
Inherently chiral belt-shaped conjugated macrocycles with strong fluorescence and circularly polarized luminescence†
Xu-Sheng
Du
ab,
Xiao-Ni
Han
a,
Ying
Han
*a and
Chuan-Feng
Chen
 *ab
*ab
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: cchen@iccas.ac.cn; hanying463@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 28th November 2022
Abstract
A pair of inherently chiral belt-shaped conjugated macrocycles were conveniently synthesized starting from 2,7-fluoren[3]arene triflate, and they not only exhibited green fluorescence, but also showed circularly polarized luminescence with a |glum| of 2.0 × 10−3.
Belt-shaped conjugated macrocycles (BCMs) have attracted increasing interest for their aesthetically appealing molecular architectures, specific physical properties and wide potential applications in supramolecular chemistry and materials science.1 Although some BCMs have been designed and constructed in the last few years,2–6 developing new strategies for the synthesis of BCMs and then exploring their properties and applications are still some of the most important topics in this research area.1b Recently, Wang et al. reported a new kind of BCM starting from resorcin[4]arenes.7 Soon after, an alternative methylene bridged[6]cycloparaphenylene from pillar[6]arene was reported by Itami's group.8 In 2021, we developed a highly efficient method for the synthesis of [6]cycloparaphenylene and its derivatives starting from fluoren[3]arenes.9a Starting from the calix[3]carbazole, a nitrogen-doped BCM with strong green fluorescence properties was also obtained.9b These recent successful examples reveal that macrocyclic arenes10 can be excellent precursors for the design and synthesis of BCMs.
Recently, chiral macrocycles with circularly polarized luminescence (CPL) properties have been of particular interest owing to their potential applications in the design and construction of chiral supramolecular materials11–13 and chiral optoelectronic materials.14 However, such macrocycles, especially, BCMs with CPL properties are very limited.15 Constructing new chiral BCMs with strong fluorescence and CPL properties is still challenging, and it is also becoming one of the key topics in such research areas.
Previously, we reported a series of pillar-shaped BCMs from fluoren[3]arene but found that they showed no fluorescence because of the symmetry forbidden HOMO–LUMO transitions (Scheme 1).9a Herein, we report a pair of inherently chiral belt-shaped conjugated macrocycles, which are easily synthesized starting from 2,7-fluoren[3]arene triflate (Scheme 1). Moreover, it was also found that these enantiomeric belt-shaped conjugated macrocycles not only exhibit strong green fluorescence, but also show circularly polarized luminescence properties.
 | ||
| Scheme 1 Design of BCMs starting from 2,7-fluoren[3]arene triflate. | ||
The synthesis of the enantiomeric P- and M-BCM is depicted in Scheme 2. Starting from fluoren[3]arene triflate 1,9aBCM 3 was conveniently obtained in good yields by the Wang's synthetic method.7b Subsequently, the post-modification of 3 in the presence of the propionyl chloride and AlCl3 at 0 °C was carried out, which gave the mono-propionyl substituted BCM 4 in 56% yield. Finally, the racemic 4 was subjected to chirality resolution via HPLC, and the enantiopure isomers P-4 and M-4 were obtained, respectively (Fig. S19, ESI†). The chemical structures of enantiomeric BCMs and their precursors were confirmed using the 1H NMR, 13C NMR and MALDI-TOF HRMS spectra (Fig. S14, ESI†). The absolute configurations of the enantiomers were determined using the simulated ECD spectra (Fig. S36 and S37, ESI†).
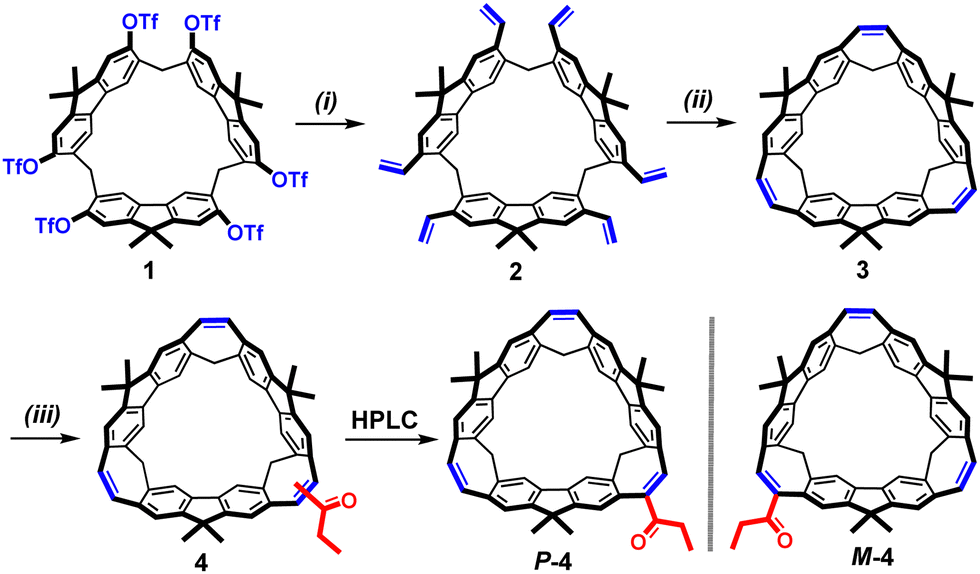 | ||
Scheme 2 Synthesis of 3 and 4. Conditions for (i) PdCl2, PPh3, BHT, LiCl, DMF, 80 °C, n-Bu3SnCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, 12 h, 85%; (ii) Grubbs-II catalyst, DCM, reflux, 12 h, 70%; (iii) CH3CH2COCl, AlCl3, DCM, 0 °C, 56%. CH2, 12 h, 85%; (ii) Grubbs-II catalyst, DCM, reflux, 12 h, 70%; (iii) CH3CH2COCl, AlCl3, DCM, 0 °C, 56%. | ||
By a slow diffusion of methanol into CH2Cl2 solution of 2 at room temperature, single crystals of 2 were obtained. The X-ray crystal structure showed three of the fluorene subunits in 2 positioned at the opposite side to form a saddle-shaped conformation (Fig. S20, ESI†). The 1H NMR spectrum of BCM 3 indicated that the macrocycle had C3 symmetry (Fig. 1a). Three kinds of aromatic protons could be found with chemical shifts at 7.57, 7.08 and 7.07 ppm, respectively. The methylene protons split into two doublets, and the methyl groups inside and outside of the bowl-shaped cavity were distinguishable in the 1H-13C HSQC spectrum (Fig. S9, ESI†). Surprisingly, it was found that the methylene proton showed an NOE signal with the lower rim aromatic proton Ha located in down field, which was quite different from the previous work.9a To elucidate the difference and give a thorough assignment of the 1H NMR spectrum, the optimization of the structure of 3 was carried out by using the B3LYP/6-31G(d) level of theory (Fig. 1a). Obviously, the shortest space distances of Hb and CH3(i) and CH3(o) were measured to be 2.437 Å and 2.867 Å, respectively, which correspond to the NOE signals (Fig. S9, ESI†). The distance of Ha and CH2(i) was 2.334 Å, but the distance of Ha and CH2(o) was 3.561 Å, which further confirmed that the observed cross-signal came from Ha and CH2(i). The reason that CH2(o) located in the up-field could be attributed to the shielding effect from the vinyl plane (Fig. S43, ESI†). In contrast, CH2(i) is positioned almost in the parallel direction to Ha, which led to no influence from the shielding effect of the bowl-shaped cavity.
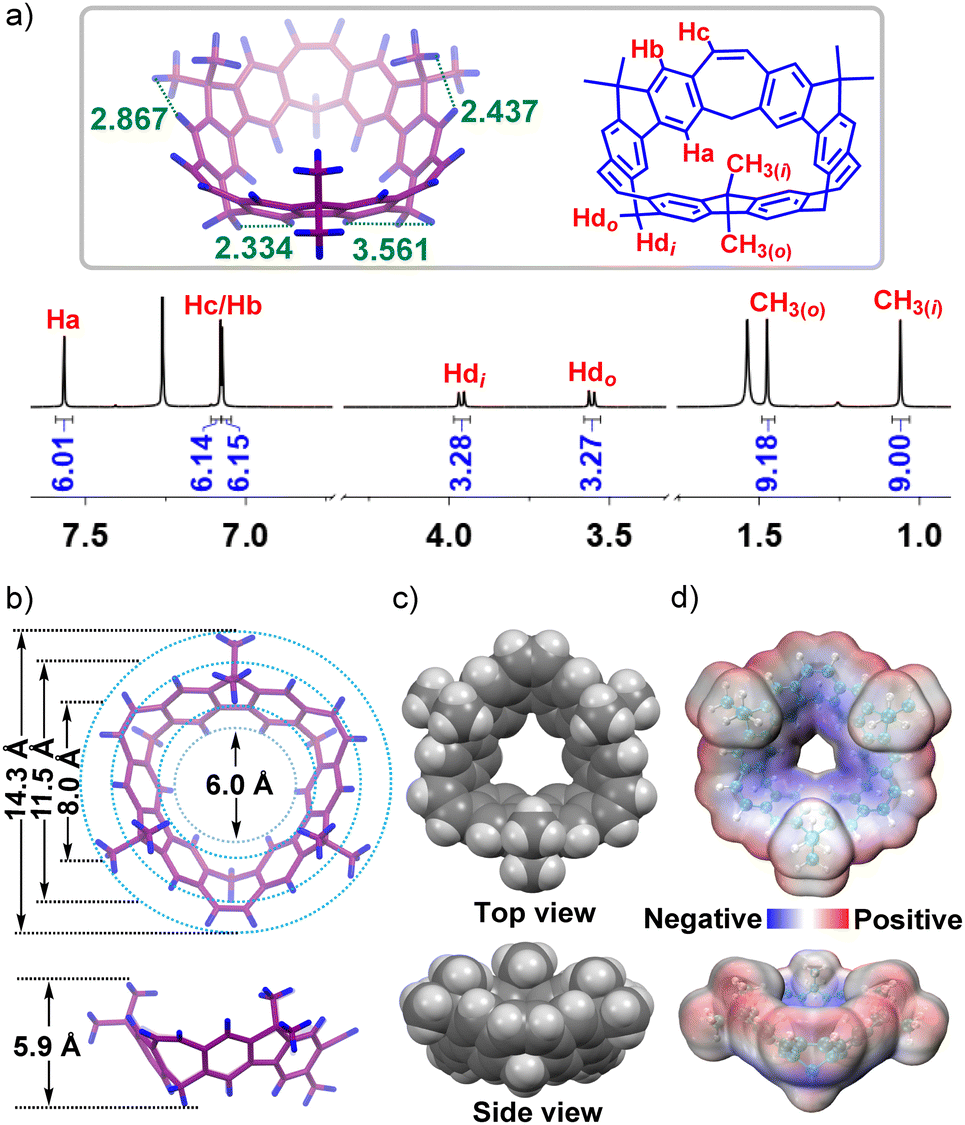 | ||
| Fig. 1 (a) 1H NMR spectrum (700 MHz, CDCl3, 298 K) of 3; (b) and (c) crystal structure of 3 in top and side views; (d) calculated potential profile of 3 in top and side views. | ||
By a slow diffusion of methanol into the CH2Cl2 solution of 3, single crystals were obtained (Fig. 1b). The crystal structure of 3 showed a bowl-shaped structure with an approximately maximum diameter of 14.3 Å, and the upper and lower rim of the bowl had an approximate diameter of 11.5 Å and 6.0 Å, respectively. Meanwhile, the cavity depth was measured to be about 5.9 Å. Besides, owing to the existence of inner side methyl groups, the efficient diameter of 3 was measured to be 8.0 Å (Fig. 1c). The fluorene plane had a curvature of 24° (Fig. S24, ESI†). Moreover, the potential profile was determined to evaluate the cavity potential distribution, which showed the electron-rich properties of 3 close to its lower rim of the bowl (Fig. 1d). The intermolecular C–H⋯π interactions between CH2Cl2 and 3 were further observed (Fig. S25, ESI†).
Compared to 3, the 1H NMR spectrum of BCM 4 showed an extraordinary complexity (Fig. S10 and S11, ESI†). To clearly assign the 1H NMR signals, the 2D NMR spectra of M-4 were obtained. First, combining the 1H–1H COSY spectrum (Fig. S15, ESI†) with the 1H–13C HSQC spectrum (Fig. S17, ESI†), the hydrogens of the ethyl group and all of –CH2 belonging to the seven-member ring, with chemical shift values from 3.55–4.05 ppm, were accurately assigned (Fig. 2d and e). Coupled with the optimized structure (Fig. 2a), the multiple peaks from 3.1–2.8 ppm were assigned to the protons Hb and Hb′, which were located at two sides of the propionyl group (Fig. 2d). According to the 2D NOESY spectrum (Fig. S16, ESI†), the observed NOE signals of aromatic hydrogen with a chemical shift value at 7.92 ppm and –CH2 belonging to the propionyl group allowed assignment of the Hc. The NOE signals between the aromatic hydrogens and –C(CH3)2 could allow the upper rim aromatic hydrogens, Hd–Hi, to be determined with a chemical shift value from 7.25 to 7.05 ppm (Fig. 2c). The NOE signals between aromatic hydrogens and –CH2 belonging to the seven-member ring allowed the lower rim aromatic hydrogens, Hj–Ho, to be determined with chemical shift values from 7.70 to 7.40 ppm (Fig. 2c). The other two groups of ethylene hydrogens with chemical shift values at 7.05 and 5.62 in ppm were assigned to Hs & Hp and Hr & Hq, which was further confirmed using the 1H–13C HMBC spectrum (Fig. S18, ESI†). The NOE signals between the lower rim aromatic protons and –CH2 belonging to the seven-member ring further allowed the protons Ht–Hv to be assigned (Fig. 2d). And the NOE signals between the upper rim aromatic hydrogens and –C(CH3)2 could allow the protons Hw–Hy to be assigned (Fig. 2e).
 | ||
| Fig. 2 (a) Optimized structure and (b) chemical structure of M-4; (c) expanded aromatic part, (d) and (e) expanded aliphatic parts of the 1H NMR spectrum (700 MHz, CD2Cl2, 298 K) of M-4. | ||
The photophysical properties of 3 and 4 were measured in CHCl3. As shown in Fig. 3a, BCM 3 displayed three major sharp absorption peaks at 245, 280 and 345 nm, respectively, and two broad absorption shoulder peaks at 325 and 375 nm. The energy gap calculated from the absorption spectrum was approximately 3.01 eV.16 The fluorescence spectrum of 3 showed a maximum emission peak at around 445 nm, and its fluorescence quantum yield was 0.58. In the case of 4, the UV-vis spectrum showed two sharp absorption peaks at 245 and 348 nm, and three broad absorption shoulder peaks at 280, 325 and 395 nm, respectively. The fluorescence spectrum of 4 contains one broad band with a maximum at ca. 500 nm, and its quantum yield was determined to be 0.18. The chiroptical properties of the enantiomeric BCMs were also studied. As shown in Fig. 3b, the CD spectra of the enantiomers showed a mirror-imaged Cotton effect with an average |gabs| of 3.1 × 10−3. Moreover, the enantiomers also exhibited a moderate CPL with the absolute |glum| of 2.0 × 10−3.
 | ||
| Fig. 3 (a) UV-vis and fluorescence spectra of 3 and 4, [3] = [4] = 1.0 × 10−5 M; (b) CD and CPL spectra of enantiomeric 4; (c) calculated frontier molecular orbital profiles, energy diagram, and major electronic transitions of P-4. | ||
To achieve an in-depth understanding of the absorption spectra of 3 and 4, time-dependent density functional theory (TD-DFT) calculations were also carried out. Initially, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of 3 and 4 were analysed comparatively. For BCM 3, the HOMO (H) and LUMO (L) showed that the frontier molecular orbitals were all delocalized over the conjugated phenylene–ethylene backbone (Fig. S39, ESI†). The HOMO–LUMO energy gap of 3 was calculated to be 3.38 eV. And the HOMO−1 and HOMO, LUMO and LUMO+1 were degenerate owing to the high symmetry of 3. In comparison, the frontier molecular orbitals of (P)-4 were delocalized over the conjugated phenylene–ethylene backbone and carbonyl group (Fig. 3c and Fig. S40, ESI†). The HOMO–LUMO energy gap of 4 was theoretically 2.72 eV, which was close to that of pillar-shaped BCM8,9a and even C60,17 implying a promising application of 4 in optoelectronic materials. Moreover, the absorption spectra of 3 and 4 were also simulated (Fig. S32 and S34, ESI†).18 For 3, the observed absorption bands at around 345 and 375 nm could be mainly assigned to electronic transitions from H−1 → L and H−1 → L+1, and from H → L and H → L+1. In the case of 4, four absorption bands were observed (Fig. 3c), in which the band I could be assigned to electronic transitions from H → L and H–1 → L, and the band II to IV were assigned to H → L + 1, H–1 → L + 1 and H → L + 2, respectively. Furthermore, the major electronic transition analysis of the main absorption bands for 3 and 4 allowed us to assign the observed maximum absorption band and the strongest absorption band (Fig. S33 and S35, ESI†). For 3, the maximum absorption of 375 nm was assigned to electronic transitions from S0 → S2, where the oscillator strength (f) was 0.0016, and the strongest absorption of 345 nm was assigned to S0 → S3 and S0 → S4, where the f value was around 1.0360. For 4, the maximum absorption of 395 nm (band I) was assigned to electronic transitions from S0 → S1 and S0 → S2, with an f value of 0.1833; and the strongest absorption of 348 nm (band II) was assigned to S0 → S3, with an f value 0.9215; the absorption band III at 325 nm was assigned to S0 → S5 (f = 0.5207) and the absorption band IV at 280 nm was assigned to S0 → S8 (f = 0.3897). Finally, the strong fluorescence of 3 and 4 was also theoretically analysed (Fig. S41 and S42, ESI†), in which the f values at 414 nm for 3 and at 488 nm for 4 were 0.6517 and 0.1068 (single electron calculation in a vacuum), respectively, confirming the efficient S1 → S0 transition.
In summary, we have conveniently synthesized a belt-shaped conjugated macrocycle with a highly symmetrical rigid structure and strong blue fluorescence starting from 2,7-fluoren[3]arene triflate. The BCM then transferred to a pair of inherently chiral belt-shaped conjugated macrocycles by Friedel–Crafts acylation and followed by chiral resolution via HPLC on a chiral column. Interestingly, it was found that the enantiomeric BCMs not only showed strong green fluorescence and narrow energy gaps, but also displayed CPL properties with a |glum| of 0.002. This work provides an efficient approach for the design and construction of inherently chiral conjugated macrocycles with strong fluorescence and CPL properties, which will be beneficial for their wide applications in supramolecular chemistry and materials science.
We thank the National Natural Science Foundation of China (22031010 and 91856117) and the Youth Innovation Promotion Association CAS (2021035) for financial support.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- (a) X. F. Lu and J. S. Wu, Chemistry, 2017, 2, 619–620 CrossRef CAS; (b) T. H. Shi and M. X. Wang, CCS Chem., 2020, 2, 916–931 Search PubMed; (c) Q. H. Guo, Y. Y. Qiu, M. X. Wang and J. F. Stoddart, Nat. Chem., 2021, 13, 402–419 CrossRef CAS PubMed; (d) K. Y. Cheung, Y. Segawa and K. Itami, Chem. – Eur. J., 2020, 26, 14791–14801 CrossRef CAS PubMed; (e) H. Chen and Q. Miao, J. Phys. Org. Chem., 2020, e4145 CAS.
- (a) Z. Sun, P. Sarkar, T. Suenaga, S. Sato and H. Isobe, Angew. Chem., Int. Ed., 2015, 54, 12800–12804 CrossRef CAS; (b) Z. Sun, T. Suenaga, P. Sarkar, S. Sato, M. Kotani and H. Isobe, Proc. Natl. Acad. Sci. USA, 2016, 113, 8109–8114 CrossRef CAS PubMed.
- (a) G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, Science, 2017, 356, 172–175 CrossRef CAS PubMed; (b) K. Y. Cheung, K. Watanabe, Y. Segawa and K. Itami, Nat. Chem., 2021, 13, 255–259 CrossRef CAS PubMed.
- (a) K. Y. Cheung, S. J. Gui, C. F. Deng, H. F. Liang, Z. M. Xia, Z. Liu, L. F. Chi and Q. Miao, Chemistry, 2019, 5, 838–847 CrossRef CAS; (b) Z. Xia, S. H. Pun, H. Chen and Q. Miao, Angew. Chem., Int. Ed., 2021, 60, 10311–10318 CrossRef CAS PubMed.
- Y. Han, S. Q. Dong, J. W. Shao, W. Fan and C. Y. Chi, Angew. Chem., Int. Ed., 2021, 60, 2658–2662 CrossRef CAS PubMed.
- W. Fan, T. Matsuno, Y. Han, X. H. Wang, Q. F. Zhou, H. Isobe and J. S. Wu, J. Am. Chem. Soc., 2021, 143, 15924–15929 CrossRef CAS PubMed.
- (a) T. H. Shi, Q. H. Guo, S. Tong and M. X. Wang, J. Am. Chem. Soc., 2020, 142, 4576–4580 CrossRef CAS PubMed; (b) Q. Zhang, Y. Zhang, S. Tong and M. X. Wang, J. Am. Chem. Soc., 2020, 142, 1196–1199 CrossRef CAS PubMed.
- Y. M. Li, Y. Segawa, A. Yagi and K. Itami, J. Am. Chem. Soc., 2020, 142, 12850–12856 CrossRef CAS PubMed.
- (a) X.-S. Du, D.-W. Zhang, Y. Guo, J. Li, Y. Han and C.-F. Chen, Angew. Chem., Int. Ed., 2021, 60, 13021–13028 CrossRef CAS PubMed; (b) F. Zhang, X.-S. Du, D.-W. Zhang, Y.-F. Wang, H.-Y. Lu and C.-F. Chen, Angew. Chem., Int. Ed., 2021, 60, 15291–15295 CrossRef CAS PubMed.
- (a) C.-F. Chen and Y. Han, Acc. Chem. Res., 2018, 51, 2093–2106 CrossRef CAS PubMed; (b) J. Li, Y. Han and C.-F. Chen, Chin. J. Org. Chem., 2020, 40, 3714–3737 CrossRef CAS; (c) Z. Y. Zhang and C. J. Li, Acc. Chem. Res., 2022, 55, 916–929 CrossRef CAS PubMed.
- (a) J. Li, H.-Y. Zhou, Y. Han and C.-F. Chen, Angew. Chem., Int. Ed., 2021, 60, 21927–21933 CrossRef CAS PubMed; (b) X.-N. Han, Y. Han and C.-F. Chen, J. Am. Chem. Soc., 2020, 142, 8262–8269 CrossRef CAS PubMed; (c) X.-N. Han, Y. Han and C.-F. Chen, Nat. Commun., 2021, 12, 6378 CrossRef CAS PubMed; (d) X.-N. Han, Q.-S. Zong, Y. Han and C.-F. Chen, CCS Chem., 2022, 4, 318–330 CrossRef CAS.
- (a) Z. Chen, Q. Wang, X. Wu, Z. Li and Y. B. Jiang, Chem. Soc. Rev., 2015, 44, 4249–4263 RSC; (b) J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 12228–12239 CrossRef CAS PubMed.
- (a) X. Huang, X. Wang, M. Quan, H. Yao, H. Ke and W. Jiang, Angew. Chem., Int. Ed., 2021, 60, 1929–1935 CrossRef CAS PubMed; (b) A. H. G. David, R. Casares, J. M. Cuerva, A. G. Campaña and V. Blanco, J. Am. Chem. Soc., 2019, 141, 18064–18074 CrossRef CAS PubMed.
- (a) D.-W. Zhang, M. Li and C.-F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC; (b) J. Yao, W. Wu, W. Liang, Y. Feng, D. Zhou, J. J. Chruma, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, Angew. Chem., Int. Ed., 2017, 56, 6869–6873 CrossRef CAS PubMed; (c) Y.-F. Wang, H.-Y. Lu, Y.-F. Shen, M. Li and C.-F. Chen, Chem. Commun., 2019, 55, 9559–9562 RSC; (d) W.-L. Zhao, Y.-F. Wang, S.-P. Wan, H.-Y. Lu, M. Li and C.-F. Chen, CCS Chem., 2022, 4, 3540–3548 CrossRef.
- (a) J. Nogami, Y. Tanaka, H. Sugiyama, H. Uekusa, A. Muranaka, M. Uchiyama and K. Tanaka, J. Am. Chem. Soc., 2020, 142, 9834–9842 CAS; (b) J. Nogami, Y. Nagashima, H. Sugiyama, K. Miyamoto, Y. Tanaka, H. Uekusa, A. Muranaka, M. Uchiyama and K. Tanaka, Angew. Chem., Int. Ed., 2022, 61, e202200800 CAS; (c) J. Wang, G. Zhuang, M. Chen, D. Lu, Z. Li, Q. Huang, H. Jia, S. Cui, X. Shao, S. Yang and P. Du, Angew. Chem., Int. Ed., 2020, 59, 1619–1626 CrossRef CAS PubMed.
- J. C. S. Costa, R. J. S. Taveira, C. F. R. A. C. Lima, A. Mendes and L. M. N. B. F. Santos, Opt. Mater., 2016, 58, 51–60 CrossRef CAS.
- M. K. Shukla and J. Leszczynski, Chem. Phys. Lett., 2006, 428, 317–320 CrossRef CAS.
- (a) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed; (b) Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 461–467 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, NMR spectra, HRMS spectra, UV-vis spectra, fluorescence spectra and DFT calculation details. CCDC 2192655 and 2192656. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc05504j |
| This journal is © The Royal Society of Chemistry 2023 |