
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reduction chemistry yields stable and soluble divalent lanthanide tris(pyrazolyl)borate complexes†
Tajrian
Chowdhury
 a,
Matthew J.
Evans
b,
Martyn P.
Coles
b,
Anna G.
Bailey
a,
William J.
Peveler
a,
Claire
Wilson
a and
Joy H.
Farnaby
*a
a,
Matthew J.
Evans
b,
Martyn P.
Coles
b,
Anna G.
Bailey
a,
William J.
Peveler
a,
Claire
Wilson
a and
Joy H.
Farnaby
*a
aSchool of Chemistry, Joseph Black Building, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: Joy.Farnaby@glasgow.ac.uk
bSchool of Chemical and Physical Sciences, Victoria University of Wellington, PO Box 600, Wellington 6140, New Zealand
First published on 30th January 2023
Abstract
Reduction of the heteroleptic Ln(III) precursors [Ln(Tp)2(OTf)] (Tp = hydrotris(1-pyrazolyl)borate; OTf = triflate) with either an aluminyl(I) anion or KC8 yielded the adduct-free homoleptic Ln(II) complexes dimeric 1-Eu [{Eu(Tp)(μ-κ1:η5-Tp)}2] and monomeric 1-Yb [Yb(Tp)2]. Complexes 1-Ln have good solubility and stability in both non-coordinating and coordinating solvents. Reaction of 1-Ln with 2 Ph3PO yielded 1-Ln(OPPh3)2. All complexes are intensely coloured and 1-Eu is photoluminescent. The electronic absorption data show the 4f–5d electronic transitions in Ln(II). Single-crystal X-ray diffraction data reveal first μ-κ1:η5-coordination mode of the unsubstituted Tp ligand to lanthanides in 1-Eu.
Divalent lanthanide Ln(II) ions have remarkable physical and chemical properties.1 Complexes of Ln(II) are electron-transfer reagents in important synthetic transformations.1a,2 The physical properties of Ln(II) include single-molecule magnetism,3 and unique optical properties.4 Judicious choice of ancillary ligand is essential to supporting Ln(II) ions.5
The tridentate nitrogen donor scorpionate ligand hydrotris(1-pyrazolyl)borate (Tp) ligand has been shown to support Ln(II) in homoleptic [Ln(TpR)2] complexes where TpR = 3 and/or 5-substituted-pyrazolyl.6 These [Ln(TpR)2] complexes display a wide range of chemical reactivity,7 and are also useful synthons for organometallic Ln complexes,8 and heterometallic Ln and transition metal complexes.9 The [Ln(TpiPr2)2] (Ln = Eu, Yb) complexes exhibit properties intermediate between Ln nitrides and metallocenes in light-emitting diodes (LEDs).10 The first molecular Eu(II)-based electroluminescent thin film device was synthesised using [Eu(Tp3,5-R)2] (R = Me).11 Recently the potential of [Eu(Tp3-R)2] (R = Me, CF3) complexes have been demonstrated in organic LEDs (OLEDs).6h
In contrast to CpR Ln(II) chemistry (CpR = substituted cyclopentadienyl), the synthetic routes to [Ln(TpR)2] complexes, with one exception,6d have always utilised Ln(II) precursors.6,7 Lanthanide chemistry of the unsubstituted Tp ligand has remained largely unexplored. Moreover the THF-adduct complexes [Ln(Tp)2(THF)2] were reported to be unstable in solution.6b,12 Here we report the reduction chemistry of the heteroleptic Ln(III) precursors [Ln(Tp)2(OTf)],13 to synthesise stable adduct-free homoleptic Ln(II) complexes [Ln(Tp)2] 1-Ln (Ln = Eu, Yb), and Ph3PO adduct complexes 1-Ln(OPPh3)2.
The formal reduction potentials (E0) of Ln3+/Ln2+ for Ln = Eu −0.35 V and Yb −1.15 V in aqueous solution vs. the Normal Hydrogen Electrode (NHE) make them the most accessible Ln(III) candidates for reduction.14 Upon reduction of the parent Ln(III), the stability of these classical Ln(II) is achieved by either the attainment of half-filled 4f orbitals in Eu(II) or full 4f orbitals in Yb(II). It is of note that the redox potentials of Ln(III) depend on the ancillary ligand environment, for example [Ln(C5H4SiMe3)3] (Ln = Eu, Yb) were recently reported to be ca. 1 V more difficult to reduce than their formal reduction potentials.15 There are no data in the literature for the E0 of Ln3+/Ln2+ in a Tp ligand environment, but the reduction potentials are expected to be intermediate between the above examples. We therefore explored the reduction chemistry of our Ln(III) (Ln = Eu, Yb) precursors with various reductants.
Complex 1-Yb was first isolated by reduction of [Yb(Tp)2(OTf)] with [{K[Al(NONDipp)]}2]16 (NONDipp = {O(SiMe2NDipp)2}2−, Dipp = 2,6-iPr2C6H3, see ESI,† SIIa). This demonstrates the utility of aluminium(I)17 in lanthanide reduction chemistry. However, the reaction of [Y(Tp)2(OTf)] with [{K[Al(NONDipp)]}2] yielded the Y(III) ‘ate’-salt [Y(Tp)2(μ-OTf)2K(18-crown-6)] 2-Y (see ESI,† SV).
Our previous work on the metathesis chemistry of [Ln(Tp)2(OTf)]13 had shown that elimination of K(OTf) in non-coordinating solvents was most effective, and this is also the case for the reduction chemistry. Neither clean reduction nor K(OTf) elimination could be achieved in ethereal solvents (see Fig. S68 for the crystal structure of 1-Yb(DME) co-crystallised with KOTf, DME = 1,2-dimethoxyethane, ESI†). While metallic K does reduce [Ln(Tp)2(OTf)] the reaction conditions could not be adequately controlled to achieve clean formation of Ln(II). Therefore, the adduct-free divalent lanthanide complexes 1-Ln were synthesised by the reduction of [Ln(Tp)2(OTf)] with excess KC8 (Scheme 1) at ambient temperature in toluene with stirring, followed by filtration and subsequent removal of volatiles in vacuo (1-Eu [{Eu(Tp)(μ-κ1:η5-Tp)}2] 94%, 1-Yb [Yb(Tp)2] 87%). Complexes 1-Ln have good solubility and stability in both coordinating and non-coordinating solvents. Furthermore, reaction of 1-Ln with two equivalents of Ph3PO in toluene resulted in the isolation of the bis-adduct complexes 1-Ln(OPPh3)2 (1-Eu(OPPh3)2 91%, 1-Yb(OPPh3)2 87%). Elemental analyses of 1-Ln and 1-Ln(OPPh3)2 are consistent with their respective formulations.
 | ||
| Scheme 1 (a) Synthesis of [{Eu(Tp)(μ-κ1:η5-Tp)}2] 1-Eu and [Yb(Tp)2] 1-Yb complexes by reduction of the Ln(III) precursor complexes [Ln(Tp)2(OTf)]. (b) Synthesis of bis-adduct complexes [Ln(Tp)2(OPPh3)2] 1-Ln(OPPh3)2 (Ln = Eu, Yb) by reaction of two Ph3PO with 1-Ln. | ||
No resonances were observed by NMR spectroscopy for the paramagnetic complexes 1-Eu and 1-Eu(OPPh3)2, consistent with the half-filled 4f7 electronic configuration of Eu(II). The corrected Evans’ method magnetic moments (μeff) for 1-Eu and 1-Eu(OPPh3)2 in d6-benzene at room temperature were found to be in the ranges of 7.62–7.78 μB and 7.19–7.46 μB, respectively. These data are consistent with the calculated spin-only magnetic moment (μso) of 7.94 μB for 4f7 Gd(III),18 and Eu(II) literature examples, for both organometallics,19 and coordination compounds.20 Complexes 1-Yb and 1-Yb(OPPh3)2 are full-shell 4f14 and diamagnetic as expected, with NMR data similar to [Yb(TpR)2] complexes.6b–d,f In d6-benzene the Tp-pyrazolyl protons in 1-Yb are observed by 1H NMR spectroscopy at δ = 5.97, 7.42 and 7.67 ppm and the Tp-borohydride at δ = 4.96 ppm, in the expected 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:6:2 ratio. The Tp-pyrazolyl protons are observed at very similar chemical shifts in the 1H NMR of 1-Yb(OPPh3)2.
:6:6:2 ratio. The Tp-pyrazolyl protons are observed at very similar chemical shifts in the 1H NMR of 1-Yb(OPPh3)2.
Diffusion-ordered NMR spectroscopy (1H 2D-DOSY) of 1-Yb, and 1-Yb with 1 or 2 equivalents of Ph3PO show that 1-Yb is monomeric, and that in solution there is an equilibrium between Ph3PO and 1-Yb(OPPh3)x (see ESI,† S1.6). The resonances assigned to 1-Yb(OPPh3)2 in the 1H and 31P NMR data are therefore an equilibrium average. The phenyl protons of Ph3PO, appear at δ = 6.92, 7.02 and 7.49 ppm in the expected 12:6:12 ratio for two equivalents of Ph3PO, slightly shifted from free Ph3PO (δ = 6.97–7.08 and 7.72–7.82 ppm in a 9:6 ratio). By 31P NMR the singlet resonance at δ = 27.63 ppm assigned to 1-Yb(OPPh3)2, is shifted from free Ph3PO (δ = 24.72 ppm). The Tp-borohydrides of both 1-Yb and 1-Yb(OPPh3)2 were observed as doublet resonances at δ = −1.85 ppm by 11B NMR. The pyrazolyl carbon resonances of 1-Yb and 1-Yb(OPPh3)2 were assigned via 2D 1H–13C HSQC NMR experiments (see ESI†). The IR spectra of 1-Ln are near-identical, with weak absorptions between 2350–2560 cm−1 assigned to the νBH of the Tp ligands.6d,f,13 The IR data for 1-Ln(OPPh3)2 are consistent with 1-Ln but additionally display strong νP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1117 cm−1. The νPO is shifted from 1184 cm−1 in free Ph3PO to lower wavenumbers upon coordination to the Ln(II).21
O at 1117 cm−1. The νPO is shifted from 1184 cm−1 in free Ph3PO to lower wavenumbers upon coordination to the Ln(II).21
Complexes 1-Ln and 1-Ln(OPPh3)2 are intensely coloured due to Laporte allowed 4fn−1–5d1 transitions characteristic of Ln(II).22 The electronic absorption spectra of 1-Ln in both non-coordinating (a) and coordinating (b) solvents, and 1-Ln(OPPh3)2 in MeCN (c) are shown in Fig. 1. In toluene (Fig. 1a) or hexane (Fig. S53 and S54 in ESI†) complexes 1-Ln display broad and strong absorptions (Ln = Eu ε = 1.45–3.67 × 103 M−1 cm−1; Ln = Yb ε = 0.68–1.93 × 103 M−1 cm−1) in the near UV and visible, with λmax of 395 nm for 1-Eu, and λmax of 341 nm and 520 nm for 1-Yb.
 | ||
| Fig. 1 Overlay of the electronic absorption spectra of 1-Ln in either toluene (a) or in MeCN (b) and overlay of the electronic absorption spectra of 1-Ln(OPPh3)2 (c) in MeCN, all spectra recorded at room temperature. The traces are coloured in the colour of the complex. Data (λmax and ε) are tabulated in ESI,† Table S3. Photoluminescence of a solid sample of 1-Eu under a UV lamp (d). Excitation (LHS, λEm = 590 nm) and emission (RHS, λEx = 389 nm) spectra of 1-Eu, recorded in toluene (e). | ||
Photoluminescence (PL) of 1-Eu is also shown in Fig. 1, in the solid-state under a UV lamp (d) and the excitation and emission spectra in toluene solution (e). Complex 1-Eu shows emission at 590 nm (λEx of 389 nm), and the Excitation-Emission Matrix data (EEM, Fig. S61, ESI†) demonstrate that the emission of 1-Eu originates only from the parity-allowed 4f65d1 to 4f7 transition of Eu(II). The PL data for 1-Eu are very similar to those reported for [Eu(Tp3-CH3)2]6h and consistent with other [Eu(TpR)2] examples.6f–h,10 The λmax and λEm values in [Eu(TpR)2] complexes correlate well to the relative donor-strength of the Tp ligand. The energy of the excitation and emission from molecular Ln(II) complexes in solution follows the spectrochemical series. The energy gap between the 4f and the 5d reduces in magnitude as the interaction with the ancillary ligands increases O < N < C, for example the data for [Ln(CpR)2] are significantly red-shifted from [Ln(TpR)2].22a In acetonitrile, the transition envelopes for 1-Ln (Fig. 1b) are in very similar spectral positions. In 1-Yb(MeCN) there is also no change in magnitude of molar extinction co-efficient, however, in 1-Eu(MeCN)ε decreases significantly. The increased solvent cut-off in MeCN allows for the observation of additional absorptions in the UV for 1-Ln. No spectral features in these data originate from the Tp ligand. The data for 1-Ln(OPPh3)2 in acetonitrile are near-identical to 1-Ln, with the addition of the π–π* transitions arising of Ph3PO (λmax = 265–272, ε = 2.6–5.3 × 103 M−1 cm−1).23
The solid-state molecular structures of 1-Eu (a), 1-Yb (b), 1-Yb(THF) (c), and 1-Eu(OPPh3)2 (d) are shown in Fig. 2. The structures of 1-Eu(THF)2 are shown in Fig. S63 and S64 and 1-Yb(OPPh3) in Fig. S69 (ESI†). Important structural metrics are tabulated in the ESI† (see S4), data comparison in Table S4 and crystallographic information in Table S5. The Ln–N(κ3-Tp),6a,d,f,10 Eu–(η5-Tp),24 Ln-O(THF),1a,b and Ln–O(OPPh3)21c,23 bond distances in 1-Ln, 1-Ln(THF)x and 1-Ln(OPPh3)x (Ln = Eu, x = 2; Yb, x = 1) fall within the expected ranges. All data are consistent with the lanthanide contraction25 and the increase in ionic radius upon reduction from Ln(III) to Ln(II).13 In the absence of coordinating solvents, complex 1-Eu (a) is an unusual example of a dimeric structure, whereas complex 1-Yb (b) is monomeric as expected.6a,c,d,f In 1-Eu (a) each Eu(II) is bound by a κ3-Tp ligand and an μ-κ1:η5 Tp ligand. The μ-κ1:η5 binding mode has been seen in Eu(II) pyrazolyl complexes,24 but this is the first example of μ-κ1:η5 Tp binding with bridging of lanthanide metal centres.6f–h,d,10 In 1-Eu a significantly longer bond distance is observed for Eu–N(μ-κ1:η5-Tp). Either dissolution in THF of 1-Ln or the addition of 2 Ph3PO to 1-Ln in toluene, and recrystallisation resulted in single-crystals of the Lewis base adduct complexes 1-Ln(THF)x (c) or 1-Ln(OPPh3)x (d) (Ln = Eu, x = 2; Yb, x = 1). Complexes 1-Ln(THF)x and 1-Ln(OPPh3)x are all monomeric, with two axial κ3-coordinated Tp ligands bound to each Ln metal centre, and either one (Yb) or two (Eu) adduct molecules bound in the equatorial plane. In the single-crystal of 1-Yb(OPPh3) the binding of one Ph3PO ligand is attributed to the specific crystallisation conditions.
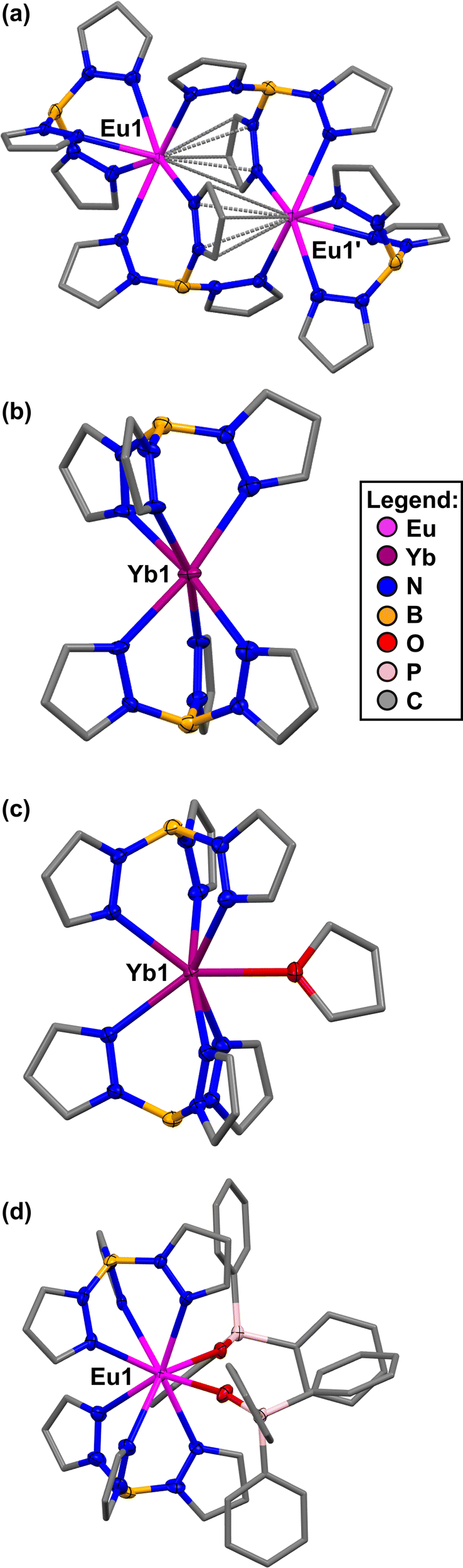 | ||
| Fig. 2 Crystal structures of 1-Eu (a), 1-Yb (b), 1-Yb(THF) (c), 1-Eu(OPPh3)2 (d). Hydrogen atoms and lattice solvent molecules omitted for clarity and pyrazolyl carbon atoms of Tp, backbone carbon atoms of THF, phenyl carbon atoms of Ph3PO displayed in wireframe. Displacement ellipsoids drawn at 50% probability. Data and crystallographic information can be found in the ESI,† S4 and Tables S4, S5. | ||
Reduction of Ln(III) in [Ln(Tp)2(OTf)] has been demonstrated to be an excellent route to Ln(II) in [Ln(Tp)2] (Ln = Eu, Yb). The binding strength in combination with ability of the unsubstituted Tp ligand to bridge, bend and flex, results in very stable and soluble Ln(II) complexes. Enabling in turn the collection of both solid-state and solution-state spectroscopic data. Highlights include the direct observation of the effect of solvent on 4f–5d electronic transitions in Ln(II), PL from Eu(II) and first example of μ-κ1:η5 Tp binding to lanthanides. Exploration of the chemistry of [Ln(Tp)2] is ongoing in our laboratory, and we anticipate future study of the chemical and physical properties of these Ln(II) complexes.
The authors acknowledge the University of Glasgow for funding and the EPSRC ECR Capital Award Scheme (EP/S017984/1). WJP acknowledges the Royal Society (RGS\R2\192190) for funding. The authors also acknowledge the awards of a College of Science and Engineering PhD Scholarship to TC, and a Victoria University of Wellington PhD Scholarship to MJE.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. C. Wedal and W. J. Evans, J. Am. Chem. Soc., 2021, 143, 18354–18367 CrossRef CAS PubMed; (b) H. M. Nicholas and D. P. Mills, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, John Wiley & Sons, Ltd, 2017, pp. 1–10 Search PubMed; (c) F. Nief, Dalton Trans., 2010, 39, 6589 RSC; (d) M. N. Bochkarev, Coord. Chem. Rev., 2004, 248, 835–851 CrossRef CAS.
- M. Szostak and D. J. Procter, Angew. Chem., Int. Ed., 2012, 51, 9238–9256 CrossRef CAS PubMed.
- W. Zhang, A. Muhtadi, N. Iwahara, L. Ungur and L. F. Chibotaru, Angew. Chem., Int. Ed., 2020, 59, 12720–12724 CrossRef CAS PubMed.
- (a) T. C. Jenks, A. N. W. Kuda-Wedagedara, M. D. Bailey, C. L. Ward and M. J. Allen, Inorg. Chem., 2020, 59, 2613–2620 CrossRef CAS PubMed; (b) J. Jiang, N. Higashiyama, K.-I. Machida and G.-Y. Adachi, Coord. Chem. Rev., 1998, 170, 1–29 CrossRef CAS; (c) R. M. Diaz-Rodriguez, D. A. Gálico, D. Chartrand, E. A. Suturina and M. Murugesu, J. Am. Chem. Soc., 2022, 144, 912–921 CrossRef CAS PubMed.
- S. Schulz, Chem. – Eur. J., 2010, 16, 6416–6428 CrossRef CAS PubMed.
- (a) G. H. Maunder, A. Sella and D. A. Tocher, J. Chem. Soc., Chem. Commun., 1994, 885–886 RSC; (b) Â. Domingos, J. Marçalo, N. Marques, A. P. D. Matos, A. Galvão, P. C. Isolani, G. Vicentini and K. Zinner, Polyhedron, 1995, 14, 3067–3076 CrossRef; (c) J. Takats, J. Alloys Compd., 1997, 249, 52–55 CrossRef CAS; (d) A. C. Hillier, Zhang, G. H. Maunder, S. Y. Liu, T. A. Eberspacher, M. V. Metz, R. McDonald, Â. Domingos, N. Marques, V. W. Day, A. Sella and J. Takats, Inorg. Chem., 2001, 40, 5106–5116 CrossRef CAS PubMed; (e) A. Momin, L. Carter, Y. Yang, R. McDonald, S. Essafi, F. Nief, I. Del Rosal, A. Sella, L. Maron and J. Takats, Inorg. Chem., 2014, 53, 12066–12075 CrossRef CAS PubMed; (f) M. Kühling, C. Wickleder, M. J. Ferguson, C. G. Hrib, R. McDonald, M. Suta, L. Hilfert, J. Takats and F. T. Edelmann, New J. Chem., 2015, 39, 7617–7625 RSC; (g) H. Qi, Z. Zhao, G. Zhan, B. Sun, W. Yan, C. Wang, L. Wang, Z. Liu, Z. Bian and C. Huang, Inorg. Chem. Front., 2020, 7, 4593–4599 RSC; (h) G. Zhan, L. Wang, Z. Zhao, P. Fang, Z. Bian and Z. Liu, Angew. Chem., Int. Ed., 2020, 59, 19011–19015 CrossRef CAS PubMed.
- (a) J. Takats, X. W. Zhang, V. W. Day and T. A. Eberspacher, Organometallics, 1993, 12, 4286–4288 CrossRef CAS; (b) X. Zhang, G. R. Loppnow, R. McDonald and J. Takats, J. Am. Chem. Soc., 1995, 117, 7828–7829 CrossRef CAS; (c) A. C. Hillier, S.-Y. Liu, A. Sella and M. R. J. Elsegood, Inorg. Chem., 2000, 39, 2635–2644 CrossRef CAS PubMed; (d) A. C. Hillier, A. Sella and M. R. J. Elsegood, J. Organomet. Chem., 2002, 664, 298–305 CrossRef CAS; (e) Â. Domingos, I. Lopes, J. C. Waerenborgh, N. Marques, G. Y. Lin, X. W. Zhang, J. Takats, R. McDonald, A. C. Hillier, A. Sella, M. R. J. Elsegood and V. W. Day, Inorg. Chem., 2007, 46, 9415–9424 CrossRef PubMed; (f) M. Kühling, R. McDonald, P. Liebing, L. Hilfert, M. J. Ferguson, J. Takats and F. T. Edelmann, Dalton Trans., 2016, 45, 10118–10121 RSC.
- (a) I. Lopes, G. Y. Lin, A. Domingos, R. McDonald, N. Marques and J. Takats, J. Am. Chem. Soc., 1999, 121, 8110–8111 CrossRef CAS; (b) G. Lin, R. McDonald and J. Takats, Organometallics, 2000, 19, 1814–1816 CrossRef CAS.
- (a) A. C. Hillier, S. Y. Liu, A. Sella, O. Zekria and M. R. J. Elsegood, J. Organomet. Chem., 1997, 528, 209–215 CrossRef CAS; (b) A. C. Hillier, A. Sella and M. R. J. Elsegood, J. Organomet. Chem., 1999, 588, 200–204 CrossRef CAS.
- M. Suta, M. Kühling, P. Liebing, F. T. Edelmann and C. Wickleder, J. Lumin., 2017, 187, 62–68 CrossRef CAS.
- C. P. Shipley, S. Capecchi, O. V. Salata, M. Etchells, P. J. Dobson and V. Christou, Adv. Mater., 1999, 11, 533–536 CrossRef CAS.
- M. A. J. Moss, R. A. Kresinski, C. J. Jones and W. J. Evans, Polyhedron, 1993, 12, 1953–1955 CrossRef CAS.
- T. Chowdhury, S. J. Horsewill, C. Wilson and J. H. Farnaby, Aust. J. Chem., 2022, 75, 660–675 CrossRef CAS.
- (a) L. J. Nugent, R. D. Baybarz, J. L. Burnett and J. L. Ryan, J. Phys. Chem., 1973, 77, 1528–1539 CrossRef CAS; (b) L. R. Morss, Chem. Rev., 1976, 76, 827–841 CrossRef CAS.
- M. T. Trinh, J. C. Wedal and W. J. Evans, Dalton Trans., 2021, 50, 14384–14389 RSC.
- (a) R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed; (b) M. D. Anker and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 18261–18265 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- (a) T. D. Tilley, R. A. Andersen, B. Spencer, H. Ruben, A. Zalkin and D. H. Templeton, Inorg. Chem., 1980, 19, 2999–3003 CrossRef CAS; (b) M. H. Kuiper and H. Lueken, Z. Anorg. Allgem. Chem., 2007, 633, 1407–1409 CrossRef CAS.
- J. Garcia and M. J. Allen, Eur. J. Inorg. Chem., 2012, 4550–4563 CrossRef CAS PubMed.
- (a) F. A. Cotton, R. D. Barnes and E. Bannister, J. Chem. Soc., 1960, 2199–2203 RSC; (b) A. G. Matveeva, A. V. Vologzhanina, E. I. Goryunov, R. R. Aysin, M. P. Pasechnik, S. V. Matveev, I. A. Godovikov, A. M. Safiulina and V. K. Brel, Dalton Trans., 2016, 45, 5162–5179 RSC; (c) R. D. Bannister, W. Levason, M. E. Light and G. Reid, Polyhedron, 2018, 154, 259–262 CrossRef CAS; (d) R. D. Bannister, W. Levason and G. Reid, Chemistry, 2020, 2, 947–959 CrossRef CAS; (e) G. B. Deacon, G. D. Fallon, C. M. Forsyth, B. M. Gatehouse, P. C. Junk, A. Philosof and P. A. White, J. Organomet. Chem., 1998, 565, 201–210 CrossRef CAS.
- (a) T. C. Jenks and M. J. Allen, in Modern Applications of Lanthanide Luminescence, ed. A. de Bettencourt-Dias, Springer International Publishing, Cham, 2021, pp. 67–92 Search PubMed; (b) M. Suta and C. Wickleder, J. Lumin., 2019, 210, 210–238 CrossRef CAS.
- A. W. G. Platt, Coord. Chem. Rev., 2017, 340, 62–78 CrossRef CAS.
- (a) C. C. Quitmann, V. Bezugly, F. R. Wagner and K. Müller-Buschbaum, Z. Anorg. Allgem. Chem., 2006, 632, 1173–1186 CrossRef CAS; (b) J. Hitzbleck, G. B. Deacon and K. Ruhlandt-Senge, Eur. J. Inorg. Chem., 2007, 592–601 CrossRef CAS.
- (a) R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef; (b) R. E. Cramer, J. M. Rimsza and T. J. Boyle, Inorg. Chem., 2022, 61, 6120–6127 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2161063–2161071 and 2195976. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc03189b |
| This journal is © The Royal Society of Chemistry 2023 |