
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight into the structures of unusual base pairs in RNA complexes containing a primer/template/adenosine ligand†
Yuliya
Dantsu
 ,
Ying
Zhang
and
Wen
Zhang
*
,
Ying
Zhang
and
Wen
Zhang
*
Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, IN 46202, USA. E-mail: wz15@iu.edu
First published on 30th August 2023
Abstract
In the prebiotic RNA world, the self-replication of RNA without enzymes can be achieved through the utilization of 2-aminoimidazole activated nucleotides as efficient substrates. The mechanism of RNA nonenzymatic polymerization has been extensively investigated biophysically and structurally by using the model of an RNA primer/template complex which is bound by the imidazolium-bridged or triphosphate-bridged diguanosine intermediate. However, beyond the realm of the guanosine substrate, the structural insight into how alternative activated nucleotides bind and interact with the RNA primer/template complex remains unexplored, which is important for understanding the low reactivity of adenosine and uridine substrates in RNA primer extension, as well as its relationship with the structures. Here we use crystallography as a method and determine a series of high-resolution structures of RNA primer/template complexes bound by ApppG, a close analog of the dinucleotide intermediate containing adenosine and guanosine. The structures show that ApppG ligands bind to the RNA template through both Watson–Crick and noncanonical base pairs, with the primer 3′-OH group far from the adjacent phosphorus atom of the incoming substrate. The structures indicate that when adenosine is included in the imidazolium-bridged intermediate, the complexes are likely preorganized in a suboptimal conformation, making it difficult for the primer to in-line attack the substrate. Moreover, by co-crystallizing the RNA primer/template with chemically activated adenosine and guanosine monomers, we successfully observe the slow formation of the imidazolium-bridged intermediate (Ap-AI-pG) and the preorganized structure for RNA primer extension. Overall, our studies offer a structural explanation for the slow rate of RNA primer extension when using adenosine-5′-phosphoro-2-aminoimidazolide as a substrate during nonenzymatic polymerization.
Introduction
According to the RNA world hypothesis proposed by Rich,1 Crick,2 Orgel3 and Woese,4 RNA was the essential molecule to construct the first cell in the prebiotic world with the functions of storing genetic information and catalyzing biochemical reactions.5,6 The idea of the RNA world hypothesis rationalizes how life began in its early stages, including the transition from simple molecules into basic RNA, and the later evolvement into complex ribozymes to catalyze primitive metabolism. For the emergence of RNA-based life, oligomerized RNA strands had to be replicated without enzymes.5,7,8 For RNA's self-replication, the building blocks or short chains need to be chemically activated, bind to the complementary template, arrange themselves in a specific conformation and undergo a polymerization reaction to produce a new copy of RNA.9 Considerable efforts have been dedicated over the years to the optimization of RNA nonenzymatic polymerization, like enhancing the fidelity and efficiency of the reaction, exploring alternative chemical activations, searching for effective catalysts, and augmenting the rate of polymerization.10 One successful example is the discovery of derivatized-imidazole-activated ribonucleotides as reaction substrates, including nucleoside-5′-phosphoro-(2-aminoimidazolide)11,12 and nucleoside-5′-phosphoro-(2-methylimidazolide),13 which can mediate the fast copying of RNA sequences in the presence of a RNA primer, template, and divalent metal ion.11,14 The thermodynamics, kinetics and reaction mechanism of primer extension using this superior substrate have been extensively explored.15–18 The nonenzymatic polymerization reaction is purely chemically catalyzed, and it requires the formation of a 5′–5′ phospho-imidazolium-phospho bridged dinucleotide intermediate.19 The dinucleotide intermediate binds the RNA template with great affinity and well-defined conformation, including multiple inter- and intramolecular hydrogen bonds to preorganize the complex.20 Consequently, the RNA primer extension proceeds stepwise with the 3′ hydroxyl group of the adjacent primer orientated to in-line attack the incoming phosphorus atom of the imidazolium-bridge and replace an activated nucleotide as the leaving group of the nucleophilic reaction.19To ensure the propagation of primitive life, the accurate transfer of complete genetic information is critical.7,8 Enzyme-free RNA replication will require the efficient incorporation of all four nucleotides and the replication of RNA strands containing mixed sequences. Generally, the error rate in nonenzymatic copying could easily lead to a standstill of primer extension and the truncated information that is passed on from generation to generation.21 The incorporation of G and C nucleotides occurs with a higher rate and fidelity than A and U monomers.22–25 The stalling effect arising from A and U monomers is possibly due to the weak binding of adenosine and uridine-containing substrates (including monomers or dinucleotide intermediates) to the template, or the local geometry of RNA primer/template/A or U substrate complexes, which is less favorable for the nucleophilic attack of the 3′-OH group of the RNA primer. Recently, the frequency of mismatches in RNA nonenzymatic polymerization and its relationship with the dinucleotide intermediate mechanism has been investigated,26 and the method of using deep-sequencing to identify mismatches in primer extension is developed.27 Overall, a more in-depth understanding of how adenosine or uridine substrates affect nonenzymatic polymerization is important and demands further exploration. Therefore, we decided to investigate the structural insights into the nonenzymatic primer extension when there is adenosine substrate binding to the RNA template and forming a primer/template complex, asking if there are nonconventional binding motifs existing to affect the primer extension.
To understand how nucleotide substrates bind to the RNA template and get preorganized for primer extension, many high-resolution crystal structures have been determined using stable reaction substrate analogs, including the guanosine-5′-phosphoro-pyrazole monomer28 and the symmetrical 5′–5′ triphosphate-linked dinucleotide GpppG.29 These studies indicate that the guanosine monomers bind the template through various modes of base pairing.30 In contrast, as the molecular analog of the imidazolium-bridged intermediate, GpppG was observed to bind to the RNA template only through Watson–Crick base-pairs, with the 3′-hydroxyl of the primer positioned for nucleophilic attack.29 Additionally, the time-resolved crystal structures also provided the atomic resolution insight into the binding motif of the real activated substrate and the mechanism of nonenzymatic primer extension.20 These structural observations suggest that Watson–Crick base pairing between the substrate and the template results in a favorable preorganized geometry and high reactivity. Here we use a similar approach of crystallography and report the structural insight into the mechanism of nucleotide substrate binding and interacting with RNA when adenosine is present.
Results and discussion
Primer extension assays with the adenosine-5′-phosphoro-(2-aminoimidazolide) (2-AIpA) substrate
We first ask whether the RNA primer can be nonenzymatically extended when the chemically activated adenosine monomer serves as the reaction substrate in solution (Fig. 1A). We synthesize an RNA primer containing Cy3 fluorophore at its 5′ end (5′-Cy3-GUAGACUGACUGA-3′, Fig. 1B), as well as different templates that contain uridine or cytidine residues at the template to bind to 2-AIpA and 2-AIpG monomers. We then examine rates of RNA-templated primer extension by one nucleotide, when there are different activated nucleotides bound and incorporated at +1 and +2 positions. In the group of RNA primer/template systems containing 3′-CC as a template and 2-AIpG as a substrate, primer extension proceeded quickly as expected, with an observed rate of 0.7 h−1. However, when the mixture of 2-AIpA and 2-AIpG was used as a substrate in solution, with the 3′-UC or 3′-CU as a template, the observed rates were dramatically reduced to 0.07 and 0.13 h−1, nearly 10-fold and 5-fold slower than the RNA extension with a 3′-CC template (Fig. 1C–E). In contrast, with the 3′-UU as a template and 2-AIpA as a substrate, the reaction became extremely slow (est. less than 0.01 h−1). Thus, our result suggests that once the A:U base pair is involved during primer extension, the reaction rate will significantly be diminished. With the exclusive presence of an A:U pair between the template and the substrate, the polymerization can hardly proceed to the end of the template.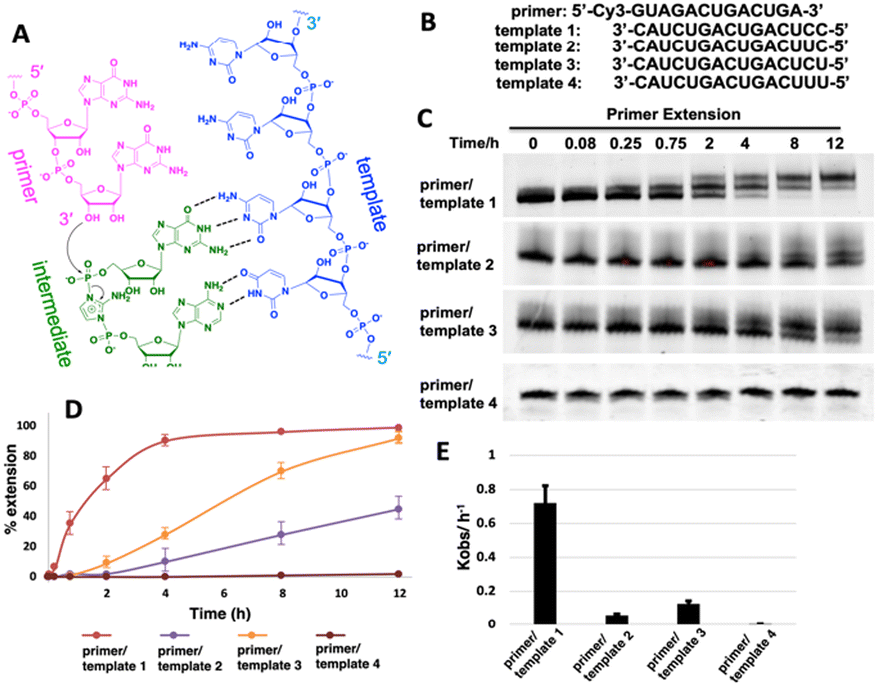 | ||
| Fig. 1 (A) Chemical representation of hypothetical template-directed polymerization with an RNA primer, RNA template and 2-aminoimidazole bridged intermediate containing adenosine and guanosine (Ap-AI-pG). (B) Sequences of the primer and templates used in the primer extension. (C) PAGE analysis of the reaction products over different times using different templates. The gels depicted are representative of triplicate experiments. (D) Comparison of primer extension with different templates. (E) Pseudo-first order rates of the reactions with different templates. | ||
It is also noteworthy that, in our primer extensions containing a mixture of 2-AIpA and 2-AIpG substrates, there could be three different dinucleotide intermediates, Ap-AI-pG, Gp-AI-pG and Ap-AI-pA. In contrast, in the reaction solution only containing 2-AIpG, there is only one possible dinucleotide intermediate of Gp-AI-pG. Therefore, the slower reaction rates in template 2 and template 3 are also possibly because of the lower concentration of correct reaction intermediate Ap-AI-pG. To explore the possibility, we performed one more control experiment of primer extension, using 3′-CC as a template and a mixture of 2-AIpG and 2-AIpC as substrates (Fig. S1, ESI†). We observed that, when 2-AIpC was added to the reaction, the reaction rate was dropped by ∼35% (0.7 h−1vs. 0.45 h−1). This reduced reaction rate could be explained by the lower concentration of Gp-AI-pG, due to the formation of Cp-AI-pG and Cp-AI-pC in solution as competitors. A similar phenomenon possibly exists in solutions containing 2-AIpG and 2-AIpA. However, the decreased concentration of the correct intermediate is unlikely the only reason for the dramatic drop in reaction rates we observed (10-fold and 5-fold difference). Therefore, we decided to explore other possibilities that causes the diminished primer extension.
Structures of the RNA primer–template complexes co-crystallized with AMP
We then decide to structurally explore how adenosine substrate binds to RNA template in nonenzymatic polymerization. We first cocrystallized AMP mononucleotide with self-complementary RNA oligonucleotide 5′-TTAGACUUAAGUCU-3′, which is flanked on both ends by two locked thymidine nucleotides serving as the binding sites for AMP (Fig. 2A). We first tried to crystallize the RNA/monomer complex, but no crystallization was observed. The locked nucleic acid (LNA) modification (denoted as bold and italic nucleotides) in current structures constrains the sugar into the 3′-endo conformation and helps the complex crystallization for high-resolution X-ray structures. The key crystallographic parameters are listed in Supporting Information. There is one RNA duplex/AMP complex per asymmetric unit. Like the structures determined previously,20 the individual double helices are slip-stacked with one another end-to-end. Groups of three RNA duplex-ligand complexes form triangular prisms, and the central channel accommodates water molecules to bridge the neighboring complexes (Fig. 2B).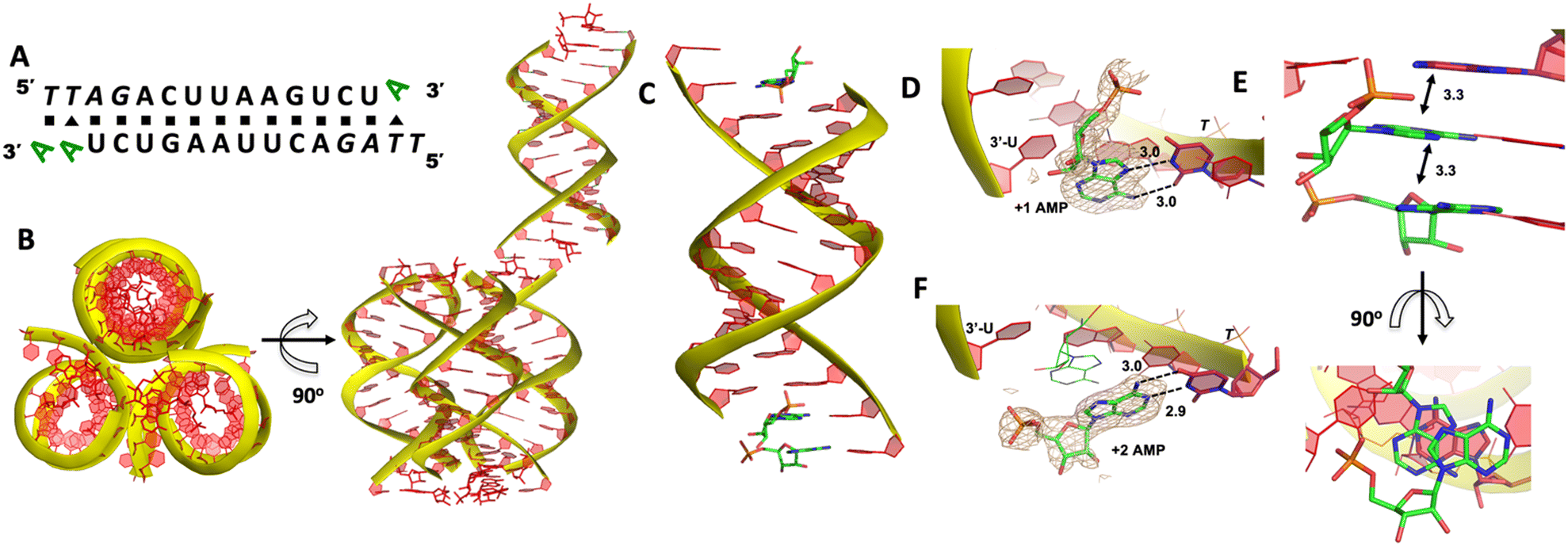 | ||
| Fig. 2 Crystal structure of the RNA/AMP complex. (A) Diagram and designed duplex structure of the RNA/AMP complex. AMP monomers (green) are bound at each end. Black square: Watson–Crick pairs. Black triangle: noncanonical base pairs. (B) The adjacent three RNA/AMP complexes assemble for crystal formation. The bound AMPs are stacked between two coaxial RNA duplexes. (C) Overall structure of the RNA/AMP duplex. (D) The local structure of AMP binding to the RNA template at the +1 position. The corresponding Fo − Fc omit map contoured at 1.5σ (wheat mesh) indicates the noncanonical base pair. (E) The bound AMP at +1 position is well stacked with AMP at the +2 position and upstream adenine. (F) The local structure of AMP binding to the RNA template at the +2 position, forming a Watson–Crick base pair. All the significant distances are labelled. | ||
At one end of the duplex, there are two AMP monomers bound to the templating locked thymidines consecutively (Fig. 2C). At the +1 position, a noncanonical A:T base pair was observed, mediated by two hydrogen bonds: the N7 of the adenine was 3.0 Å from the N3 of templating thymine, and the exocyclic amine of the AMP was 3.0 Å from the exocyclic oxygen atom of thymine (Fig. 2D). Interestingly, by forming this unique base pairing, the adenine nucleobase at the +1 position was sandwiched between the AMP monomer bound at the +2 position and the adenine nucleobase which is located at the templating strand and adjacent to the templating thymidine (Fig. 2E). The internucleobase distance between the two bound adenine bases is approximately 3.3 Å based on analysis using the CONTACT program from CCP4,31 and the distance between the adenine bound at +1 position and “upstream” adenine is also 3.3 Å. This binding motif of AMP is different from what was observed in the structures of template/primer/guanosine substrate complexes, and the distance between the 3′-OH group of primer and the phosphorus atom of bound AMP was over 10 Å. This local structure may explain the slow primer extension rate when using adenine monomer as substrate. At the +2 position, the AMP is forming a Watson–Crick base pair with the thymine nucleobase (hydrogen-bond lengths, 3.0 and 2.9 Å, Fig. 2F). Unfortunately, the sugar moieties of both bound AMP monomers are not ordered enough to predict the sugar conformation. At the other end of the duplex, only one AMP monomer was observed to bind the template at the +1 position via the noncanonical base pairing. The overall binding motif remains the same as the other +1 bound AMP.
To explore whether the noncanonical A:T pair widely exist when binding to RNA primer/template, we next cocrystallized another self-complementary RNA 5′-TmCmCGACUUAAGUCG-3′ with both AMP and GMP monomers to bind the two nucleotides overhang 5′-TmC at both ends (Fig. 3A, mC represents the locked 5-methyl cytidine residue). The structure was determined to 1.45 Å resolution by molecular replacement using the previously determined RNA duplex as a search model.20 As in the structures with similar sequences, the individual double helices are slip-stacked with one another end-to-end. At one end, one AMP and one GMP nucleotides are clearly bound to the RNA, fully occupying the two-nucleotide binding sites. The adenine and guanine nucleobases of both monomers were defined with clear electron densities (Fig. 3B), while the sugars and monophosphate groups were highly disordered. At +1 position of the primer/template complex, the guanine nucleobase of GMP was forming Watson–Crick base pair with mC LNA nucleotide (hydrogen-bond distances, 3.1, 3.0 and 2.8 Å, Fig. 3C). At +2 position, the unusual base-recognition motif was observed again, via the previously described noncanonical A:T interactions (hydrogen-bond distances, 2.7 and 3.1 Å, Fig. 3D). Overall, B factor and electron density fitting indicated that the bound GMP monomer at +1 position was more structurally ordered than AMP bound at +2 position, which is consistent with the previous structural and biophysical data,28,32 that the downstream bound substrate enhances the binding affinity of upstream monomer.
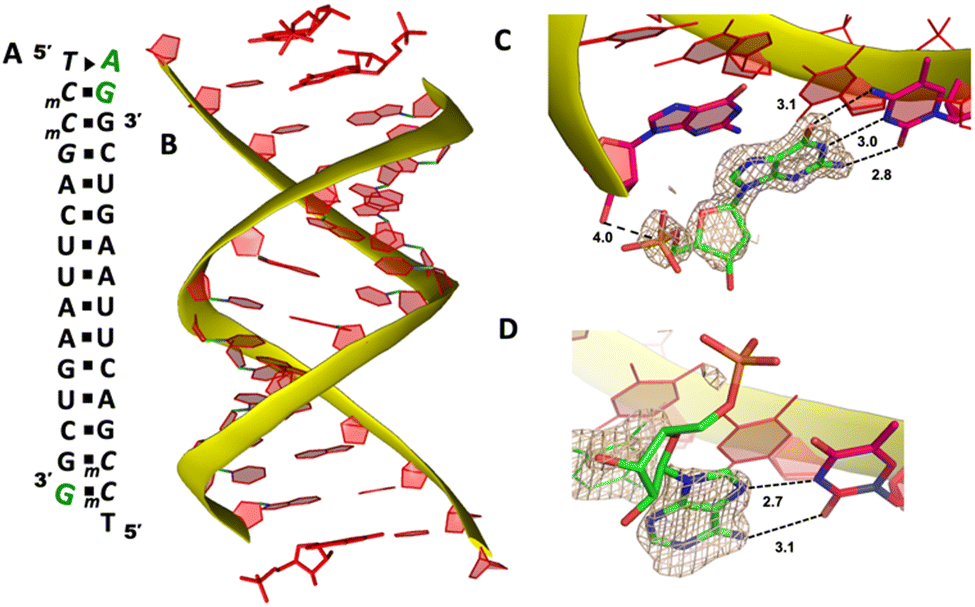 | ||
| Fig. 3 The structure of the RNA/AMP/GMP complex. (A) Diagram and designed duplex structure of the RNA/AMP/GMP complex. AMP and GMP monomers (green) are bound at each end. Black squares: Watson–Crick base pairs. Black triangle: noncanonical base pair. (B) Overall structure of the RNA/AMP/GMP duplex. (C) The local structure of GMP binding to the RNA template at the +1 position. The corresponding Fo − Fc omit map contoured at 1.5σ (wheat mesh) indicates the Watson–Crick base pair. (D) The local structure of AMP binding to the RNA template at the +2 position, forming a noncanonical base pair. All the significant distances are labelled. | ||
It is noteworthy that, the unique A:T base pair was observed in both structures of RNA/mononucleotides complexes. By forming the unusual base pair, the sugar and phosphate groups of the AMP point to the spacious major groove of the helix and avoid the potential steric hindrance with the neighbouring bound monomer. A similar binding motif was also observed when two guanosine monomers bound to the consecutive cytosine templates.28 However, in previously determined structures, half of the monomer-template binding motifs were consecutive Watson–Crick base pairing, which is different from AMP-template binding. According to the current two structures, it's more frequent to observe AMP forming the noncanonical base pair with the templating thymidine when another neighbouring monomer is present. After forming the noncanonical base pair, the distance between the phosphate groups of two bound monomers is much longer (over 10 Å) than usual, which makes it difficult for the two monomers to have nucleophilic interaction between each other. Considering this information, we propose that in RNA nonenzymatic polymerization using chemically activated adenosine as a substrate, there is a higher likelihood of forming the noncanonical base pair between the substrate and template. This, in turn, inhibits the formation of the imidazolium-bridged dinucleotide substrate on the template. This structure highly disfavours the RNA primer extension, which probably explains the slow reaction rate when using 2-AIpA as a substrate.
Structure of GpppA bound to the RNA template
Our RNA/AMP complex structures indicate that when the activated adenosine monomer is included in the polymerization reaction and bound to the template, the formation of an imidazolium-bridged intermediate on the template will be challenging. Next, we explore whether the adenosine-containing dinucleotide intermediate, which is a product assumably formed in the solution, can indeed bind to the RNA template in a favorable conformation for the primer extension process. To study the complex structures when the imidazolium-bridged dinucleotide substrate binds to RNA template, the P1,P3-diguanosine-5′-triphosphate (GpppG) had been used as a close analog and co-crystallized with RNA primer/template complex to interpret the preorganization and reactivity of dinucleotide intermediate.29 Therefore, we decide to use the commercially available P1-5′-adonesine-P3-5′-guanosine triphosphate (ApppG) to mimic the imidazolium-bridged intermediate (Ap-Im-pG) in nonenzymatic polymerization, and structurally study how the dinucleotide binds to RNA template via Watson–Crick or noncanonical base pairs.We first cocrystallized ApppG with RNA 5′-TmCmCGACUUAAGUCG-3′, where the 5′-TmC overhang served as the binding site for ApppG (Fig. 4A). The locked residues restrain the sugars into the 3′-endo conformation and facilitate crystallization. The pKa of N3 of mC is close to that of native cytidine (4.45 for C and 4.6 for mC),33 and mC can form a Watson–Crick base pair with a guanine nucleobase much like canonical C. The RNA crystallized with ApppG with the same symmetry as in the RNA-AMP complexes. The space group is P3121, and there is one RNA duplex with two bound ApppG molecules per asymmetric unit. At each end of the RNA duplex, the overhanging 5′-TmC binding site is fully occupied by the dinucleotides (Fig. 4B). As in the RNA–AMP complex structures, the RNA double helices are A-form and the duplexes slip-stack on each other to form extended columns. At one end, the ApppG ligand forms two Watson–Crick base pairs with the 5′-TmC overhang (Fig. 4C, hydrogen bonds distances 2.8–3.1 Å). The guanine and adenine nucleobases are coplanar and stacked with the upstream primer and the neighboring duplex. The electron density suggests that the ribose sugar of guanosine at the +1 position is in the C3′-endo A-form conformation, while the sugar of adenosine is disordered at the +2 position. This observation is similar to the structure of the RNA/GMP/AMP complex, which is consistent with the conclusion that the downstream bound substrate enhances the binding of the sandwiched monomer. The electron density indicates that, the triphosphate group of ApppG dinucleotide exhibits a moderate level of order, enabling us to determine its geometric properties.
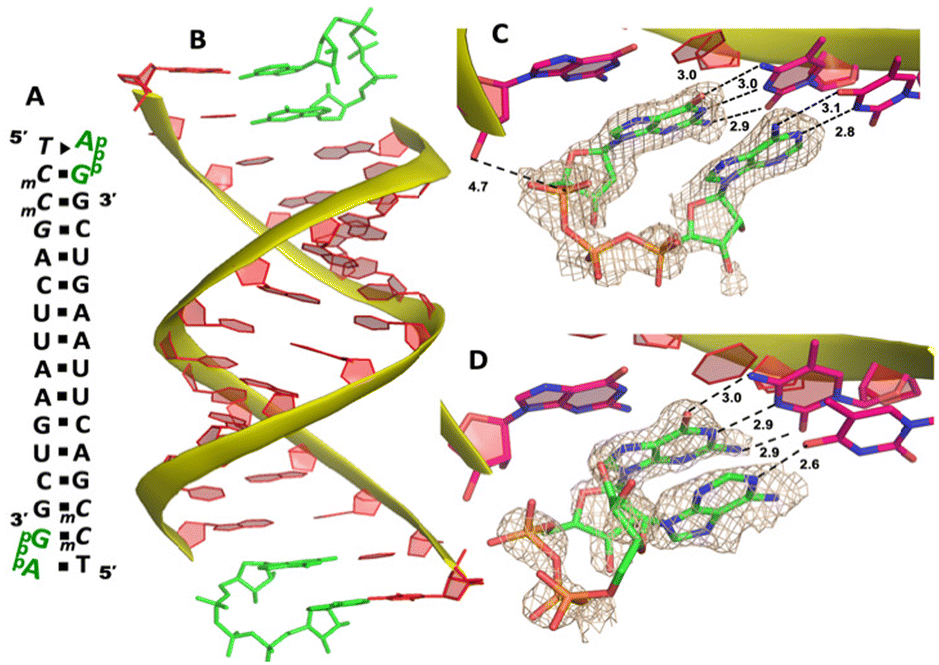 | ||
| Fig. 4 The structure of the RNA/ApppG complex. (A) Diagram and designed duplex structure of the RNA/ApppG complex. The ApppG ligand (green) is bound at each end. Black squares: Watson–Crick pairs. Black triangle: noncanonical base pair. (B) Overall structure of the RNA/ApppG duplex. (C) At one end, ApppG forms two Watson–Crick base pairs with the template with a moderately ordered linkage. (D) At the other end of the duplex, ApppG forms one Watson–Crick and one noncanonical base pairs with the template. The wheat color mesh indicates the corresponding Fo − Fc omit map of ApppG contoured at 1.5σ. All the significant distances are labelled. | ||
As the close analog of the imidazolium-bridged intermediate, it is important that the triphosphate linkage of ApppG is preorganized in a favorable conformation for the SN2 primer extension reaction. The distance between the primer 3′-hydroxyl and the phosphorus atom of the closest phosphate of ApppG is 4.7 Å, and the angle between the 3′-OH and the bridging P–O bond of GpppG is 110° (Fig. 4C). This distance is significantly longer than what we observed in RNA-GpppG structure (4.1 Å).29 Unlike the RNA-GpppG structure that a Mg2+ ion was coordinating the triphosphate linkage in a well-defined conformation, no metal ion is observed surrounding the triphosphate group of ApppG and the reaction angle for the in-line attack by the primer is less favorable (110° vs. 126°). Overall, the local structure of ApppG binding to the RNA template suggests that the dinucleotide intermediate containing adenosine nucleotide is unlikely to be optimal for the RNA primer extension.
At the other end of the duplex, ApppG binds to the template in a distinctly different manner (Fig. 4D). The guanosine adjacent to the primer is Watson–Crick base paired with the template mC through three hydrogen bonds. However, the adenosine nucleobase forms a noncanonical A:T “pair”. The adenine nucleobase is well stacked with the upstream guanine, and doesn’t form any hydrogen bond with the templating thymine. However, the distance between N1 of adenosine and O4 of T residue is measured to be 2.6 Å, close enough to form a hydrogen bonding. Considering the potential tautomeric equilibrium in adenosine and the barely fitted electron density on the nucleobase, it is possible that the adenosine forms a weak base pair with the templating T by one hydrogen bond. The triphosphate linkage and sugar of adenosine are highly disordered, making it difficult to define the geometry of the linkage and how it benefits the SN2 primer attachment. The overall structure suggests that ApppG likely binds to the RNA template with a weaker affinity than GpppG, and the primer/template/ApppG complex is unlikely to be structurally optimal for the primer extension.
To explore whether the downstream bound ligand can enhance the binding of adenine-containing intermediate and better preorganize the adenosine nucleotide, we then cocrystallized ApppG with another RNA 14mer, 5′-mCTmCGACUUAAGUCG-3′, where the locked thymidine is adjacent to the primer and serves as the binding site of adenosine (Fig. 5A). The structure was determined to 1.95 Å, and the crystal grew with the same symmetry as other RNA-ligand complexes. The overall crystal structure remains the same molecular packing patterns, and there is one ApppG dinucleotide ligand binding to the 5′-mCT template at each end (Fig. 5B). However, with the different template, the binding mode of ApppG is different from that of 5′-TmC template. At both ends, the electron density of adenine and guanine nucleobases strongly indicates that the ligand binds to the template only via Watson–Crick base pairing and the hydrogen bonds range from 2.8 to 3.1 Å (Fig. 5C). At both ends, the triphosphate linkage is moderately ordered, and the distance between the 3′-hydroxyl group of the primer and the phosphorus center of triphosphate linkage is measured to be 4.7 Å, also significantly longer than the distance in RNA-GpppG complex. Interestingly, one important water molecule is observed close to ApppG (Fig. 5D). This well-ordered water molecule is forming two hydrogen bonds with N7 of adenosine (2.7 Å) and one of the nonbridging oxygens of the triphosphate linkage (3.0 Å), which is likely to contribute to stabilizing ApppG. Based on our structural analysis, we suggest that the downstream G:C base pair plays a crucial role in impeding the formation of noncanonical A:T pairs. Instead, it enforces the A:T Watson–Crick base pair at the +1 position of the primer extension. Nevertheless, it's worth noting that the RNA-ApppG complex typically adopts a suboptimal conformation for the SN2 nucleophilic attack, despite this beneficial effect of the G:C base pair.
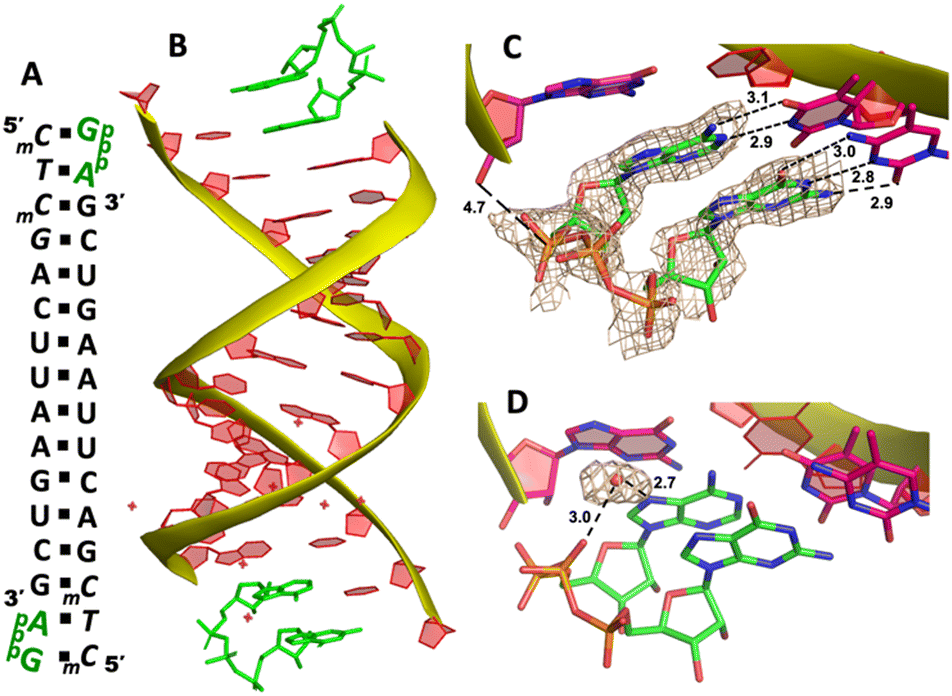 | ||
| Fig. 5 The structure of the RNA/ApppG complex. (A) Diagram and designed duplex structure of the RNA/ApppG complex. The ApppG ligand (green) is bound at each end. Black squares: Watson–Crick pairs. (B) Overall structure of the RNA/ApppG duplex. (C) At both ends, ApppG forms two Watson–Crick base pairs with the template with a moderately ordered linkage. (D) One water molecule forms two hydrogen bonds to bridge the triphosphate linkage and adenosine nucleobase. The wheat color mesh indicates the corresponding Fo − Fc omit map of ApppG contoured at 1.5σ. All the significant distances are labelled. | ||
Structure of RNA-GpppA containing A:C mismatch
In replication, maintenance of fidelity in genetic information transfer is critical for the accurate replication of life. In the modern world, the genetic molecule of DNA undergoes replication with remarkable precision, thanks to the regulation of DNA polymerase. This enzyme is capable of discerning and correcting mismatched base pairs through proofreading mechanisms.34 In comparison, the nonenzymatic polymerization is error-prone, because it lacks mismatch-repairing enzymes and the fidelity entirely depends on base pairing. The error rate in nonenzymatic copying could easily lead to a standstill of primer extension and the truncated/mutated genetic information.21 Therefore, it is critical to understand the origin and chemistry of mismatches in prebiotic nonenzymatic copying.In this study, we decided to explore the possibility of an A:C mispair in nonenzymatic polymerization. In natural biology, DNA is able to tautomerize via proton transfer to form an irregular A:C mispair,35,36 which is important for the origin of spontaneous point mutations. An A:C mismatched pair is stabilized by three hydrogen bonds, and it's believed to play an important role in gene replication errors.37 To investigate whether the A:C mismatch could occur and affect the fidelity of RNA enzyme-free replication, we decided to perform structural studies on the ApppG dinucleotide when it binds to the RNA template through mismatched base pair.
We cocrystallized the ApppG ligand with RNA 5′-mCmCmCGACUUAAGUCG-3′, in which two locked methylcytidine served as binding sites (Fig. 6A). The complex successfully crystallized and the structure was determined to 1.5 Å. There is one ApppG dinucleotide ligand binding to 5′-mCmC template at each end (Fig. 6B). In the structure, ApppG binds to the template through an unusual conformational motif. At the +1 position, guanine nucleobase is binding to the templating mCvia Watson–Crick base pair (3.2, 2.9, 2.8 Å), and the ribose sugar is in 3′-endo conformation. At the +2 position, the electron density indicates that the adenine nucleobase is well stacked with the guanine base at +1 position, and the adenine nucleobase is forming only one hydrogen bond with mC (3.1 Å, between N3 of adenine and N4 of mC). The sugar of adenosine is highly disordered. The electron density associated with the triphosphate linkage is ordered, and the distance between the 3′-hydroxyl group of the primer and the adjacent phosphorus center is about 4.6 Å (Fig. 6C). The overall structure indicates that when there are mismatched nucleotides in the RNA template, the adenosine-containing dinucleotide intermediate is prone to forming a noncanonical weak base pair. This local complex is then stabilized by stacking interactions, resulting in a suboptimal structure for the primer extension reaction.
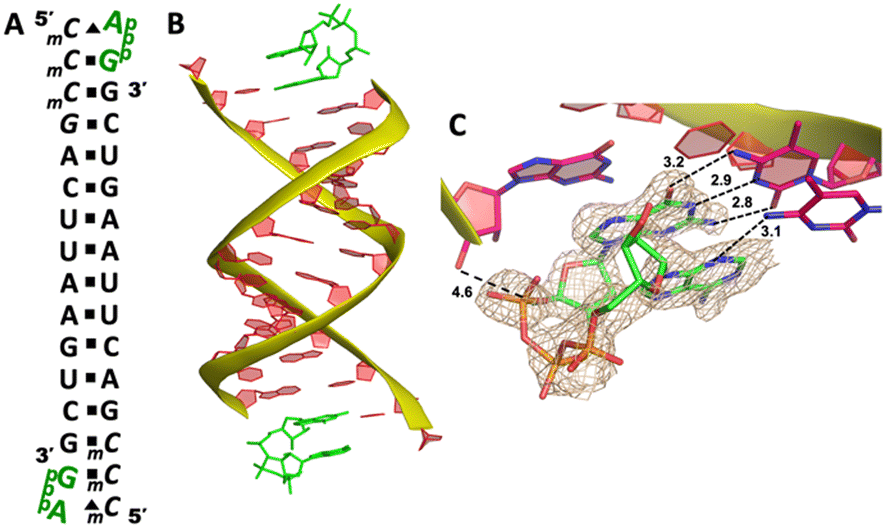 | ||
| Fig. 6 The structure of the RNA/ApppG complex containing A:C mismatch. (A) Diagram and designed duplex structure of the RNA/ApppG complex. The ApppG ligand (green) is bound at each end. Black squares: Watson–Crick pairs. Black triangles: noncanonical base pairs. (B) Overall structure of the RNA/ApppG duplex. (C) At both ends, ApppG forms one Watson–Crick and one noncanonical base pairs with the template with a moderately ordered linkage. The wheat color mesh indicates the corresponding Fo − Fc omit map of ApppG contoured at 1.5σ. All the significant distances are labelled. | ||
Structure of imidazolium-bridged adenosine, guanosine intermediate (Ap-AI-pG) bound to RNA template
It has been previously shown that nonenzymatic RNA primer extension is mediated by a highly reactive imidazolium-bridged dinucleotide intermediate, which binds to the template with great affinity and well-preorganized conformation to benefit the nucleophilic attack of the primer.20,38 We therefore attempt to explore whether the dinucleotide intermediate containing different nucleobases can be formed and how this intermediate binds to RNA primer/template complex in nonenzymatic polymerization.We first followed the previously reported method in time-resolved structure determination,20 by cocrystallizing GMP and AMP monomers with RNA 5′-TmCmCGACUUAAGUCG-3′ to form the primer/template/monomers complex (Fig. 7A). After obtaining the crystals, we tried to exchange the inactivated nucleotides with the activated guanosine-5′-phosphoro-2-aminoimidazolide (2-AIpG) and adenosine-5′-phosphoro-2-aminoimidazolide (2-AIpA) by soaking the crystals in a solution of activated monomers. By soaking the crystals for different periods, we sought to observe the formation of imidazolium-bridged Ap-AI-pG intermediate on the 5′-TmC binding site. However, even after screening a wide range of soaking times (up to 10 days) and activated monomer concentrations (up to 50 mM), we didn’t observe the formation of dinucleotide intermediate in the crystal structures. The sugar and phosphate moieties of both monomers are highly disordered, so it is impossible to conclude whether the bound ligands are 2-aminoimidazole-activated monomers or the nucleoside-5′-monophosphate molecules.
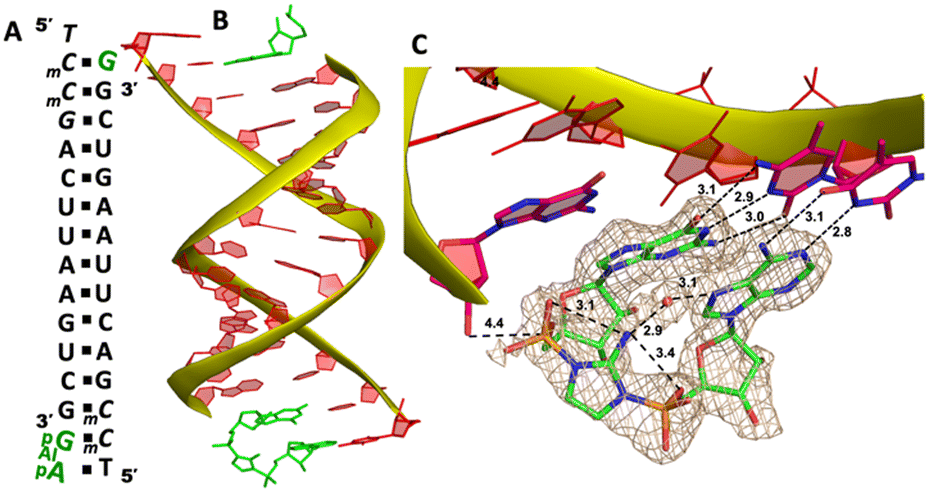 | ||
| Fig. 7 The structure of the RNA/Ap-AI-pG complex. (A) Diagram and designed duplex structure of the RNA/Ap-AI-pG complex. The Ap-AI-pG ligand (green) is bound at one end. Black squares: Watson–Crick pairs. (B) Overall structure of the RNA/Ap-AI-pG duplex. (C) At one end, Ap-AI-pG is formed and binds the template via two Watson–Crick base pairs. One water molecule forms two hydrogen bonds to bridge the 2-aminoimidazole linkage and adenosine nucleobase. The wheat color mesh indicates the corresponding Fo − Fc omit map of ApppG contoured at 1.5σ. All the significant distances are labelled. | ||
To structurally visualize the RNA/Ap-AI-pG complex, we then decided to cocrystallize RNA with 2-AIpG and 2-AIpA monomers. In the crystallization drop, the activated monomers ought to bind to the corresponding RNA template when we set up the crystallization. We expect that, during crystal growth, intermolecular interaction between 2AIpG and 2AIpA could occur on a template to form the intermediate, or the slowly emerging imidazolium-bridged dimer in solution can exchange with 2-AIpG and 2-AIpA monomers bound to the template. It took about 40 days from setting up crystallization to the diffraction data collection. The optimal structure was determined to be 1.7 Å, and there are two RNA/ligands complexes observed in one asymmetric unit. At one end of the A-helical duplex, we observe electron density consistent with imidazolium-bridged Ap-AI-pG bound to the template (Fig. 7B). Both guanosine and adenosine are well ordered and form canonical Watson–Crick base pairs with the templating mC and T residues (hydrogen bonds from 2.8 to 3.1 Å, Fig. 7C). Both ribose sugars are restrained to 3′-endo conformations. Critically, the electron density between the two nucleotides agrees with the formation of an imidazolium bridge, and the ordered structure enables us to estimate the SN2 attack geometry. The distance between the 3′-OH of the primer and the incoming P atom of the Ap-AI-pG molecule is 4.4 Å. This distance is shorter than all the distances we observed in RNA/ApppG complex structures where a noncanonical A:T mismatch pair is present. It is even slightly shorter than the distance observed in the RNA complex containing a diguanosine intermediate Gp-AI-pG (4.6 Å).20 Furthermore, the important O-P-N angle of primer attack is 122.4°, which is less than the previously observed angles of 132° and 170° in the RNA/Gp-AI-pG structure. The phosphate-aminoimidazole-phosphate bridge is stabilized by hydrogen bonds between the 2-amino group of the imidazolium moiety and the non-bridging oxygen atoms of the flanking phosphates (3.1 and 3.4 Å), and this unique geometry was also observed in Gp-AI-pG structure. In the structure, we also clearly observe electron density near the N7 of the adenine nucleobase that appears to represent a water molecule. This water molecule is located in the major groove of the RNA duplex, and forms two hydrogen bonds with an N7 atom of adenine and a 2-amino group of the imidazolium-bridge (distances 3.1 and 2.9 Å). This proposed water mediates the intramolecular hydrogen bonds and bridges the imidazolium linkage and nucleobase. It potentially stabilizes the weakly bound adenosine ligand and preorganizes the structure of the primer/template/intermediate complex for RNA polymerization. At the other end of the duplex, only one guanosine monomer is observed bound to the mC template, and the Ap-AI-pG dinucleotide is not formed. Table 1 summarizes all the RNA/ligand structures, for comparing the important conformational parameters for primer extension.
| Entry | Template | Bound ligands | Base pair at +1 position | Base pair at +2 position | Distance | 3′-O-P-O angle | Additional interaction |
|---|---|---|---|---|---|---|---|
| 1 | 5′-TT | AMP | Noncanonical | Watson–Crick | >10 | n.d. | na |
| 2 | 5′-TmC | AMP/GMP | Watson–Crick | noncanonical | 4.0 | n.d. | na |
| 3 | 5′-TmC | ApppG | Watson–Crick | Watson–Crick/noncanonical | 4.7 | 110° | na |
| 4 | 5′-mCT | ApppG | Watson–Crick | Watson–Crick | 4.7 | 131° | Water mediated bridge |
| 5 | 5′-mCmC | ApppG | Watson–Crick | noncanonical | 4.6 | 125° | na |
| 6 | 5′-TmC | Ap-AI-pG | Watson–Crick | Watson–Crick | 4.4 | 122.4° | Water mediated bridge |
Discussion
In modern biology, during the replication of genetic information, the Watson–Crick base pairing geometry plays a dominant role in DNA polymerization. This is particularly significant due to the numerous hydrophobic, electrostatic, and hydrogen-bonding interactions that facilitate the discrimination of mismatched pairs. This recognition motif, evolved over time, serves as the foundation for precise and efficient transfer of genetic information. In contrast, in the prebiotic world, nonenzymatic gene replication lacks the constraints imposed by enzymes to aid in substrate/template recognition. As a result, a wide range of base pairing possibilities is allowed, enabling the full utilization of the hydrogen bond donor and acceptor groups on the nucleobases. The previous study suggests that the nonconventional base pairing motifs are possible to occur between the activated guanosine monomer and cytidine template during nonenzymatic RNA copying in solution and in crystals.28 In the current work, we observe that adenosine substrates can also form various unusual base pairs with the thymidine template under nonenzymatic conditions. Moreover, in specific structures, the A:T base pair exhibits exceptional weakness, relying only on a single hydrogen bond and the stacking interaction between neighboring nucleobases. Indeed, if such weak A:T base pairs occur during RNA polymerization in solution, the process with the adenosine substrate could lead to errors and significant stalling effects during primer extension. This phenomenon explains the reduction in the polymerization rate observed when we attempt to extend the primer in the presence of adenosine at the +1 or +2 position. To overcome the problem, one possible strategy in the RNA world could be the evolvement of G/C-rich ribozymes which can enforce the A:U Watson–Crick base pair and thereby significantly enhance the RNA template replication accuracy and efficiency, thus expanding the genetic code in primitive RNA genome.The RNA enzyme-free polymerization relies on the involvement of the imidazolium-bridged dinucleotide intermediate, which can bind the RNA template with great affinity and interact with the RNA primer with high reactivity. Through the structural analysis of the actual substrates Gp-AI-pG and its closely related analog GpppG, compelling evidence has been obtained. The high-resolution structures demonstrate that the dinucleotide intermediate, particularly the imidazolium-bridge, plays a crucial role in preorganizing the primer/template/substrate complex, which significantly enhances the reaction rate.20 Many inter- and intramolecular hydrogen bonds contribute to stabilize the structure. If a similar mechanism occurs with the imidazolium-bridged G, A-dinucleotide substrate, a question arises: why does the presence of the adenosine residue result in such a significant retardation effect during primer extension? In our structure, the only difference when replacing the Gp-AI-pG intermediate with Ap-AI-pG is the loss of one hydrogen bond when binding to template (A:U vs. G:C). Ap-AI-pG intermediate can still form 5 hydrogen bonds with template via Watson–Crick base pairs, and the hydrogen bonds between the 2-amino group of the imidazolium bridge and the flanking non-bridging phosphate oxygen atoms remain. The distance between the 3′-OH of the primer and the incoming P atom of Ap-AI-pG is also comparable to that in the RNA/Gp-AI-pG structure (4.4 Å vs. 4.6 Å). However, observing the formation of the imidazolium bridge in Ap-AI-pG takes significantly more time compared to Gp-AI-pG. One potential explanation is that the dinucleotide intermediate is formed after the mononucleotide binds the pairing template. Frequent noncanonical base pairing occurs when the 2-AIpA monomer binds to the uridine residue on the template, which makes the intermolecular interaction between 2-AIpA and 2-AIpG extremely challenging (distance between the phosphorus atoms >10 Å). The Ap-AI-pG intermediate we observed was the product when both 2-AIpA and 2-AIpG were bound via Watson–Crick base pairing, which is uncommon.
In addition, our RNA/ApppG structures exhibit the presence of multiple A:T base pairing motifs, which serve as substitutes for the weak A:T Watson–Crick base pair. Once adenosine forms the noncanonical base pair, the linkage between the nucleotides shifts towards the major groove of the duplex. As a result, the distance critical for the primer attack to occur increases. In contrast, in all the structures of RNA/GpppG and RNA/Gp-AI-pG, the dinucleotide ligands bind to the template exclusively through Watson–Crick base pairs, leading to a preorganized local geometry necessary for the SN2 in-line attack. Based on the obtained structural information, there is another possibility to consider. If the adenosine-containing intermediate is formed in solution without the aid of an RNA template, it could adopt multiple binding motifs simultaneously when approaching and binding to the template. Among the various motifs, only the Watson–Crick fashion possesses the favorable structure for primer extension. However, this motif faces competition from numerous unfavorable structures at the binding site, creating a challenging environment for primer extension. It eventually leads to a slow reaction rate.
It is also important to note that in our primer extension reactions, there is a possibility of alternative mismatched base pairs forming. For example, the guanosine nucleobase has the ability to form a wobble pair with a uridine residue on the template. This means that the Gp-AI-pG intermediate could potentially bind to the CU template, which might be one of the reasons for the slow rate observed in our primer extension experiments. All the structures presented here only represent a single instance of the primer/template/intermediate complex in the presence of an adenosine substrate in solution. To address the questions regarding the structural binding of the Gp-AI-pG intermediate to the CU template, as well as the competitive nature between wobble-paired Gp-AI-pG and Watson–Crick-paired Ap-AI-pG substrates, further investigation in the future is required.
Moreover, our measured primer extension rates are also possibly affected by the dinucleotide substrate concentration in solution. In the reaction solution containing 2-AIpG and 2-AIpA, there are three possible dinucleotide intermediates formed, Ap-AI-pG, Ap-AI-pA and Gp-AI-pG. The concentration of the actual substrate, Ap-AI-pG, which binds the template and reacts, is likely lower than Gp-AI-pG in the solution only containing 2-AIpG monomer. This is also proved by our primer extension experiments using the same template and different substrates. With the same –CC residues as the binding template, the primer extension rate is dropped by ∼35% when using mixed monomers of 2-AIpG and 2-AIpC (Fig. S1, ESI†), compared to the pure 2-AIpG substrate. In the solution with mixed monomers, the concentration of “correct” dinucleotide must be lower than the one in the solution with pure monomer. Therefore, the reaction rates we observed in the primer extensions using the 2-AIpG and 2-AIpA substrates are the results of disfavorable binding structures, low concentration of “correct” intermediate and its competition with “incorrect” intermediate at the template. The insights into the kinetic with different intermediates will be the future direction.
Materials and methods
All the oligonucleotides were chemically synthesized in a 1.0 μmol scale using an ABI394 Synthesizer. The RNA and locked nucleotide phosphoramidites used in this work were purchased from Glen Research and ChemGenes Corporation. All the oligonucleotides were prepared with DMTr-on, and in-house deprotected using concentrated ammonium hydroxide solution for 16h at 55 °C. The RNA strands were additionally desilylated with Et3N·3HF solution to remove TBDMS groups. 5′-DMTr deprotection was either carried on using a commercial Glen-Pak purification cartridge (Glen Research Inc.), or performed by adding 3% trichloroacetic acid solution, followed by neutralization to pH 7.0 with 1 M TEAAc buffer.The oligonucleotides were purified by reversed phase HPLC on a Hamilton PRP-C18 HPLC Column (250 × 21.2 mm) at a flow rate of 10 ml min−1. The oligonucleotides were collected, lyophilized, re-dissolved in water, and concentrated as appropriate for downstream experiments. Data were collected at the LS-CAT beamlines at Argonne National Laboratory. Datasets were processed using HKL2000 and DENZO/SCALEPACK.39 All structures were solved by molecular replacement. The refinement protocol includes simulated annealing, positional refinement, restrained B-factor refinement, and bulk solvent correction.40 The topologies and parameters for locked nucleotides mC(LCC), G(LCG), T(LNT), GpppA (GA3), and dinucleotide intermediate (GMA) were constructed and applied. Detailed experimental protocols are provided in the ESI.†
Accession numbers
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank under accession codes 8SXL, 8SX6, 8SWO, 8SX5, 8SWG, and 8SY1.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the Zhang lab for helpful discussions, insightful commentary and careful revision of the manuscript. We thank Dr Zdzislaw Wawrzak from the Life Sciences Collaborative Access Team beamline 21-ID-D at the Advanced Photon Source, Argonne National Laboratory (USA). This research used resources from the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).References
- A. Rich, Horizons in Biochemistry, Academic Press: New York, NY, 1962, pp. 103–126 Search PubMed.
- F. H. Crick, J. Mol. Biol., 1968, 38, 367–379 CrossRef CAS PubMed.
- L. E. Orgel, J. Mol. Biol., 1968, 38, 381–393 CrossRef CAS PubMed.
- C. R. Woese, The genetic code, 1967 Search PubMed.
- G. F. Joyce, Nature, 2002, 418, 214–221 CrossRef CAS PubMed.
- T. R. Cech, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 4360–4363 CrossRef CAS PubMed.
- J. W. Szostak, Angew. Chem., Int. Ed., 2017, 56, 11037–11043 CrossRef CAS PubMed.
- G. Joyce and J. Szostak, Biol, 2018, 10, a034801 Search PubMed.
- L. E. Orgel, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 99–123 CrossRef CAS PubMed.
- J. W. Szostak, J. Syst. Chem., 2012, 3, 2 CrossRef CAS.
- L. Li, N. Prywes, C. P. Tam, D. K. O’Flaherty, V. S. Lelyveld, E. C. Izgu, A. Pal and J. W. Szostak, J. Am. Chem. Soc., 2017, 139, 1810–1813 CrossRef CAS PubMed.
- T. Walton and J. W. Szostak, Biochemistry, 2017, 56, 5739–5747 CrossRef CAS PubMed.
- T. Inoue and L. E. Orgel, J. Am. Chem. Soc., 1981, 103, 7666–7667 CrossRef CAS.
- T. Inoue and L. E. Orgel, J. Mol. Biol., 1982, 162, 201–217 CrossRef CAS PubMed.
- E. C. Izgu, A. C. Fahrenbach, N. Zhang, L. Li, W. Zhang, A. T. Larsen, J. C. Blain and J. W. Szostak, J. Am. Chem. Soc., 2015, 137, 6373–6382 CrossRef CAS PubMed.
- E. Kervio, B. Claasen, U. E. Steiner and C. Richert, Nucleic Acids Res., 2014, 42, 7409–7420 CrossRef CAS PubMed.
- A. Kanavarioti, C. F. Bernasconi, D. J. Alberas and E. E. Baird, J. Am. Chem. Soc., 1993, 115, 8537–8546 CrossRef CAS PubMed.
- A. Kanavarioti, C. F. Bernasconi and E. E. Baird, J. Am. Chem. Soc., 1998, 120, 8575–8581 CrossRef CAS.
- T. Walton, W. Zhang, L. Li, C. P. Tam and J. W. Szostak, Angew. Chem., Int. Ed., 2019, 58, 10812–10819 CrossRef CAS PubMed.
- W. Zhang, T. Walton, L. Li and J. W. Szostak, eLife, 2018, 7, e36422 CrossRef PubMed.
- S. Rajamani, J. K. Ichida, T. Antal, D. A. Treco, K. Leu, M. A. Nowak, J. W. Szostak and I. A. Chen, J. Am. Chem. Soc., 2010, 132, 5880–5885 CrossRef CAS PubMed.
- E. Hänle and C. Richert, Angew. Chem., Int. Ed., 2018, 57, 8911–8915 CrossRef PubMed.
- E. Kervio, A. Hochgesand, U. E. Steiner and C. Richert, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12074–12079 CrossRef CAS PubMed.
- B. D. Heuberger, A. Pal, F. Del Frate, V. V. Topkar and J. W. Szostak, J. Am. Chem. Soc., 2015, 137, 2769–2775 CrossRef CAS PubMed.
- K. Leu, B. Obermayer, S. Rajamani, U. Gerland and I. A. Chen, Nucleic Acids Res., 2011, 39, 8135–8147 CrossRef CAS PubMed.
- D. Duzdevich, C. E. Carr, D. Ding, S. J. Zhang, T. S. Walton and J. W. Szostak, Nucleic Acids Res., 2021, 49, 3681–3691 CrossRef CAS PubMed.
- D. Duzdevich, C. E. Carr and J. W. Szostak, Nucleic Acids Res., 2020, 48, e70 CrossRef CAS PubMed.
- W. Zhang, C. P. Tam, J. Wang and J. W. Szostak, ACS Cent. Sci., 2016, 2, 916–926 CrossRef CAS PubMed.
- W. Zhang, C. P. Tam, T. Walton, A. C. Fahrenbach, G. Birrane and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7659–7664 CrossRef CAS PubMed.
- W. Zhang, C. P. Tam, L. Zhou, S. S. Oh, J. Wang and J. W. Szostak, J. Am. Chem. Soc., 2018, 140, 2829–2840 CrossRef CAS PubMed.
- E. Krissinel, M. Winn, C. Ballard, A. Ashton, P. Patel, E. Potterton, S. McNicholas, K. Cowtan and P. Emsley, Acta Crystallogr. D, 2004, 60, 2250–2255 CrossRef CAS PubMed.
- C. P. Tam, A. C. Fahrenbach, A. Björkbom, N. Prywes, E. C. Izgu and J. W. Szostak, J. Am. Chem. Soc., 2017, 139, 571–574 CrossRef CAS PubMed.
- J. J. Fox, D. Van Praag, I. Wempen, I. L. Doerr, L. Cheong, J. E. Knoll, M. L. Eidinoff, A. Bendich and G. B. Brown, J. Am. Chem. Soc., 1959, 81, 178–187 CrossRef CAS.
- K. A. Johnson, Annu. Rev. Biochem., 1993, 62, 685–713 CrossRef CAS PubMed.
- M. K. Shukla and J. Leszczynski, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 637–649 CAS.
- O. H. O. Brovarets and D. M. Hovorun, J. Biomol. Struct. Dyn., 2015, 33, 28–55 CrossRef CAS PubMed.
- O. B. Ol'ha, I. M. Kolomiets and D. M. Hovorun, Quantum chemistry-molecules for innovations, IntechOpen, 2012, pp. 69–84 Search PubMed.
- T. Walton and J. W. Szostak, J. Am. Chem. Soc., 2016, 138, 11996–12002 CrossRef CAS PubMed.
- Z. Otwinowski and W. Minor, Meth. Enzymol., 1997, 276, 307–326 CrossRef CAS PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr. D, 1997, 53, 240–255 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00137g |
| This journal is © The Royal Society of Chemistry 2023 |