15N–13C NCA spectrum of rehydrated freeze-dried RSL (Fig. 1) was assigned by comparison with the assignments for microcrystalline RSL – sclx8, and confirmed by 3D ssNMR spectral analysis (Table S3, ESI†). Similar to the microcrystalline sample, all of the 2D 15N–13C NCA resonances were assigned, except for the N-terminus (S1-Q4), K34 and the C-terminus (T89-N90). It is intriguing that the K34 resonance was also unassigned in this sample. The other two lysines, K25 and K83, were assigned (Fig. 1). Interestingly, some signals belonging to residues in loops or flexible regions (G33, L54, V55, G56, G68, T69, A88) had a lower intensity.
The assigned 2D 15N–13C spectra for both rehydrated freeze-dried RSL and microcrystalline RSL – sclx8 enabled an analysis of the effects of the calixarene and different packing (protein–macrocycle versus protein–protein) in the two materials. Fig. 3 illustrates the chemical shift perturbations (CSP, Δδ) between the 15N and 13Cα resonances of rehydrated freeze-dried RSL and microcrystalline RSL – sclx8, evaluated according to  . The resonances with perturbations above the cut-off (mean + std. dev. ≥0.16 ppm) were Arg17, Glu28, Trp36, Tyr37, Thr38, Ala40, Phe41, Asp46, Trp81, Gly84, Ala85, Tyr86, and Ala88. Of these 13 residues, all (except Arg17) occur near the protein surface with either or both the N and Cα groups in proximity with solvent. Thus, these groups may be sensing different protein–macrocycle, protein–protein and/or protein–solvent packing that occur in the two materials. Only three of the residues (Arg17, Tyr37 and Asp46) make contact with sclx8 in the X-ray crystal structure (PDB 6Z5Q) that was obtained under similar conditions to the microcrystalline sample used for ssNMR analysis. Interestingly, the largest CSP occurred for Asp46, which undergoes a substantial side chain conformation change to complex sclx8.11 Other important sclx8-binding residues such as Val13, Asn23 and Thr67 did not exhibit significant Δδ effects.
. The resonances with perturbations above the cut-off (mean + std. dev. ≥0.16 ppm) were Arg17, Glu28, Trp36, Tyr37, Thr38, Ala40, Phe41, Asp46, Trp81, Gly84, Ala85, Tyr86, and Ala88. Of these 13 residues, all (except Arg17) occur near the protein surface with either or both the N and Cα groups in proximity with solvent. Thus, these groups may be sensing different protein–macrocycle, protein–protein and/or protein–solvent packing that occur in the two materials. Only three of the residues (Arg17, Tyr37 and Asp46) make contact with sclx8 in the X-ray crystal structure (PDB 6Z5Q) that was obtained under similar conditions to the microcrystalline sample used for ssNMR analysis. Interestingly, the largest CSP occurred for Asp46, which undergoes a substantial side chain conformation change to complex sclx8.11 Other important sclx8-binding residues such as Val13, Asn23 and Thr67 did not exhibit significant Δδ effects.
 | ||
| Fig. 3 Plot of CSP between the 15N and 13Cα resonances of rehydrated freeze-dried RSL and microcrystalline RSL – sclx8. Residues with Δδ ≥ 0.16 ppm are highlighted purple and mapped to the RSL – sclx8 crystal structure (PDB 6Z5Q). The RSL trimer is rendered as a light grey surface. Residues for which data were unavailable (1–4, K34 and 89–90) are dark grey. sclx8 and D-fructose are shown as sticks. | ||
In addition to CSP effects, there were also significant differences in line widths. Signal intensity variations in the 2D 15N–13C NCA spectra of microcrystalline RSL – sclx8 and rehydrated freeze-dried RSL were analysed as follows: the peak intensities in each spectrum were normalized versus the average signal intensity; the normalized peak intensities were subtracted to give a difference plot (Fig. 4) yielding 13 and 18 higher intensity resonances (based on a cut-off of 0.36) in the microcrystalline and rehydrated freeze-dried spectra, respectively. This analysis indicates structural heterogeneity across the protein including surface patches and features involved in calixarene binding. Intensity differences were evident also in the 2D 13C–13C DARR spectrum, in particular, for the carboxylate groups. RSL contains six acidic residues, some of which had sharper signals in the rehydrated freeze-dried RSL compared with microcrystalline RSL – sclx8 (Fig. S6, ESI†). In particular, Asp32 and Asp46 that bind sclx8 were significantly broadened in the microcrystalline sample. CSP and line broadening were evident also for the carboxylate resonances of homologous Glu28 and Glu73, which form hydrogen bonds to D-fructose in the sugar binding sites. Glu28 (near Lys34) might sense calixarene binding via the sugar as this site makes contact with sclx8, while the site containing Glu73 does not bind the calixarene. Previous comparative ssNMR analyses of protein dynamics in crystalline and rehydrated freeze-dried samples indicated no significant differences in backbone or side chain mobility.16
 | ||
| Fig. 4 Plot of the normalised intensity changes between the 15N–13Cα resonances of rehydrated freeze-dried RSL and microcrystalline RSL – sclx8. Higher intensity signals are indicated by positive bars for rehydrated freeze-dried RSL and by negative bars for microcrystalline RSL – sclx8. Residues with significant difference in signal intensity (highlighted blue and red, respectively) were mapped on to the RSL – sclx8 crystal structure (PDB 6Z5Q). | ||
In summary, high-quality ssNMR spectra were acquired for both microcrystalline RSL – sclx8 and rehydrated freeze-dried RSL. The microcrystalline sample exhibits slightly better resolved spectra, as expected for a crystalline sample, but fewer cross-peaks. This seemingly contradictory finding is explained by a larger structural heterogeneity arising from residue masking by sclx8 that resulted in signal broadening, in some cases beyond detection. The quality of the multidimensional spectra was sufficient to allow near complete resonance assignment and the identification of restraints useful for structural calculation. Therefore, calixarene-mediated protein precipitation may be a straightforward and rapid route to high-quality ssNMR samples. Despite the differences in intensity and the CSP affecting some signals, the spectra of microcrystalline RSL – sclx8 and rehydrated freeze-dried RSL are largely superimposable (Fig. 1 and 2). A good match occurred also between the cross-peaks in the solution-state29 and solid-state spectra. Furthermore, the data show the versatility of ssNMR and its virtual independence from a highly ordered distribution of the proteins in the material as required for X-ray crystallography. This feature is particularly relevant for the characterization of new protein-based materials where the presence of additional guest molecules can lead to decreased crystallinity, thus preventing X-ray characterization.4 This advance is relevant to ferritin, which has been characterized by ssNMR17 and is prevalent in protein framework studies.1,3
This work was supported by Regione Toscana (CERM-TT and BioEnable), the Italian Ministero dell’Istruzione, dell’Università e della Ricerca (PRIN 2017A2KEPL), the Recombinant Proteins JOYNLAB laboratory, the project FISR2021_SYLCOV, University of Galway, NUI Travelling Studentship, Teagasc Walsh fellowship and Science Foundation Ireland (grant 13/CDA/2168). We acknowledge also Instruct-ERIC, specifically CERM/CIRMMP, H2020 INFRAIA iNEXT-Discovery (contract 871037), H2020 PANACEA (grant 101008500), EOSC-Life (contract 824087) and H2020-MSCA-ITN-2020 (contract 956758).
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Kojima, S. Abe and T. Ueno, Biomater. Sci., 2022, 10, 354–367 RSC.
- R. Fernández-Penas, C. Verdugo-Escamilla, S. Martínez-Rodríguez and J. A. Gavira, Cryst. Growth Des., 2021, 21, 1698–1707 CrossRef PubMed.
- J. B. Bailey, L. Zhang, J. A. Chiong, S. Ahn and F. A. Tezcan, J. Am. Chem. Soc., 2017, 139, 8160–8166 CrossRef CAS PubMed.
- K. Han, Y. Na, L. Zhang and F. A. Tezcan, J. Am. Chem. Soc., 2022, 144, 10139–10144 CrossRef CAS PubMed.
- M. Uchida, K. McCoy, M. Fukuto, L. Yang, H. Yoshimura, H. M. Miettinen, B. LaFrance, D. P. Patterson, B. Schwarz, J. A. Karty, P. E. Prevelige, B. Lee and T. Douglas, ACS Nano, 2018, 12, 942–953 CrossRef CAS PubMed.
- M. Uchida, N. E. Brunk, N. D. Hewagama, B. Lee, P. E. Prevelige, V. Jadhao and T. Douglas, ACS Nano, 2022, 16, 7662–7673 CrossRef CAS PubMed.
- F. Sakai, G. Yang, M. S. Weiss, Y. Liu, G. Chen and M. Jiang, Nat. Commun., 2014, 5, 4634 CrossRef CAS PubMed.
- B. E. Partridge, P. H. Winegar, Z. Han and C. A. Mirkin, J. Am. Chem. Soc., 2021, 143, 8925–8934 CrossRef CAS PubMed.
- L. Vandebroek, H. Noguchi, K. Kamata, J. R. H. Tame, L. van Meervelt, T. N. Parac-Vogt and A. R. D. Voet, Chem. Commun., 2020, 56, 11601–11604 RSC.
- F. Guagnini, S. Engilberge, K. O. Ramberg, J. Pérez and P. B. Crowley, Chem. Commun., 2020, 56, 360–363 RSC.
- K. O. Ramberg, S. Engilberge, T. Skorek and P. B. Crowley, J. Am. Chem. Soc., 2021, 143, 1896–1907 CrossRef CAS PubMed.
- K. O. Ramberg, F. Guagnini, S. Engilberge, M. A. Wrońska, M. L. Rennie, J. Pérez and P. B. Crowley, Chem. – Eur. J., 2021, 27, 14619–14627 CrossRef CAS PubMed.
- S. D. Kennedy and R. G. Bryant, Biopolymers, 1990, 29, 1801–1806 CrossRef CAS PubMed.
- J. Pauli, B. van Rossum, H. Förster, H. J. de Groot and H. Oschkinat, J. Magn. Reson., 2000, 143, 411–416 CrossRef CAS PubMed.
- F. Castellani, B. van Rossum, A. Diehl, M. Schubert, K. Rehbein and H. Oschkinat, Nature, 2002, 420, 98–102 CrossRef CAS PubMed.
- A. Krushelnitsky, Y. Gogolev, R. Golbik, F. Dahlquist and D. Reichert, Biochim. Biophys. Acta., 2006, 1764, 1639–1645 CrossRef CAS PubMed.
- I. Bertini, C. Luchinat, G. Parigi, E. Ravera, B. Reif and P. Turano, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10396–10399 CrossRef CAS PubMed.
- M. Chan-Huot, L. Duma, J.-B. Charbonnier, J.-E. Herbert-Pucheta, L. Assairi, Y. Blouquit, D. Abergel and G. Bodenhausen, Cryst. Growth Des., 2012, 12, 6199–6207 CrossRef CAS.
- M. Fragai, C. Luchinat, G. Parigi and E. Ravera, J. Biomol. NMR, 2013, 57, 155–166 CrossRef CAS PubMed.
- M. Fragai, C. Luchinat, T. Martelli, E. Ravera, I. Sagi, I. Solomonov and Y. Udi, Chem. Commun., 2014, 50, 421–423 RSC.
- E. Ravera, S. Ciambellotti, L. Cerofolini, T. Martelli, T. Kozyreva, C. Bernacchioni, S. Giuntini, M. Fragai, P. Turano and C. Luchinat, Angew. Chem., Int. Ed., 2016, 55, 2446–2449 CrossRef CAS PubMed.
- L. Cerofolini, S. Giuntini, A. Carlon, E. Ravera, V. Calderone, M. Fragai, G. Parigi and C. Luchinat, Chem. – Eur. J., 2019, 25, 1984–1991 CrossRef CAS PubMed.
- J. Mao, V. Aladin, X. Jin, A. J. Leeder, L. J. Brown, R. C. D. Brown, X. He, B. Corzilius and C. Glaubitz, J. Am. Chem. Soc., 2019, 141, 19888–19901 CrossRef CAS PubMed.
- B. R. Sahoo, S. J. Cox and A. Ramamoorthy, Chem. Commun., 2020, 56, 4627–4639 RSC.
- K. Jaudzems, A. Kirsteina, T. Schubeis, G. Casano, O. Ouari, J. Bogans, A. Kazaks, K. Tars, A. Lesage and G. Pintacuda, Angew. Chem., Int. Ed., 2021, 60, 12847–12851 CrossRef CAS PubMed.
- S. Sarkar, B. Runge, R. W. Russell, K. T. Movellan, D. Calero, S. Zeinalilathori, C. M. Quinn, M. Lu, G. Calero, A. M. Gronenborn and T. Polenova, J. Am. Chem. Soc., 2022, 144, 10543–10555 CrossRef CAS PubMed.
- D. Rizzo, L. Cerofolini, S. Giuntini, L. Iozzino, C. Pergola, F. Sacco, A. Palmese, E. Ravera, C. Luchinat, F. Baroni and M. Fragai, J. Am. Chem. Soc., 2022, 144, 10006–10016 CrossRef CAS PubMed.
- S. Ahlawat, K. R. Mote, N. A. Lakomek and V. Agarwal, Chem. Rev., 2022, 122, 9643–9737 CrossRef CAS PubMed.
- P. M. Antonik, A. N. Volkov, U. N. Broder, D. Lo Re, N. A. J. van Nuland and P. B. Crowley, Biochemistry, 2016, 55, 1195–1203 CrossRef CAS PubMed.
- N. Kostlánová, E. P. Mitchell, H. Lortat-Jacob, S. Oscarson, M. Lahmann, N. Gilboa-Garber, G. Chambat, M. Wimmerová and A. Imberty, J. Biol. Chem., 2005, 280, 27839–27849 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Sample preparation, NMR data acquisition, spectra and tables of assignments. See DOI: https://doi.org/10.1039/d2cc05725e |
| This journal is © The Royal Society of Chemistry 2023 |