
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnique Li deposition behavior in Li3PS4 solid electrolyte observed via operando X-ray computed tomography†
Jaehee
Park
 a,
Toshiki
Watanabe
*a,
Kentaro
Yamamoto
ab,
Tomoki
Uchiyama
a,
Tsuyoshi
Takami
a,
Atsushi
Sakuda
c,
Akitoshi
Hayashi
c,
Masahiro
Tatsumisago
c and
Yoshiharu
Uchimoto
a
a,
Toshiki
Watanabe
*a,
Kentaro
Yamamoto
ab,
Tomoki
Uchiyama
a,
Tsuyoshi
Takami
a,
Atsushi
Sakuda
c,
Akitoshi
Hayashi
c,
Masahiro
Tatsumisago
c and
Yoshiharu
Uchimoto
a
aGraduate School of Human and Environmental Studies, Kyoto University, Kyoto, Kyoto 606-8501, Japan
bFaculty of Engineering, Nara Women's University, Nara, Nara 630-8506, Japan
cDepartment of Applied Chemistry, Graduate School of Engineering, Osaka Metropolitan University, Sakai, Osaka 599-8531, Japan
First published on 22nd May 2023
Abstract
The problem of lithium dendrites must be addressed for practical lithium metal all-solid-state batteries. Herein, three-dimensional morphological changes within Li3PS4 electrolyte away from the anode were observed using operando X-ray computed tomography. We revealed that the electronic conduction of decomposition and the electrolyte/void interface cause the lithium deposition within the Li3PS4.
The application of Li-ion secondary batteries has expanded from small-scale electronic devices to middle- to large-scale devices for electric vehicles, and further improvement of energy density is required. Li metal is an attractive anode candidate for this goal owing to its high theoretical capacity (3860 mA h g−1) and low potential (−3.04 V vs. standard hydrogen electrode).1 However, the use of Li metal anodes with traditional liquid or polymer electrolytes has been unsuccessful because of difficulties in suppressing internal short circuits caused by Li dendrite formation.2–4 This problem leads to degradation and safety concerns during charging/discharging cycles. Compared to liquid or polymer electrolytes, solid electrolytes have the advantage of being able to prevent the Li dendrites owing to their high mechanical strength and high Li transference number.5 Among solid electrolytes, sulfide solid electrolytes have received particular attention because of their formability and ionic conductivities that are comparable or superior to those of liquid electrolytes.6 However, the use of sulfide solid electrolytes has not suppressed Li dendrites to the extent required for practical application.7,8
Non-uniform contact at the interface between Li metal and sulfide solid electrolyte, leading to non-uniform current at the interface, has been reported to be the main reason for Li dendrite formation.9–14 Although various interfacial modifications have been developed to reduce the non-uniform interfacial contact, Li dendrites still have not been suppressed completely.5,15–21 Several studies have reported that Li dendrite formation occurs not from the Li/sulfide solid electrolyte interface, but in the sulfide solid electrolyte, isolated from the interface, suggesting that improving only the heterogeneous structure at the interface is insufficient for the suppression of Li dendrite formation.7,22,23 The formation of Li dendrites within the sulfide solid electrolyte layer has been attributed to electron conduction in the sulfide solid electrolyte and at the sulfide solid electrolyte/void interface.24 However, fundamental information on the mechanism of Li dendrite formation in a sulfide solid electrolyte caused by its electron conduction is lacking.
The formation of Li dendrites in sulfide solid electrolytes is caused by a complex combination of factors, including non-uniform contact at the Li/sulfide solid electrolyte interface,13,25 the density of the sulfide solid electrolyte layer,26 and reductive decomposition products from the sulfide solid electrolyte that exists in a non-equilibrium state on the Li metal surface.27–29 Therefore, observation of continuous morphological changes in Li dendrites during the Li plating process using ex situ analysis is difficult, and operando analysis is important.
The Li concentration distribution and morphology changes in the cross-section of Li/solid electrolytes have been examined using operando neutron depth profiling and operando microscopic observation.23,24 Although these techniques are useful for analyzing the Li dendrite formation mechanism, the obtained information is limited to averages or two-dimensional. Three-dimensional (3D) morphological analysis with nanometer-order spatial resolution is necessary for a comprehensive understanding of the mechanism of the Li dendrite formation because dendrites are generated heterogeneously in the sulfide solid electrolyte. X-ray computed tomography (CT) is a useful technique for 3D morphology analysis and can be conducted at the same applied pressure as that of actual cells, and it has been used to elucidate the mechanism of Li dendrite growth in sulfide solid electrolytes.8,13,29,30 In addition, it is necessary to distinguish between solid electrolytes, which are composed primarily of light elements, Li, and voids. The phase-contrast CT method using monochromatized synchrotron radiation X-rays satisfies these requirements, and Ning et al. have succeeded in directly observing micrometer-order Li dendrites in cracks using phase-contrast CT with synchrotron radiation X-rays.8
In this study, the three-dimensional morphological changes of the Li dendrites in Li3PS4, which is a typical sulfide solid electrolyte, were observed directly using multimodal/multiscale operando CT under an applied pressure. At the BL20XU CT beamline of SPring-8, where this experiment was performed, CT measurement using transmission X-ray microscopy (nano CT) can be accomplished with a high spatial resolution of 150 nm in any field of view in a projection-type CT (nominal pixel size of 0.5 μm) image. Furthermore, nano CT can enhance the contrast of low-absorption materials using the Zernike phase-contrast method with a phase plate. In addition, the reductive decomposition product at the Li/Li3PS4 interface was examined using soft X-ray absorption spectroscopy (XAS) and electrochemical impedance measurements. The combined results of these analyses revealed that the electronic conduction of the reductive decomposition products and that at the solid electrolyte/void interface cause the Li deposition not at the Li/Li3PS4 interface, but in the Li3PS4 solid electrolyte.
Li3PS4 glass, as a solid electrolyte, was prepared using the mechanical milling method with stoichiometric amounts of Li2S and P2S5 (details in the ESI†). The characteristics of the prepared Li3PS4 are summarized in Fig. S1 (ESI†). Powder X-ray diffraction revealed that Li3PS4 was amorphous, and Raman spectroscopy confirmed the presence of the PS43− unit.31 The ionic conductivity was 5.0 × 10−4 S cm−1 at 25 °C, which was calculated based on the Nyquist plot in Fig. S1c (ESI†). The particle size was 10–50 μm, as confirmed by scanning electron microscopy. These results were consistent with those from previous studies.31
Prior to the operando CT test, we performed Li deposition tests with the operando cell (Fig. S2a, ESI†) at various current densities (0.6, 0.8 and 1.0 mA cm−2) to determine the appropriate experimental conditions for the operando CT measurements. Cell failure was very slow at a current density of 0.6 mA cm−2, whereas the cell showed an extremely large polarization at 1.0 mA cm−2, indicating that these current densities are not suitable for operando measurements. Therefore, operando CT measurements were conducted at a current density of 0.8 mA cm−2. The charge/discharge behavior of the operando cell at this current density was 0.8 mA cm−2 and was similar to that of a typical bulk-type cell with a diameter of ∅ 10 mm (Fig. S2b, ESI†), which confirms that an electrochemical test similar to that for a typical pellet-type cell can be used for the operando cell. Operando CT measurements with the operando cell were performed using the setup at beamline BL20XU, SPring-8, as shown in Fig. S3 (ESI†). To prevent complications from the incomplete dissolution of Li during the stripping process, CT images were collected every 0.5 h during the Li plating process at a current density of 0.8 mA cm−2 until the voltage could not be measured.
Before the Li deposition reaction, we captured a CT image of the Li3PS4 layer in the operando cell (Fig. 1a). In the Li3PS4 layer, a small X-ray absorption region due to voids, with different amounts in the depth direction to the Li metal electrode, was observed. Quantitative analysis of the void volume at positions perpendicular to the Li/Li3PS4 interface revealed that a relatively large number of voids, 0.5% of the total volume of the solid electrolyte, exists around the position of 100–120 μm away from the Li/Li3PS4 interface (Fig. 1b). The impact of the secondary particle system on the Li3PS4 is discussed in detail in the ESI.†
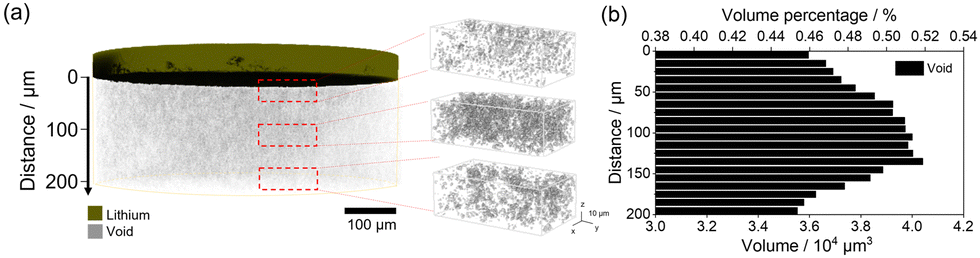 | ||
| Fig. 1 (a) 3D image of voids in the Li3PS4 layer and magnified views of voids at different depths against the Li plating electrode. (b) Histogram of the volume percentages and volumes of voids in Li3PS4 located perpendicular to the Li electrode and Li3PS4 interface. | ||
Fig. 2 shows the charge curve of the Li/Li3PS4/Li symmetric cell during operando CT measurements and the CT renderings obtained for the Li metal anode and Li3PS4. Polarization of the operando cell decreased gradually with increasing Li deposition time, and the voltage could not be measured after Li deposition for 3.2 h (Fig. 2a) owing to a short circuit. For clearer visualization of the morphological changes of the cell during plating, the 3D images showed only the top Li electrode and electrodeposited Li metal in Li3PS4 (Fig. 2b), whereas images of voids observed in the pristine state were excluded. The original Li metal electrode (without electroplating) was only observed in the CT rendering in the pristine state, and no morphological changes were observed after charging for 1.0 h (Fig. 2b). After charging for 1.5 h, a region with the same contrast as Li metal was not observed at the interface between the Li metal anode and the Li3PS4 electrolyte, but was observed in the Li3PS4 electrolyte with a relatively large number of voids in the pristine state (Fig. 1a). After charging for 3.2 h, the region with the same contrast as Li metal grew around the region formed after charging for 1.5 h. As the X-ray absorption coefficient of Li metal is similar to that of voids,32 the region formed in the Li3PS4 electrolyte cannot be attributed to Li metal or large voids using the absorption-contrast CT method. To investigate whether the region formed in the Li3PS4 electrolyte is attributed to large voids or electrodeposited Li metal, we performed nano CT with an absorption-contrast method and a phase-contrast method that uses the density difference of the objects in the region (Fig. 2c). With the absorption-contrast method, a clear contrast was not observed between a needle-like region, which might be attributed to Li deposition and shows a black color similar to that of the void region (Fig. 2c-ii). With the phase-contrast method, the needle-like region was gray, while the void was black in color (Fig. 2c-i). These results indicate that the region formed in Li3PS4 is attributed to the electrodeposited Li metal. Fig. 2d shows a plot of the volume of the electrodeposited Li metal obtained through the CT measurements and the theoretical volume of electrodeposited Li metal against plating time. The volume obtained through CT measurement after plating for 1.5 h was significantly smaller than the theoretical volume. After plating for 3.2 h, the volume obtained through CT measurement increased and was closer to the theoretical value. These results indicate that the side reactions other than Li deposition occur at an early stage of plating.
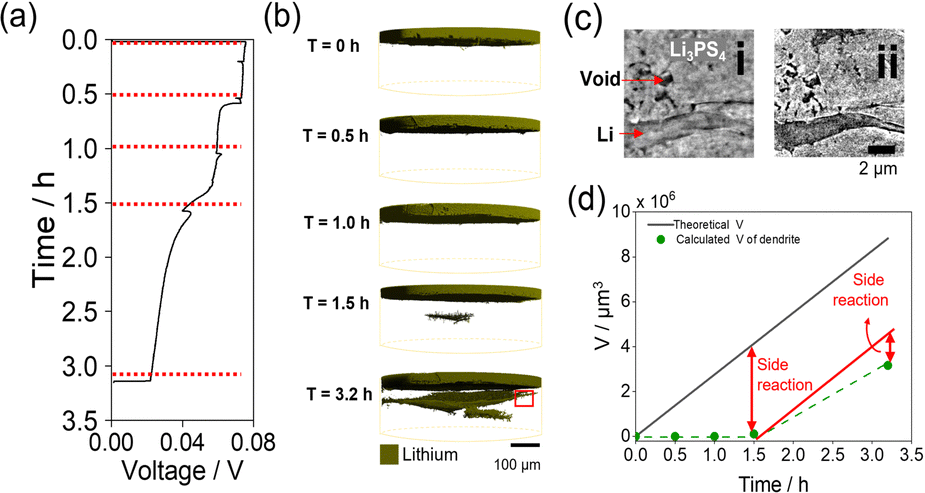 | ||
| Fig. 2 (a) Galvanostatic voltage curves of the Li/Li3PS4/Li cell measured during plating. (b) 3D renderings of Li metal of the operando cell during Li deposition. (c) Phase-contrast (i) and absorption-contrast (ii) nano CT image at T = 3.2 h of red demarcation in Fig. 2b. (d) Theoretical volumes of Li (black) at various times during charging and the calculated volume of Li dendrites (green). | ||
To clarify the side reactions during Li plating, XAS for S and P K-edges was performed for the Li/Li3PS4/Li symmetric cell before and after electrochemical measurements (Fig. 3a and b). The XAS measurements were performed after removing only the Cu current collector from the cells (details in Fig. S4, ESI†). The S K-edge XAS spectrum changed significantly after electrochemical cycling, consistent with Li2S (Fig. 3a). The P K-edge XAS spectrum changed slightly after the electrochemical cycling in the range between 2140 and 2147 eV, which is consistent with Li3P (Fig. 3b). These results indicate that Li3PS4 underwent reductive decomposition, generating Li2S and Li3P during the Li plating reaction, which is consistent with our previous reports.1 When Li3PS4 is reductively decomposed into Li3P and Li2S, the volume expands about 1.5 times (see the ESI† for details). Fig. S5 (ESI†) shows the porosity of the solid electrolyte layer from 0.0 to 1.0 hour calculated from the CT images. This result suggests that the electrolyte is undergoing reductive decomposition.
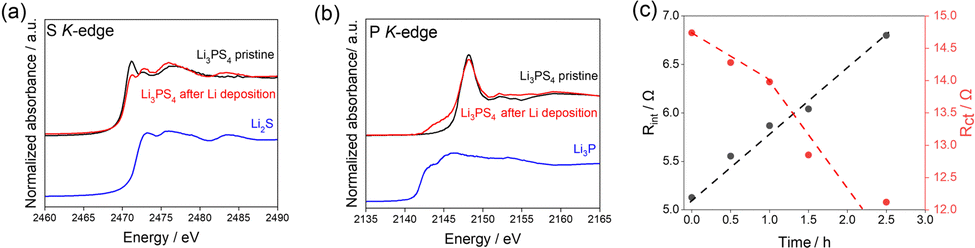 | ||
| Fig. 3 (a) S K-edge and (b) P K-edge partial fluorescence yield XAS spectra of Li3PS4 before and after the galvanostatic cycling tests. (c) Obtained resistance values of the Li/Li3PS4/Li cell for the surface layer and charge transfer based on Fig. S6 (ESI†). | ||
Additionally, to evaluate the change in interfacial resistance between Li metal and Li3PS4 during Li deposition, electrochemical impedance spectroscopy experiments were carried out in the 3.0 MHz to 0.1 Hz frequency range (Fig. 3c). Two semicircles were observed, consistent with the results of previous studies.1,33 The impedance spectra were fitted with a previously reported equivalent circuit34 (Fig. S6) and the results are presented in Table S1 (ESI†).
Comparing the obtained capacitance values of ∼0.01 and ∼10 μF to those from previous reports33,35 revealed that the semicircles in the high- and low-frequency regions correspond to interfacial resistance (Rint) and charge transfer resistance (Rct), respectively. Rint increased with increasing Li deposition time, whereas Rct decreased gradually with increasing Li deposition time up to 1.0 h and then decreased drastically afterward. The increase in the interfacial layer resistance was caused by the reductive decomposition products, Li2S and Li3P, because of their low ionic conductivities.1 The gradual decrease in the charge transfer resistance until 1.0 h may be attributed to the increase in the surface area due to the electrodeposited Li on the Li metal electrode. The drastic decrease beginning at 1.0 h is attributed to the increased surface area of the electrodeposited Li metal in the Li3PS4, which was revealed by operando CT measurements (Fig. 2).
The Li nucleation and growth mechanism in the Li/Li3PS4/Li symmetric cell, based on the results of electrochemical measurements, operando CT, and XAS, is depicted in Fig. 4. In the early to middle stages of the Li plating process, operando CT measurements did not indicate Li deposition. Because the Li deposition potential is beyond the reduction limit of Li3PS4,6 Li3PS4 undergoes reductive decomposition to Li2S and Li3P, resulting in a discrepancy between the observed and theoretical Li volumes.
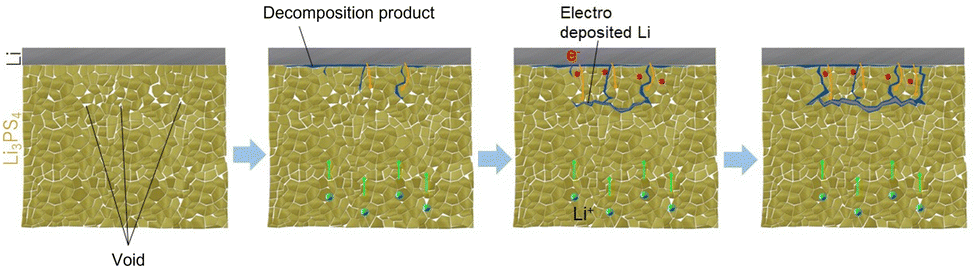 | ||
| Fig. 4 Schematic diagram of dendrite growth within Li3PS4 caused by the electronic conduction in the reductively decomposed layer and the void/Li3PS4 interface. | ||
In the middle to late stages of the charging process, Li nucleation and growth in the Li3PS4 solid electrolyte are caused by electronic conduction in the reductively decomposed layer and the void/Li3PS4 interface. Li3P, a reductive decomposition product, has been reported to have a relatively high electronic conductivity owing to its small band gap of 1.25 eV.36,37 The electronic conduction in the Li3P in the reductively decomposed layer results in Li nucleation and growth in the Li3PS4 solid electrolyte layer. A relatively large number of voids exist 100–120 μm from the Li/Li3PS4 interface, which was confirmed by operando CT (Fig. 1). A previous study reported that the band gap of a solid electrolyte at the void/solid electrolyte interface is smaller than that of the bulk solid electrolyte because of the space charge layer, which enhances the electronic conduction in the solid electrolyte. Li3PS4 showed an electronic conductivity of 3.2 × 10−8 S cm−1 at 25 °C (Fig. S7b, ESI†), which is high compared to those of other sulfide solid electrolytes.23 Therefore, the relatively high electronic conductivity of Li3PS4 enhanced by the space charge effect leads to Li nucleation around the voids at the position of 100–120 μm from the Li/Li3PS4 interface, as shown in Fig. 4. In the subsequent Li deposition process, additional Li is deposited from the deposited Li nuclei in the Li3PS4 layer. In summary, the electronic conduction of the reductive decomposition products and that at the solid electrolyte/void interface cause Li deposition not at the Li/Li3PS4 interface, but in the Li3PS4 solid electrolyte. Reducing these electronic conductions is a key in suppressing the Li dendrite growth in all-solid-state batteries. This finding will be beneficial for the use of Li metal anodes in all-solid-state batteries. In our previous study, we found that the addition of LiI to Li3PS4 improves the CCD, suggesting that LiI blocks the electronic conduction at the Li/Li3PS4 interface and suppresses Li dendrite growth in the electrolyte layer.29
In this study, we performed a 3D visualization of Li dendrite formation in a Li3PS4 solid electrolyte using operando CT and clarified the mechanism by combining electrochemical measurements with soft XAS with it. In the early stages of the Li plating process, reductive decomposition of the Li3PS4 solid electrolyte mainly occurs. Later in the Li plating process, Li nucleation occurs in the Li3PS4 solid electrolyte owing to the electronic conduction of the reductive decomposition products and that at the solid electrolyte/void interface, from which the growth of deposited Li occurs. Reducing the electronic conductions is key in suppressing the Li dendrite growth in all-solid-state batteries.
This work was financially supported by the JST, ALCA-SPRING Project (Grant Number: JPMJAL1301). Synchrotron radiation experiments were performed at beam line BL20XU and BL27SU at SPring-8 with the approval of the JASRI (Proposal no. 2020A1014, 2020A1015, 2021A1026).
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Yang, M. Takahashi, K. Yamamoto, K. Ohara, T. Watanabe, T. Uchiyama, T. Takami, A. Sakuda, A. Hayashi, M. Tatsumisago and Y. Uchimoto, Solid State Ion., 2022, 377, 115869 CrossRef CAS.
- X. B. Cheng, R. Zhang, C. Z. Zhao, F. Wei, J. G. Zhang and Q. Zhang, Adv. Sci., 2015, 3, 1500213 CrossRef PubMed.
- X. Ke, Y. Wang, L. Dai and C. Yuan, Energy Storage Mater., 2020, 33, 309–328 CrossRef.
- X. Zhang, Y. Yang and Z. Zhou, Chem. Soc. Rev., 2020, 49, 3040–3071 RSC.
- D. Cao, X. Sun, Q. Li, A. Natan, P. Xiang and H. Zhu, Matter, 2020, 3, 57–94 CrossRef.
- J. Wu, S. Liu, F. Han, X. Yao and C. Wang, Adv. Mater., 2021, 33, 2000751 CrossRef CAS PubMed.
- Y. Guo, S. Wu, Y. He, F. Kang, L. Chen, H. Li and Q. Yang, eScience, 2022, 2, 138–163 CrossRef.
- Z. Ning, D. S. Jolly, G. Li, R. D. Meyere, S. D.Pu, Y. Chen, J. Kasemchainan, J. Ihli, C. Gong, B. Liu, D. L. R. Melvin, A. Bonnin, O. Magdysyuk, P. Adamson, G. O. Hartley, C. W. Monroe, T. J. Marrow and P. G. Bruce, Nat. Mater., 2021, 20, 1121–1129 CrossRef CAS PubMed.
- Z. Ning, D. S. Jolly, G. Li, R. D. Meyere, S. D. Pu, Y. Chen, J. Kasemchainan, J. Ihli, C. Gong, B. Liu, D. L. R. Melvin, A. Bonnin, O. Magdysyuk, P. Adamson, G. O. Hartley, C. W. Monroe, T. J. Marrow and P. G. Bruce, Adv. Mater., 2021, 33, 2000721 CrossRef PubMed.
- S. Lou, F. Zhang, C. Fu, M. Chen, Y. Ma, G. Yin and J. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 9242–9248 CrossRef PubMed.
- S. Wang, R. Fang, Y. Li, Y. Liu, C. Xin, F. H. Richter and C. W. Nan, J. Materiomics, 2021, 7, 209–218 CrossRef.
- N. Seitzman, O. F. Bird, R. Andrykowski, S. Robbins, M. M. Al-Jassim and S. Pylypenko, ACS Appl. Energy Mater., 2021, 4, 1346–1355 CrossRef CAS.
- J. A. Lewis, F. J. Q. Cortes, Y. Liu, J. C. Miers, A. Verma, B. S. Vishnugopi, J. Tippens, D. Prakash, T. S. Marchese, S. Y. Han, C. Lee, P. P. Shetty, H. W. Lee, P. Shevchenko, F. de Carlo, C. Saldana, P. P. Mukherjee and M. T. McDowell, Nat. Mater., 2021, 20, 503–510 CrossRef CAS PubMed.
- S. Chen, D. Xie, G. Liu, J. P. Mwizerwa, Q. Zhang, Y. Zhao, X. Xu and X. Yao, Energy Storage Mater., 2018, 14, 58–74 CrossRef.
- M. Rosso, C. Brissot, A. Teyssot, M. Dollé, L. Sannier, J. M. Tarascon, R. Bouchet and S. Lascaud, Electrochim. Acta, 2006, 51, 5334–5340 CrossRef CAS.
- I. Ismail, A. Noda, A. Nishimoto and M. Watanabe, Electrochim. Acta, 2001, 46, 1595–1603 CrossRef CAS.
- M. K. Aslam, Y. Niu, T. Hussain, H. Tabassum, W. Tang, M. Xu and R. Ahuja, Nano Energy, 2021, 86, 106142 CrossRef CAS.
- X. Zhang, S. Wang, C. Xue, C. Xin, Y. Lin, Y. Shen, L. Li and C. W. Nan, Adv. Mater., 2019, 31, 1806082 CrossRef PubMed.
- S. Ni, S. Tan, Q. An and L. Mai, J. Energy Chem., 2020, 44, 73–89 CrossRef.
- H. Huo, Y. Chen, R. Li, N. Zhao, J. Luo, J. G. Pereira Da Silva, R. Mücke, P. Kaghazchi, X. Guo and X. Sun, Energy Environ. Sci., 2020, 13, 127–134 RSC.
- L. N. Wu, J. Peng, F. M. Han, Y. K. Sun, T. Sheng, Y. Y. Li, Y. Zhou, L. Huang, J. T. Li and S. G. Sun, Mater. Chem. A, 2020, 8, 4300–4307 RSC.
- Q. Tu, T. Shi, S. Chakravarthy and G. Ceder, Matter, 2021, 4, 3248–3268 CrossRef CAS.
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, Nat. Energy, 2019, 4, 187–196 CrossRef CAS.
- M. Sun, T. Liu, Y. Yuan, M. Ling, N. Xu, Y. Liu, L. Yan, H. Li, C. Liu, Y. Lu, Y. Shi, Y. He, Y. Guo, X. Tao, C. Liang and J. Lu, ACS Energy Lett., 2021, 6, 451–458 CrossRef CAS.
- A. Banerjee, X. Wang, C. Fang, E. Wu and S. Meng, Chem. Rev., 2020, 120, 6878–6933 CrossRef CAS PubMed.
- M. B. Dixit, N. Singh, J. P. Horwath, P. D. Shevchenko, M. Jones, E. A. Stach, T. S. Arthur and K. B. Hatzell, Matter, 2020, 3, 2138–2159 CrossRef.
- K. N. Wood, K. X. Steirer, S. E. Hafner, C. Ban, S. Santhanagopalan, S. H. Lee and G. Teeter, Nat. Commun., 2018, 9, 2490 CrossRef PubMed.
- F. Han, J. Yue, X. Zhu and C. Wang, Adv. Energy Mater., 2018, 8, 1703644 CrossRef.
- M. Takahashi, T. Watanabe, K. Yamamoto, K. Ohara, A. Sakuda, T. Kimura, S. Yang, K. Nakanishi, T. Uchiyama, M. Kimura, A. Hayashi, M. Tatsumisago and Y. Uchimoto, Chem. Mater., 2021, 33, 4907–4914 CrossRef CAS.
- M. Otoyama, M. Suyama, C. Hotehama, H. Kowada, Y. Takeda, K. Ito, A. Sakuda, M. Tatsumisago and A. Hayashi, ACS Appl. Mater. Interfaces, 2021, 13, 5000–5007 CrossRef CAS PubMed.
- T. Yamada, S. Ito, R. Omoda, T. Watanabe, Y. Aihara, M. Agostini, U. Ulissi, J. Hassoun and B. Scrosati, Electrochem. Soc., 2015, 162, A646–A651 CrossRef CAS.
- N. Seitzman, H. Guthrey, D. B. Sulas, H. A. S. Platt, M. Al-Jassim and S. Pylypenko, J. Electrochem. Soc., 2018, 165, A3732–A3737 CrossRef CAS.
- L. M. Riegger, R. Schlem, J. Sann, W. G. Zeier and J. Janek, Angew. Chem., Int. Ed., 2021, 60, 6718–6723 CrossRef CAS PubMed.
- S. Wenzel, S. J. Sedlmaier, C. Dietrich, W. G. Zeier and J. Janek, Solid State Ion., 2018, 318, 102–112 CrossRef CAS.
- J. T. S. Irvine, D. C. Sinclair and A. R. West, Adv. Mater., 1990, 2, 132–138 CrossRef CAS.
- H. K. Tian, B. Xu and Y. Qi, J. Power Sources, 2018, 392, 79–86 CrossRef CAS.
- T. Krauskopf, F. H. Richter, W. G. Zeier and J. Janek, Chem. Rev., 2020, 120, 7745–7794 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc05224e |
| This journal is © The Royal Society of Chemistry 2023 |