
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective hydroboration of electron-rich isocyanates by an NHC-copper(I) alkoxide†
Laura E.
English
 ab,
Thomas M.
Horsley Downie
a,
Catherine L.
Lyall
a,
Mary F.
Mahon
a,
Claire L.
McMullin
a,
Samuel E.
Neale
a,
Carla M.
Saunders
a and
David J.
Liptrot
*a
ab,
Thomas M.
Horsley Downie
a,
Catherine L.
Lyall
a,
Mary F.
Mahon
a,
Claire L.
McMullin
a,
Samuel E.
Neale
a,
Carla M.
Saunders
a and
David J.
Liptrot
*a
aDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: d.j.liptrot@bath.ac.uk
bCentre for Sustainable and Circular Technologies, Bath, BA2 7AY, UK
First published on 26th December 2022
Abstract
The (IPr)CuOtBu catalysed reduction of 11 aryl and alkyl isocyanates with pinacolborane gave only the boraformamides, pinBN(R)C(O)H, in most cases. Overreduction, which hampers almost all isocyanate hydroborations, was restricted to electron poor aryl isocyanates (4-NC-C6H4NCO, 4-F3C-C6H4NCO, 3-O2N-C6H4NCO). Computational analysis showed stability of [(IPr)CuH]2, which was proposed to be the catalyst resting state, drives selectivity, suggesting an approach to prevent overreduction in future work. In the case of iPrNCO, formation of this species renders overreduction kinetically inaccessible. For 4-NC-C6H4NCO, however, the barrier height for the first step of over-reduction is much lower, even relative to [(IPr)CuH]2, resulting in unselective reduction.
Catalytic reduction of isocyanates leveraging p-block hydrides has emerged as a generalisable route to N-methyl amines via the deoxygenation of intermediate formamide derivatives. This transformation has been reported to be catalysed by zirconium, magnesium,1–3 platinum and boron-centred4 catalysts; or via the uncatalysed action of boranes5 and alanes6–8 upon these substrates. Selective generation of the partially reduced formamide derivatives containing the –N(R)C(O)H fragment remains challenging, limited to the stoichiometric action of alane and zirconium derived systems upon these substrates9 and restricted reports of hydrogenation10,11 and hydrosilylation12 under palladium catalysis. A recent report by Nembenna showed that a zinc catalyst can provide a highly chemoselective route to boraformamides through the reaction of isocyanates with pinacolborane via a zinc(II) hydride intermediate.13 This was followed by a report of a similarly selective reduction catalysed by LiHBEt3 by Ding and co-workers.14 This dearth of chemoselective transformations contrasts with another group of heteroallenes, the carbodiimides, where selective access to the formamidine derivatives, containing –N(R)C(NR)H, is facile.15
Following the initial report of [CuH(PPh3)]6 in 1971,16 and its subsequent exploration in reduction of carbonyl derivatives by Stryker and co-workers,17,18 copper(I) hydrides have emerged as increasingly popular reducing agents.19 Subsequent extension of the ancillary ligand on copper to encompass NHCs (N-heterocyclic carbenes)20 has fuelled an explosion in the catalytic and stoichiometric implication of copper(I) hydrides in productive transformations. This growth is in no small part a consequence of the facile synthesis of [(NHC)CuH]n systems via the action of main group hydrides on copper-element bonds. For example, Sadighi and co-workers described the synthesis and characterisation of [(IPr)CuH]2 (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) via the reaction of (IPr)CuOtBu with HSi(OEt)3, which also generated tBuOSi(OEt)3.20 Herein we report that the reaction of isocyanates with pinacolborane to generate boraformamides, pinBN(R)C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)H, with high chemoselectivity can be catalysed by the readily accessible, and stable precatalyst (IPr)CuOtBu.
O)H, with high chemoselectivity can be catalysed by the readily accessible, and stable precatalyst (IPr)CuOtBu.
We initially noted this reaction during our work on copper(I) catalysed hydrophosphination of isocyanates using phosphines generated in situ as a result of P–O bond reduction by pinacolborane.21 When we added 2 mol% (IPr)CuOtBu to an equimolar mixture of triphenylphosphinite, pinacolborane (pinBH) and phenyl isocyanate we observed a number of products, including peaks we attributed to pinBN(Ph)C(O)H. We initially considered that this selective monohydroboration may originate from the partial poisoning of the catalyst by the phosphines present. The reaction in the absence of phosphines, however, disproved this hypothesis. 2 mol% of (IPr)CuOtBu was added to an equimolar C6D6 solution of pinBH and PhNCO and within 30 minutes both pinBH and the isocyanate were consumed, leaving only resonances attributed to pinBN(Ph)C(O)H and a minor resonance in the 11B NMR spectrum at 21.7 ppm attributed to pinBOtBu. While the hydroboration of carbodiimides is a well-explored reaction,15 the monohydroboration of isocyanates to give pinBN(R)C(O)H is underreported due to their propensity towards over-reduction to give methylamines.22 Despite its seeming simplicity, no route has yet been described to access pinBN(Ph)C(O)H, with Nembenna reporting the reduction of only substituted arenes.13
To test if this hydroboration could be applied to other substrates, pinBH was reacted with a selection of heterocumulenes, in the presence of 2 mol% (IPr)CuOtBu (Scheme 1). Small alkyl groups were tolerated well under these reaction conditions (1a and 1bvs.1c). Under the conditions reported in Scheme 1, no hydroboration of isopropyl isocyanate was observed in the absence of (IPr)CuOtBu (see ESI†).
 | ||
Scheme 1 Catalytic hydroboration of heterocumulenes (0.10 mmol) with pinBH (14.5 μL, 0.10 mmol) and 2 mol% (IPr)CuOtBu in C6D6 (0.5 mL), room temperature unless otherwise noted, NMR yields calculated based on duplicate 1H NMR spectra compared to a 1,3,5-(MeO)3C6H3 standard. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction was conducted at 80 °C for 16 h. bEvidence of other products with inconsistent conversions, see ESI.† Reaction was conducted at 80 °C for 16 h. bEvidence of other products with inconsistent conversions, see ESI.† | ||
Electron-donating aryl substitution also provided good conversions, albeit the very bulky 2,6-di-isopropylphenyl isocyanate required 16 hours at 80 °C to provide a 94% NMR yield (1f). 1,4-Di-isocyanatobenzene could be successfully dihydroborated in 71% yield (1k). Substitution of the aryl ring with electron withdrawing trifluoromethyl, cyano- and nitro-groups (1h, 1i, 1j respectively), however, provided limited conversion to the desired boraformamide and evidence of other products in the 1H and 11B NMR spectra (vide infra). Whilst carbodiimides were also viable substrates (3a–c), providing good to excellent yields, phenyl isothiocyanate provided limited conversion (2a). Only one example of isothiocyanate hydroboration exists in the literature.23
In the cases of alkyl and electron-rich aryl isocyanates the only observable organic product was the boraformamide. Moreover, even in the presence of excess pinacolborane, no evidence of further reduction was observed for phenyl isocyanate, instead extended reaction times provided evidence of catalyst death in the form of visible metallic copper. In contrast, electron-poor arenes provided evidence of other products which could be attributed to further reduction steps, including complete deoxygenation of the isocyanate fragment to produce the corresponding N-methylaniline. In the case of 4-cyanophenyl isocyanate and 3-nitrophenyl isocyanate, competing reduction of the other arene substituents were also observed providing complex product mixtures. Nevertheless, the formation of 1h could be confirmed by the isolation of a single crystal of this material from a reaction mixture which was characterised by X-ray crystallography (Fig. 1). In contrast, reaction of 4-trifluoromethylphenyl isocyanate with one equivalent of pinBH provided only evidence of the boraformamide and methylaniline products in a ∼6:1 ratio. Addition of 3 equivalents of isocyanate provided a 99% conversion to the boraformamide and 4-F3C-C6H4N(Bpin)CH2OBpin in a ∼11:1 ratio. Heating of this reaction mixture to 80 °C for 16 hours provided 85% conversion to N-methyl-N-(pinacolbora)-4-trifluoromethylaniline.
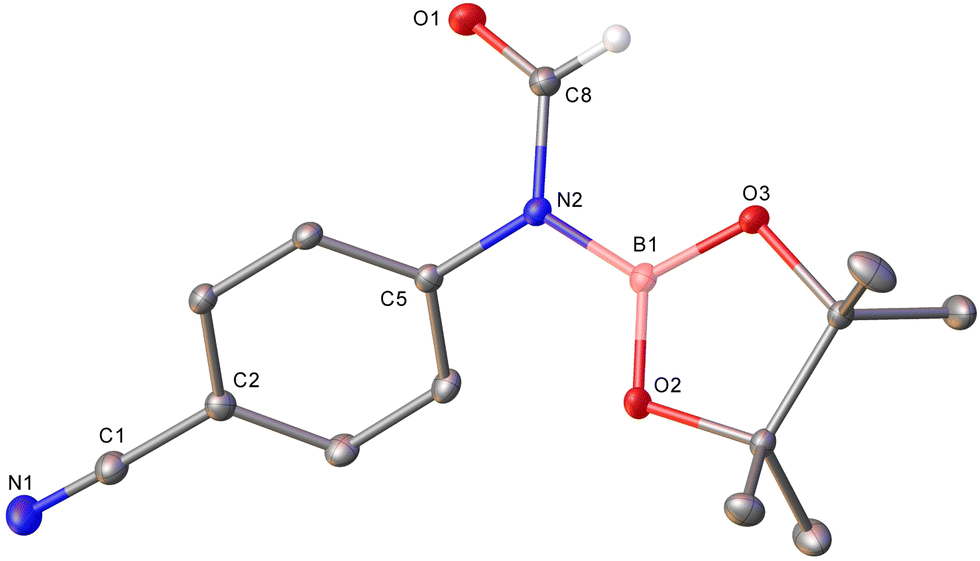 | ||
| Fig. 1 Molecular structure (30% probability ellipsoids) of 4-NC-C6H4N(Bpin)C(O)H (1h). Hydrogen atoms except on C8 are omitted for clarity. For metric parameters see ESI.† | ||
Copper(I)-catalysed hydroboration of other unsaturated compounds in the literature generally occurs via the copper(I) hydride, with insertion of the unsaturated moiety into the Cu–H bond.24 A similar mechanism was expected here, and stoichiometric reactions were used to garner evidence for this. The 6-Dipp ligand was used to ensure stability of the hydride intermediate.25 Thus one equivalent of pinBH was added to a solution of (6-Dipp)CuOtBu to give a bright orange solution, characteristic of the copper(I) hydride. To this was added one equivalent of isopropyl isocyanate. 1H and 11B NMR spectroscopy revealed the formation of a single new 6-Dipp-containing species and pinBOtBu, and crystalline material precipitated from the reaction mixture. Recrystallisation of this material yielded colourless crystals suitable for XRD (Fig. 2). This confirmed the new complex to be (6-Dipp)CuN(iPr)C(O)H, resulting from the insertion of the isocyanate into the Cu–H bond. Addition of pinacol borane to this compound reformed (6-Dipp)CuH with concomitant elimination of pinBN(iPr)C(O)H.
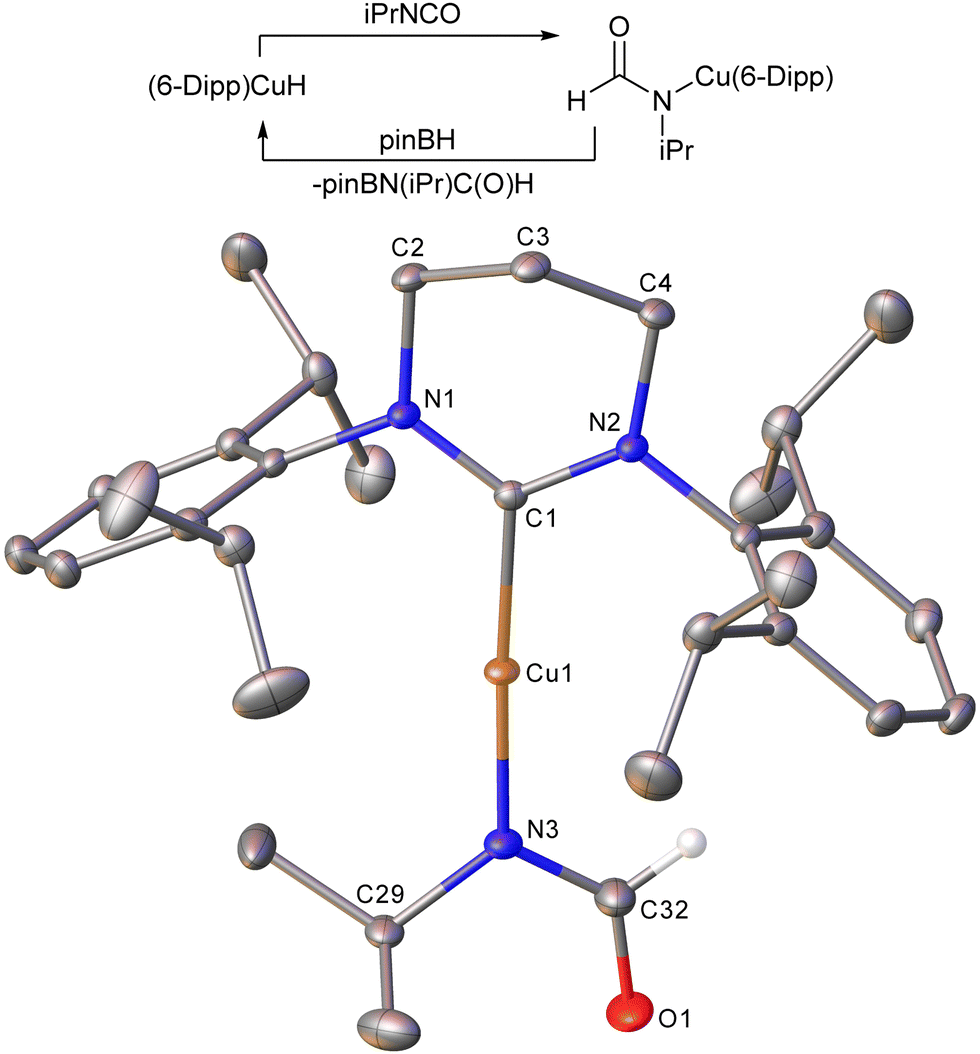 | ||
| Fig. 2 Scheme depicting the reactivity and molecular structure (30% probability ellipsoids) of (6-Dipp)CuN(iPr)C(O)H. Hydrogen atoms except on C32 and solvent molecules are omitted for clarity. For metric parameters see ESI.† | ||
With this insertion step experimentally validated, we turned our attention to understanding the specific mono-hydroboration observed with most isocyanates. In order to gain insight into the energetic parameters of this reaction, we undertook a DFT study at the BP86-D3BJ/BS2(benzene)//BP86/BS1 level of theory to investigate the proposed hydroboration pathway (Scheme 2). We initially focussed on the resting state of the copper catalyst.
 | ||
| Scheme 2 Computed pathways (free energy, kcal mol−1) for the hydroboration of iPrNCO (black/red) and 4-NC-C6H4-NCO (blue) at the BP86-D3BJ/BS2(benzene)//BP86/BS1 level of theory. For full computational details see ESI.† | ||
Dimeric [(IPr)CuH]2 was found to be −14.8 kcal mol−1 lower in energy than the monomer, (IPr)CuH, and conversion was associated with a low barrier (TS[(IPr)CuH]2 9.4 kcal mol−1). In contrast, the bridged borohydride [(IPr)CuH2Bpin] was found to be slightly higher in energy (2.2 kcal mol−1). Computational investigation of the interaction between either of these species and the isocyanate did not lead to a viable hydrocupration pathway.
We thus turned our attention to reactivity predicated on monomeric (IPr)CuH, which has been widely implicated in catalysis, but with [(IPr)CuH]2 proposed as the resting state. As shown, the stepwise formation of the hydride monomer, (IPr)CuH, and subsequent insertion of the isocyanate proceeds with a high but accessible barrier of 28.5 kcal mol−1 (TS1) relative to [(IPr)CuH]2. This step is exergonic by −9.9 kcal mol−1 towards Int1 (−24.7 kcal mol−1) and is likely to be essentially irreversible. σ-bond metathesis then proceeds with a barrier of 15.4 kcal mol−1 (TS2), associated but is slightly endergonic by 2.9 kcal mol−1 on forming the boraformamide product, 1a, and reforming (IPr)CuH (−21.8 kcal mol−1). Dimerisation of the hydride to reform [(IPr)CuH]2 (−36.6 kcal mol−1), however, provides an energy decrease overall to these steps of −11.9 kcal mol−1. For the reaction to proceed to deoxygenation, as observed with most systems, 1a could be expected to be further hydrocuprated to a borylated aminolate, Int2, which could then proceed in a stepwise fashion to an N-methylamine with net elimination of a borate ester. Investigation of this step from the [(IPr)CuH]2 resting state indicated that its barrier is 33.5 kcal mol−1 (TS3), larger than that of the first insertion and σ-bond metathesis steps suggesting this step is kinetically less accessible especially as this reaction is performed at room temperature. This insertion product, Int2, is also higher in energy than the resting state and 1a by 11.2 kcal mol−1 We thus posit that the source of selectivity in this reaction is that the first step of overreduction is both kinetically disfavoured and significantly higher in energy than dimerization of the copper(I) hydride in the presence of 1a. The copper(I) hydride dimer is also well-established to show limited stability, converting to free IPr, copper metal and hydrogen.20
With a model for the selective mono-hydroboration proposed we turned our attention to the cases where overreduction was observed, calculating a comparative pathway for 4-cyano-phenyl isocyanate. The initial reduction steps show a generally similar pattern to the isopropyl isocyanate. An accessible and stabilising initial hydrocupration is followed by a slightly uphill Cu–N/B–H σ-bond metathesis except when dimerization is considered. The second hydrocupration shows a significant deviation. In contrast to the isopropyl case, where the calculated barrier was 33.5 kcal mol−1 (TS2′), for the 4-cyanophenyl isocyanate, this second hydrocupration is only associated with an overall barrier of 26.3 kcal mol−1 (TS3′) relative to [(IPr)CuH]2, much more accessible and commensurate with that for the first hydrocupration. Moreover, the resultant borylated aminolate, Int2′, shows similar stability to 1h and [(IPr)CuH]2 (energy difference: 0.4 kcal mol−1). This ensures that the second hydroboration competes with dimer formation and consequent decomposition and contributes to the reduced selectivity observed for electron-poor arenes.
In conclusion, we have described a highly selective hydroboration system; the action of pinacol borane on a range, of isocyanates gives, in most cases, only the boraformamide with (IPr)CuOtBu as a catalyst. A computational study implied that the origin of the selectivity is the barrier height to boracupration of the boraformamide, especially in competition with dimerization of the copper(I) hydride to form [(IPr)CuH]2. In the presence of electron rich isocyanates, the formation of this species was suggested to kinetically disfavour over-reduction thus producing the observed selectivity. In future, carefully designed reaction systems might leverage oligomerisation as a source of selectivity based on this work.
DJL thanks the Royal Society for the support of a University Research Fellowship. We wish to thank MC2 for use of their analysis facilities, and the Anatra High Throughput Computing (HTC) Cluster at the University of Bath. We would like to thank the EPSRC for funding (EP/L016354/1) for a PhD studentship for LEE.
Conflicts of interest
There are no conflicts to declare.References
- D. Mukherjee, S. Shirase, T. P. Spaniol, K. Mashima and J. Okuda, Chem. Commun., 2016, 52, 13155–13158 RSC.
- Y. Yang, M. D. Anker, J. Fang, M. F. Mahon, L. Maron, C. Weetman and M. S. Hill, Chem. Sci., 2017, 8, 3529–3537 RSC.
- M. Magre, M. Szewczyk and M. Rueping, Org. Lett., 2020, 22, 3209–3214 CrossRef PubMed.
- M. Tan and Y. Zhang, Tetrahedron Lett., 2009, 50, 4912–4915 CrossRef.
- K. Turnbull and D. M. Krein, Synthesis, 1999, 391–392 CrossRef.
- J. S. Cha and H. C. Brown, J. Org. Chem., 1993, 58, 3974–3979 CrossRef.
- H. C. Brown, P. M. Weissman and N. M. Yoon, J. Am. Chem. Soc., 1966, 88, 1458–1463 CrossRef.
- A. E. Finholt, C. D. Anderson and C. L. Agre, J. Org. Chem., 1953, 18, 1338–1340 CrossRef.
- V. Pace, K. de la Vega-Hernández, E. Urban and T. Langer, Org. Lett., 2016, 18, 2750–2753 CrossRef PubMed.
- I. Ojima, S. Inaba and Y. Nagai, Tetrahedron Lett., 1973, 14, 4363–4366 CrossRef.
- H. G. Howell, Synth. Commun., 1983, 13, 635–637 CrossRef.
- I. Ojima and S.-i Inaba, J. Organomet. Chem., 1977, 140, 97–111 CrossRef.
- R. K. Sahoo, N. Sarkar and S. Nembenna, Angew. Chem., Int. Ed., 2021, 60, 11991–12000 CrossRef PubMed.
- Z. Du, B. Behera, A. Kumar and Y. Ding, J. Organomet. Chem., 2021, 950, 121982 CrossRef.
- C. D. Huke and D. L. Kays, in Advances in Organometallic Chemistry, ed. P. J. Pérez, Academic Press, 2021, vol. 75, pp. 1–54 Search PubMed.
- S. A. Bezman, M. R. Churchill, J. A. Osborn and J. Wormald, J. Am. Chem. Soc., 1971, 93, 2063–2065 CrossRef.
- D. M. Brestensky, D. E. Huseland, C. McGettigan and J. M. Stryker, Tetrahedron Lett., 1988, 29, 3749–3752 CrossRef.
- W. S. Mahoney, D. M. Brestensky and J. M. Stryker, J. Am. Chem. Soc., 1988, 110, 291–293 CrossRef.
- C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916–2927 CrossRef PubMed.
- N. P. Mankad, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 3369–3371 CrossRef.
- T. M. Horsley Downie, M. F. Mahon, J. P. Lowe, R. M. Bailey and D. J. Liptrot, ACS Catal., 2022, 12, 8214–8219 CrossRef.
- V. Pace, S. Monticelli, K. de la Vega-Hernández and L. Castoldi, Org. Biomol. Chem., 2016, 14, 7848–7854 RSC.
- M. Fischer and M. Schmidtmann, Chem. Commun., 2020, 56, 6205–6208 RSC.
- R. Shintani and K. Nozaki, Organometallics, 2013, 32, 2459–2462 CrossRef CAS.
- A. J. Jordan, C. M. Wyss, J. Bacsa and J. P. Sadighi, Organometallics, 2016, 35, 613–616 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full characterisation data, computational data, details of X-ray structures. CCDC 2195333 and 2195334. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04742j |
| This journal is © The Royal Society of Chemistry 2023 |