
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccessing unsymmetrical Ru(II) bipyridine complexes: a versatile synthetic mechanism for fine tuning photophysical properties†
Lukas
Hallen‡
 a,
Alexandra M.
Horan‡
b,
Brendan
Twamley
a,
Eoghan M.
McGarrigle
*b and
Sylvia M.
Draper
*ac
a,
Alexandra M.
Horan‡
b,
Brendan
Twamley
a,
Eoghan M.
McGarrigle
*b and
Sylvia M.
Draper
*ac
aSchool of Chemistry, Trinity College Dublin, the University of Dublin, Dublin 2, Ireland. E-mail: smdraper@tcd.ie
bSSPC, the SFI Research Centre for Pharmaceuticals, Centre for Synthesis & Chemical Biology, UCD School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: eoghan.mcgarrigle@ucd.ie
cAMBER (Advanced Materials and Bioengineering Research) Centre, Trinity College Dublin, Dublin 2, Ireland
First published on 23rd November 2022
Abstract
Three novel unsymmetrical Ru(II) bipyridine complexes were generated via a convenient, modular, convergent synthetic route. An investigation of their photophysical properties revealed solvent-dependent excited state behaviour including altered absorption and emission wavelengths, emission lifetimes and quantum yields of phosphorescence.
Transition metal complexes comprising N-coordinating ligands such as bipyridine and phenanthroline, have been actively targeted for many applications in photophysics and photochemistry.1–6 The high stability, strong absorption and low biotoxicity of ruthenium polypyridyl complexes in particular have led to the development of a wide library of such compounds.7 Applications range from photodynamic therapy (PDT) to triplet-triplet annihilation upconversion (TTA-UC), and from photovoltaics to photocatalysis,5,8–11 but in all cases the design of the ligand chromophore(s) is key to tuning the nature and emissive character of the excited states.
Two main approaches have been explored to determine the structure-property relationships that govern the important optical characteristics such as absorption wavelengths, excited-state lifetimes and singlet oxygen quantum yields. The first is to append self-contained, highly-absorbing chromophores such as Nile Red or BODIPY to the ligand, allowing population of the chromophore's triplet excited state by exploiting the heavy-atom effect of the metal centre.12–14 The second is to modify the energy levels of the N-coordinating ligand, using donor and/or acceptor moieties, so as to make these energy levels accessible via intra-ligand electronic transitions.15–17 Of particular interest in the latter, are unsymmetrical ligand systems, where different donor/acceptor groups are attached to e.g. the bipyridine rings in either the 4,4′ or 5,5′-positions, as these can reveal some very unusual optical properties.2,18
Gordon and co-workers recently reported solvent-dependent excited state switching in unsymmetrical rhenium bipyridyl systems, where two separate and distinct excited states were accessed depending on the solvent.3 Draper et al. showed that appending accepting groups can significantly alter the excited state properties of unsymmetrical Ir complexes compared to analogous symmetrical dual donor systems.2 It is clear that there is enormous potential for fine-tuning the electronic properties of unsymmetrical polypyridyl systems. However their syntheses remain challenging, not least because they typically require sequential synthetic functionalisation of each pyridine ring, complicating purification processes.18–21
A common synthetic approach involves successive palladium-catalysed reactions such as Suzuki or Sonogashira couplings on preformed symmetrical halogenated bipyridines, although Wittig reactions have also been employed.15,19,20 Such desymmetrisation methodologies suffer from stepwise reductions in yield and challenging purifications due to site-selectivity problems. A convergent, modular synthetic approach to unsymmetrical bipyridine ligands would facilitate an investigation of the effects of donor/acceptor moieties on the photophysical properties of the resulting Ru(II) complexes.
The McGarrigle group recently disclosed a sulfur-mediated ligand-coupling methodology for the preparation of unsymmetrical bipyridines, including biologically active targets (Scheme 1).22–26 In this process, a pyridyllithium or Grignard reagent is reacted with a pyridylsulfonium salt, to give a trigonal bipyramidal sulfurane intermediate, which on collapse yields the desired bipyridine. This methodology permits the modular and selective introduction of functionality to either side of an unsymmetrical bipyridine ligand, i.e., from both the organometallic reagent and the pyridylsulfonium salt.
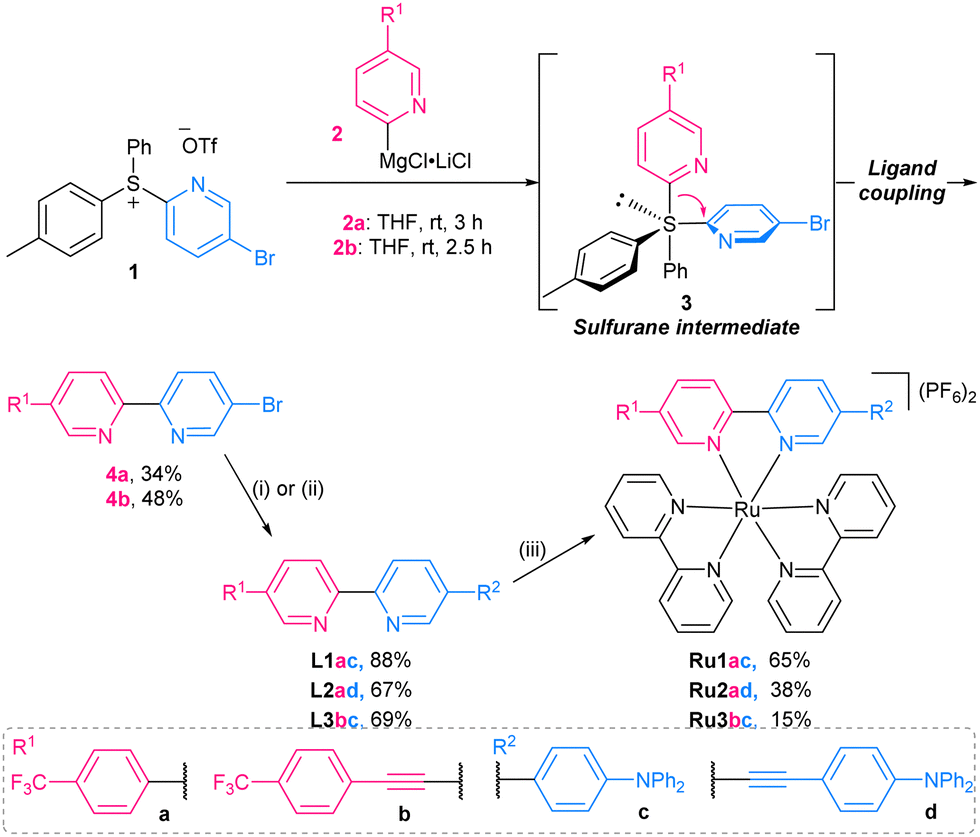 | ||
Scheme 1 Synthetic route to ligands L1–3 and complexes Ru1–3. [(i): Ph2NC6H4B(OH)2 (1.2 equiv.), Pd(PPh3)4 (3 mol%), K2CO3 (3.0 equiv.) THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (2:1), 80 °C, 22 h], [(ii): Ph2NC6H4C :H2O (2:1), 80 °C, 22 h], [(ii): Ph2NC6H4C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH (1.2 equiv.) Pd(PPh3)4 (3 mol%) CuI (3 mol%) Et3N:toluene (3:1), 80 °C, 5 h], [(iii): Ru(bpy)2Cl2 (0.9 equiv.), ethylene glycol, 140 °C, 16 h] CH (1.2 equiv.) Pd(PPh3)4 (3 mol%) CuI (3 mol%) Et3N:toluene (3:1), 80 °C, 5 h], [(iii): Ru(bpy)2Cl2 (0.9 equiv.), ethylene glycol, 140 °C, 16 h] | ||
Herein, we demonstrate the use of this reaction in the syntheses of three novel unsymmetrical functionalised 2,2′-bipyridine ligands (Scheme 1 and Fig. 2). Our standard coupling conditions using 5-bromopyridylsulfonium salt 1 and Grignard reagents 2a,b gave the brominated bipyridines 4a,b in acceptable yield (without optimisation). Subsequent Sonagashira or Suzuki coupling introduced the triphenylamine donor moieties. Complexation with [Ru(bpy)2Cl2] gave Ru1–3.
 | ||
| Fig. 1 (a) UV-vis absorption spectra Ru1–3; (b–d) Normalized emission spectra of Ru1–3 (λex = 435 nm for Ru1; 440 nm for Ru2 and Ru3). Emission spectra measured under Argon (yellow) and in air (black). MeCN, [10−5 M], 298 K. | ||
 | ||
| Fig. 2 Chemical structures of Ru1–Ru3. | ||
Ready access to L1–3 and Ru1–3 provided a platform for an investigation of the effects of the appended donor/acceptor moieties on the overall photophysical behaviour of the novel bipyridine ligands and their Ru(II) complexes. The route also enabled us to incorporate bridging linkers to either the donor or the acceptor species for the first time and to explore the opto-electronic consequences of selective conjugation.
The absorption and emission spectra of the ligands were measured in CH2Cl2 (Table 1 and Fig. S22, ESI†). The emissions are sensitive to solvent (Fig. S23–S25, ESI†) and are typical of singlet charge transfer (1CT) transitions. Low-temperature measurements confirm the 1CT nature of the excited state (Fig. S29–S31, ESI†). The fluorescence quantum yields (Φem) for L1–3 were recorded using fluorescein in 0.1 M aq. NaOH as a standard and are shown in Table 1.27,28 Due to literature inconsistencies in the quantum yield of fluorescein in 0.1 M NaOH a range of quantum yields are reported.
| λ abs [nm] | ε (×104)b [M−1cm−1] | λ em [nm] | Φ em [%] | τ em [ns] | |
|---|---|---|---|---|---|
| a In CH2Cl2 [10−5 M], 298 K. b Molar extinction coefficient at the absorption maxima. c Emission maximum in CH2Cl2, excited at the corresponding λabs value. d Fluorescence quantum yield with Fluorescein as a standard (Φf = 79% in 0.1 M aq. NaOH).27,28 e Emission lifetime in CH2Cl2. f Fitted monoexponentially. | |||||
| L1 | 375 | 3.71 | 520 | 86.9–93.3 | 3.21f |
| L2 | 390 | 3.43 | 540 | 95.4–98.7 | 3.47f |
| L3 | 380 | 3.37 | 540 | 86.8–92.9 | 3.02f |
The high quantum yields reflect the proficiency of the triphenylamine donor and trifluorotoluene acceptor to act as electronic scaffolds for 1CT transitions.29 Dilution studies performed on the three ligands eliminated the possibility of any contribution to the quantum yield of emission through aggregation -enhanced or -enabled emission (Fig. S26–S28, ESI†). The emission lifetimes (3.02–3.47 ns) are typical of singlet excited state character (for traces see Fig. S44–S46, ESI†).
The UV-Vis absorption spectra of Ru1–3 (Fig. 1a) in MeCN show high-energy transitions under 300 nm and a broad absorption band around 440 nm. The high-energy bands are assigned to π → π* transitions centred on the bipyridine ligands. The lower energy absorption around 440 nm is likely comprised of the metal-to-ligand charge transfer (1MLCT) transitions typical of polypyridyl ruthenium complexes.1 It is possible that this absorption band also contains an intra-ligand charge transfer (1ILCT) transition from the triphenylamine to the trifluorotoluene moieties. A red-shift in ligand charge-transfer absorption bands following conjugation to a metal centre is common for CT-active ligands, and similar behaviour was reported by Wang et al.2,30 There are no significant differences between the maximum absorption wavelengths, of Ru1 compared to Ru2/Ru3, suggesting that the influence of the acetylene linker on the absorption is minimal (Fig. 1a and Fig. S32–S34, ESI†). The emission spectra of Ru1–3 (Ru1 excited at 435 nm; Ru2 and Ru3 at 440 nm) in MeCN were recorded in air and argon and are shown in Fig. 1b–d. The profiles of the emission bands are broad and featureless, suggesting a CT-type excited state. The emission wavelengths are red-shifted compared to those of the ligands, with Ru1 emitting at 645 nm and Ru2/Ru3 emitting at 670 nm. The presence of the acetylene linker exerts a greater influence on the emission wavelength than on the absorption wavelength, although its position in the ligand structure does not cause any change to either. The emission intensity of the complexes is enhanced in deaerated MeCN, suggesting that the excited state has triplet character and is capable of sensitising triplet oxygen, an important prerequisite for a wide range of applications such as PDT and TTA-UC.2,11
The emission lifetimes of the complexes in deaerated MeCN, are shown in Table 2 (Traces shown in S38-S40). The short emission lifetimes of Ru2 and Ru3 are unusual for ruthenium complexes, potentially arising from rapid nonradiative relaxation pathways accessible through the acetylene moiety.7 The similarities in emission wavelength, lifetime and quantum yield of phosphorescence between Ru2 and Ru3 (Table 2) suggest that they share a common, likely metal-based emissive excited state in MeCN.
| λ abs [nm] | ε (× 104)b [M−1cm−1] | λ em [nm] | Φ em [%] | τ em [ns] | |
|---|---|---|---|---|---|
| a In MeCN [10−5 M], 298 K. b Molar extinction coefficient at λabs. c Emission maximum in MeCN, excited at the corresponding λabs value. d Phosphorescent quantum yield in deaerated MeCN with [Ru(bpy)3](PF6)2 as a standard (Φp = 9.5% in MeCN).13 e Emission lifetime in deaerated MeCN. f Fitted monoexponentially. | |||||
| Ru1 | 435 | 3.00 | 645 | 1.79 | 1.044f |
| Ru2 | 440 | 2.87 | 670 | 0.78 | 0.21f |
| Ru3 | 440 | 3.11 | 670 | 1.12 | 0.23f |
Solvatochromic absorption and emission studies were completed for the three metal complexes (Fig. S32–S37, ESI†). In the majority of ruthenium complexes, increasing solvent polarity leads to a stabilisation of the excited state dipole by solvent rearrangement.13 This stabilisation results in a reduction of LUMO energies, leading to a red shift in emission wavelength.
Absorption wavelengths are shifted bathochromically by 20–25 nm in CH2Cl2 compared to MeCN (Table 3), with Ru2 and Ru3 showing a larger shift than Ru1. Such behaviour is unusual in ruthenium tris-bipyridine complexes, although Das et al. have recorded similar trends in their ruthenium arene complexes.31 Excitation studies in CH2Cl2 and MeCN are reported in Fig. S32–S34 (ESI†).
| λ abs [nm] | ε (×104)b [M−1cm−1] | λ em [nm] | Φ em [%] | τ em [μs] | |
|---|---|---|---|---|---|
| a In CH2Cl2 [10−5 M], 298 K. b Molar extinction coefficient at the absorption maxima past 400 nm. c Emission maximum in CH2Cl2, excited at the corresponding λabs value. d Phosphorescent quantum yield in deaerated CH2Cl2 with [Ru(bpy)3](PF6)2 in MeCN as a standard (Φp = 9.5% in MeCN).13 e Emission lifetime in deaerated CH2Cl2. f Fitted monoexponentially. | |||||
| Ru1 | 455 | 3.25 | 700 | 3.40 | 2.85f |
| Ru2 | 465 | 2.87 | 745 | 1.02 | 0.42f |
| Ru3 | 465 | 2.96 | 720 | 2.38 | 1.42f |
The emission wavelengths of the complexes Ru1 to Ru3 in CH2Cl2 are shifted significantly compared to those in MeCN, however the expected trend of red-shifted emission wavelength with increasing solvent polarity is not observed e.g. the emission wavelength for Ru2 shows a red shift of 75 nm in less polar CH2Cl2. Additionally, where there was no difference in the emission wavelengths between Ru2 and Ru3 in MeCN, there is one in CH2Cl2.
Continued investigation into the excited state properties of the complexes in CH2Cl2 revealed additional changes in behaviour compared to those observed in MeCN (Table 3). (i) The emission lifetime was strongly affected, with a 600% increase in emission lifetime recorded for Ru3 and smaller % increases recorded for both Ru1 and Ru2. (ii) The quantum yields of phosphorescence are also clearly solvent-dependent and are much larger in CH2Cl2. (iii) As can be seen in Fig. S32–S34 (ESI†), the emission profiles also change significantly, with the emissions in CH2Cl2 being significantly broader compared to those in MeCN. These combined differences strongly suggest different excited states are being accessed that are dependent on the ligand structure and the solvent.
Low-temperature measurements of the complexes (Fig. S47–S49, ESI†) in an EtOH/MeOH (4:1) glass at 77 K show the presence of multiple bands at low temperatures, as seen in [Ru(bpy)3]2+.32 These coalesce to form the single emission peak observed at room temperature. While the low-temperature emission spectra are not solvent-sensitive (Fig. S51, ESI†), those at room temperature in the same solvents are (Fig. S50, ESI†). Although further studies, (including time-resolved and transient investigations of the excited states) are required to fully rationalise the observed differences, the behaviour recorded for Ru1–3 resemble those seen in rhenium complexes by Gordon and co-workers.3 The presence of two separate excited states at room temperature, one accessed in CH2Cl2 and the other in MeCN, would explain the difference in absorption and emission wavelengths, quantum yields of phosphorescence and emission lifetimes.21
In addition, this work shows that the position of the acetylene linker significantly influences the excited state in CH2Cl2, but not in MeCN. In CH2Cl2, a hypsochromic shift in emission wavelength and increases in both quantum yield of phosphorescence and emission lifetime are observed when the acetylene links to the CF3 (as in Ru3), as compared to Ru2 where it links to the NPh3. This suggests that the excited state accessed in CH2Cl2 has greater ligand character than the one accessed in MeCN. It does not rule out however the possibility of different MLCT states (Ru → bpy and Ru → bpysub) and further analysis is necessary to determine the exact nature of these two excited states.
In summary, we have demonstrated the suitability of recently developed sulfonium-enabled unsymmetrical bipyridyl synthesis for the efficient generation of unsymmetrical ruthenium triplet photosensitisers. A series of novel ruthenium complexes were generated to investigate the effect that selectively enhanced conjugation to donor or acceptor groups would have on their photophysical behaviour. The fascinating results indicate the presence of two separate excited states, one with greater metal character at higher energies and one with greater ligand character at lower energies (Fig. S52, ESI†). Continued investigations of these complexes and other related unsymmetrical systems are now possible via the application of a new synthetic route. This will allow researchers to investigate just how widespread the phenomenon of solvent-dependent excited state switching might be and could have a profound influence on how emissive excited states are investigated in the future.
This material is based upon works supported by Science Foundation Ireland under PI Research Grant 15/IA/3046, AMBER II research centre award 12/RC/2278_p2 and additional funding from the Higher Education Authority and the Department of Further and Higher Education, Research, Innovation and Science. A. M. H. thanks the Irish Research Council for a Postgraduate Scholarship (GOIPG/2017/1306) and SFI (12/RC/2275_p2) and (18/RI/5702) for MS infrastructure. We thank Drs J. O’Brien, Y. Ortin, M. Reuther, J. Muldoon and G. Hessman for spectroscopic and technical assistance.
Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed.
- Y. Lu, N. McGoldrick, F. Murphy, B. Twamley, X. Cui, C. Delaney, G. M. Ó. Máille, J. Wang, J. Zhao and S. M. Draper, Chem. – Eur. J., 2016, 22, 11349–11356 CrossRef CAS PubMed.
- J. J. Sutton, D. Preston, P. Traber, J. Steinmetzer, X. Wu, S. Kayal, X. Z. Sun, J. D. Crowley, M. W. George, S. Kupfer and K. C. Gordon, J. Am. Chem. Soc., 2021, 143, 9082–9093 CrossRef CAS PubMed.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC.
- K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS PubMed.
- M. Chandrasekharam, C. P. Kumar, S. P. Singh, V. Anusha, K. Bhanuprakash, A. Islam and L. Han, RSC Adv., 2013, 3, 26035–26046 RSC.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- Y. Lu, J. Wang, N. McGoldrick, X. Cui, J. Zhao, C. Caverly, B. Twamley, G. M. ÓMáille, B. Irwin, R. Conway-Kenny and S. M. Draper, Angew. Chem., Int. Ed., 2016, 55, 14688–14692 CrossRef CAS PubMed.
- S. C. Boca, M. Four, A. Bonne, B. van der Sanden, S. Astilean, P. L. Baldeck and G. Lemercier, Chem. Commun., 2009, 4590–4592 RSC.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef CAS PubMed.
- H. Abrahamse and M. R. Hamblin, Biochem. J., 2016, 473, 347–364 CrossRef CAS PubMed.
- C. Condon, R. Conway-Kenny, X. Cui, L. J. Hallen, B. Twamley, J. Zhao, G. W. Watson and S. M. Draper, J. Mater. Chem. C, 2021, 9, 14573–14577 RSC.
- J. Wang, Y. Lu, N. McGoldrick, C. Zhang, W. Yang, J. Zhao and S. M. Draper, J. Mater. Chem. C, 2016, 4, 6131–6139 RSC.
- R. Bodapati, M. Sarma, A. Kanakati and S. K. Das, J. Org. Chem., 2015, 80, 12482–12491 CrossRef CAS PubMed.
- H. Feng He, X. Tao Shao, L. Li Deng, J. Xin Zhou, Y. Yuan Zhu, H. Ying Xia, L. Shen and F. Zhao, Tetrahedron Lett., 2019, 60, 150968 CrossRef.
- K. A. Opperman, S. L. Mecklenburg and T. J. Meyer, Inorg. Chem., 1994, 33, 5295–5301 CrossRef CAS.
- K. R. Benson, J. Stash, K. L. Moffa, R. H. Schmehl, T. J. Dudley and J. J. Paul, Polyhedron, 2021, 205, 115300 CrossRef CAS.
- J. Lu, Y. Zheng and J. Zhang, Phys. Chem. Chem. Phys., 2015, 17, 20014–20020 RSC.
- A. Martin, A. Byrne, C. Dolan, R. J. Forster and T. E. Keyes, Chem. Commun., 2015, 51, 15839–15841 RSC.
- D. Magde, M. D. Magde and E. C. Glazer, Coord. Chem. Rev., 2016, 306, 447–467 CrossRef CAS.
- V. K. Duong, A. M. Horan and E. M. McGarrigle, Org. Lett., 2020, 22, 8451–8457 CrossRef CAS PubMed.
- A. M. Horan, V. K. Duong and E. M. McGarrigle, Org. Lett., 2021, 23, 9089–9093 CrossRef CAS PubMed.
- B. T. Boyle, M. C. Hilton and A. McNally, J. Am. Chem. Soc., 2019, 141, 15441–15449 CrossRef CAS PubMed.
- M. Zhou, J. Tsien and T. Qin, Angew. Chem., Int. Ed., 2020, 59, 7372–7376 CrossRef CAS PubMed.
- X. A. F. Cook, L. R. E. Pantaine, D. C. Blakemore, I. B. Moses, N. W. Sach, A. Shavnya and M. C. Willis, Angew. Chem., Int. Ed., 2021, 60, 22461–22468 CrossRef CAS PubMed.
- D. Magde, R. Wong and P. G. Seybold, Photochem. Photobiol., 2002, 75, 327 CrossRef CAS.
- Z. C. Yang, M. Wang, A. M. Yong, S. Y. Wong, X. H. Zhang, H. Tan, A. Y. Chang, X. Li and J. Wang, Chem. Commun., 2011, 47, 11615–11617 RSC.
- Z. Zhao, X. Yin, T. Peng, N. Wang and S. Wang, Inorg. Chem., 2020, 59, 7426–7434 CrossRef CAS PubMed.
- Y. Wang, S. Liu, M. R. Pinto, D. M. Dattelbaum, J. R. Schoonover and K. S. Schanze, J. Phys. Chem. A, 2001, 105, 11118–11127 CrossRef CAS.
- R. Bodapati, C. Sahoo, M. Gudem and S. K. Das, Inorg. Chem., 2019, 58, 11470–11479 CrossRef CAS PubMed.
- H. Yersin and E. Gallhuber, J. Am. Chem. Soc., 1984, 106, 6582–6586 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data and X-ray crystallographic data for Ru1 and Ru2. CCDC 2204851 and 2204852. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04910d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |