
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn enhanced biophysical screening strategy to investigate the affinity of ASOs for their target RNA†
Rouven
Stulz
a,
Michael
Lerche
b,
Olivia
Luige
 cd,
Agnes
Taylor
b,
Stefan
Geschwindner
e and
Alice
Ghidini
*e
cd,
Agnes
Taylor
b,
Stefan
Geschwindner
e and
Alice
Ghidini
*e
aOligonucleotide Chemistry, Discovery Sciences, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
bAdvanced Drug Delivery, Pharmaceutical Sciences, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
cDepartment of Biosciences and Nutrition, Karolinska Institutet, Neo, Huddinge, 14183, Sweden
dEarly Chemical Development, Pharmaceutical Sciences, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
eMechanistic and Structural Biology, Discovery Sciences, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
First published on 23rd October 2023
Abstract
The recent and rapid increase in the discovery of new RNA therapeutics has created the perfect terrain to explore an increasing number of novel targets. In particular, antisense oligonucleotides (ASOs) have long held the promise of an accelerated and effective drug design compared to other RNA-based therapeutics. Although ASOs in silico design has advanced distinctively in the past years, especially thanks to the several predictive frameworks for RNA folding, it is somehow limited by the wide approximation of calculating sequence affinity based on RNA–RNA/DNA sequences. None of the ASO modifications are taken into consideration, losing hybridization information particularly fundamental to ASOs that elicit their function through RNase H1-mediated mechanisms. Here we present an inexpensive and enhanced biophysical screening strategy to investigate the affinity of ASOs for their target RNA using several biophysical techniques such as high throughput differential scanning fluorimetry (DSF), circular dichroism (CD), isothermal calorimetry (ITC), surface plasmon resonance (SPR) and small-angle X-ray scattering (SAXS).
Introduction
Antisense oligonucleotides (ASOs) are chemically modified oligonucleotides whose sequence is designed to bind to a complementary region of an RNA target through Watson–Crick base pairing, modulating its function. The modulation can be through RNase H1 mediated cleavage or through steric occupation of the RNA target. RNase H1-activating oligonucleotides are usually gapmers which consist of a central DNA region flanked by “wings” bearing modified nucleotides. Due to the insufficient binding affinity and pharmacokinetics of natural, unmodified oligonucleotides, the first discovery of antisense oligonucleotides has been followed by several decades of efforts in the development of nucleotide analogues to improve their stability, biodistribution, as well as RNA binding affinity. Although the identification of molecules that can modulate RNA is nowadays a key activity in the drug discovery process, little effort has been spent characterising the mode of action of ASOs in detail. Consequently, only a small number of biophysical assays are currently of assistance in this task.In traditional small-molecule drug discovery, large libraries of chemical compounds are screened for activity in cellular, biochemical, and biophysical assays to identify genuine hits which ideally present specific activity towards a selected biological target. In contrast to the central small molecule drug discovery dogma, the identification of ASO sequences that are pharmaceutically active is based on a so-called HIT finding followed by RNA walk. First, the target mRNA regions are chosen based on the criteria of accessibility (e.g., loops and bulges), identified through a combination of several predictive bioinformatics programs like PFRED.1 Then the gapmer sequences are systematically walked along the mRNA target sequence (e.g., ±15nt from initial hit), generating a library of ASOs considered thermodynamically favourable. In this process the gapmer architecture is kept constant. One of the most used gapmer structures is the LNA–DNA–LNA, 3–10–3 configuration, where the flanking side consists of 3 LNA monomers and the central part is DNA, and the sequences is generally fully phosphorothioate.2,3 A major limitation of this strategy is that the computationally predicted hybridization properties are based only on the sequences without considering the nucleoside modifications influential for the affinity to the target. The second limitation is that most of the models consider only the most favourable secondary structure of the RNA, neglecting the fast dynamics of RNA structures and their tertiary structures. The third issue is that the relationship between the denaturation temperature of a duplex (Tm), which represents the temperature at which half of the ASO is in a double stranded conformation with its target RNA and half is free in solution, and the dissociation constant Kd is not linear. It has been suggested in fact that the correlation is parabolic and has a high variance, challenging the possibility of designing predicting tools.4
It has recently been shown experimentally that enhancing the stereochemical uniformity in the sequence, introducing modifications in one position in the DNA sequence,5,6 or changing the linkage type7 can dramatically change the toxicity and efficacy of gapmers. To date, there has not been any explanation for these phenomena, but there may be several components influencing it, including intracellular protein binding.6 Papargyri et al., at the Roche Innovation Centre Copenhagen, have recently evaluated 768 gapmers (both sequence and architecture diverse) in HeLa cells for target knockdown activity and cytotoxic potential.8 They found that ASO/RNA binding affinity and the presence of certain short sequence motifs in the gap region can explain differences between knockdown/cytotoxic ASO clusters. As a conclusion, they proposed machine-learning models to be used to predict region-specific, optimal LNA-modification architectures. A more extensive profiling of the ASO/RNA duplex formation, including binding kinetics, would provide a wealth of information that might be considerably more predictive of in vivo behaviour than Tm. High-throughput biophysical assays and target binding kinetics of oligonucleotide drugs are generally tedious to perform and are rarely reported. We therefore worked on designing a detailed biophysical screening process to generate affinity, kinetics, and structural data to be used for prospective machine learning.
Results and discussion
ASO–RNA high-throughput affinity measurement through differential scanning fluorimetry (DSF)
The state-of-the-art assay for Tm evaluation is a UV-based binding affinity assay.9,10 Since RNA absorbs ultraviolet light at a wavelength of 260 nm, a UV-Vis spectrometer with a programmable temperature gradient is often used for melting temperature measurements. This technique has several disadvantages, one of them being the limited number of samples that can be measured simultaneously (6 to 12 depending on the instrument). The second and probably bigger limitation is the length of the RNA stretch that can be measured through this assay, not allowing direct measurement with more physiologically relevant, longer RNA sequences. The data generated though UV are also limited to affinity and do not give any indication on how the introduction of modifications affects the kinetics of target binding. Hence, there is a need to enhance the scalability of existing assays and advance the level of information that can be used to feed increasingly accurate prediction models.High-throughput differential scanning fluorimetry (DSF) is a screening method normally used to analyse the stability of a protein through the temperature transitions in response to environmental factors such as pH, salts, or the presence of ligands.11–13 Upon temperature increase, the protein gradually unfolds, and more hydrophobic side chains become accessible and the fluorescent reporter dye (e.g., SYPRO orange or 1-anilinonaphthalene-8-sulfonic acid) changes its fluorescence in response to a change in hydrophobic amino acid side chains presented to the environment. We found it timely to exploit the low-volume and high-throughput format of DSF for the characterization of the thermodynamic stability of libraries of ASOs and their corresponding RNA target.
DSF was previously and successfully applied for the measurement of RNA stabilisation in addition to additives such as mono- and divalent cations as well as polyamines.14 The assay makes use of an extrinsic fluorescent dye (Quant-iT RiboGreen RIBO from Life Technologies) that fluoresces with an intensity, several orders of magnitude greater when bound to nucleic acids compared to the unbound form. RiboGreen has been shown to have specificity for structured vs. unstructured RNA, specifically in being able to distinguish between double stranded RNA14,15 and triplex structures.16,17 We thus hypothesised that this specificity might be employed to assess the unfolding process of the ASO/RNA structure. As previously described by Silvers et al., the process goes through three main fluorescence phases.14 Initially, the dye binds in a competent way to the highly stable and structured ASO/RNA duplex. That process corresponds to the initial higher fluorescence value that decays with the increase of temperature. The curve of the fluorescence intensity reaches its inflection point at a temperature corresponding to the Tmax of the ASO/RNA duplex. The melting process of ASO/RNA continues, and the binding affinity of the fluorescent dye decreases until fluorescence completely disappears when the ASO and RNA are completely unfolded.
As a model sequence to test the Tmax measurement assay, we selected a PCSK9 specific LNA antisense oligonucleotide sequence which was previously evaluated in nonhuman primate pharmacology studies.‡![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18,19 The SPC5001 gapmer, complementary to the human sequence (Fig. 1, GenBank accession no. #NM174936), was shown to potentially reduce PCSK9 mRNA levels in treated cells. Early studies suggest that PCSK9 mRNA levels were reduced by 60% within 24 hours, and the effect was sustained for over 16 days.18 In non-human primates, the hepatic PCSK9 mRNA levels and plasma LDL-C levels decreased by 85 and 50%, respectively.19 Meanwhile, subcutaneous administration of SPC5001 lowered the PCSK9 mRNA and LDL-C levels in a dose-dependent manner by ∼50 and 25%, respectively, in healthy human volunteers.
18,19 The SPC5001 gapmer, complementary to the human sequence (Fig. 1, GenBank accession no. #NM174936), was shown to potentially reduce PCSK9 mRNA levels in treated cells. Early studies suggest that PCSK9 mRNA levels were reduced by 60% within 24 hours, and the effect was sustained for over 16 days.18 In non-human primates, the hepatic PCSK9 mRNA levels and plasma LDL-C levels decreased by 85 and 50%, respectively.19 Meanwhile, subcutaneous administration of SPC5001 lowered the PCSK9 mRNA and LDL-C levels in a dose-dependent manner by ∼50 and 25%, respectively, in healthy human volunteers.
 | ||
| Fig. 1 (A) Predicted secondary structure of the terminal section of PCSK9 exon 8, which represents the binding site of ASO 1 (SPC5001) gapmer; (B) MFE structure diagram depicting base-pair probabilities for the RNA sequences used in this study (RNAfold ViennaRNA Package 2.0). | ||
The clinical development of SPC5001 was terminated due to transient renal tubular toxicity, and injection site reactions were also observed. The exact cause of acute kidney injury remains uncertain to date, reinforcing the need to collect more biophysical data on ASO sequences to generate correlation between their biophysical properties and their clinical output. The LNA–DNA–LNA gapmer sequence of SPC5001 (ASO 1) was used as a reference tool and compared to the corresponding MOE–DNA–MOE (ASO 2) and the full 2′OMe sequence (ASO 3) (Table 1, Fig. S1 and Table S1, ESI†). As expected, the substitution of LNA in the wings with MOE decreased the stability of the duplex ASO/RNA by about 14.5 °C (Fig. 2C). The full substitution of ASO 1 with 2′OMe retained its affinity. Presumably, the loss of affinity due to the substitution of LNA with 2′OMe was compensated through the gain of affinity due to the substitution of DNA. RNAs and ASOs alone did not show any fluorescent curve, confirming that the interaction of the dye is specific and not influenced by the chemistry of the ASOs. When looking at different RNA lengths, no difference in affinity was detected for the chosen ASOs. The 48-mer long RNA was designed to have a target site with a similar secondary structure compared to the 24-mer RNA, as a result the melting temperature curves and the Tm values resulted in being comparable, highlighting the flexibility of the DSF assay in terms of RNA sequence length. The melting experiment was repeated removing the salts from the buffer, but unexpectedly the Tm values were not significantly affected by the change (Fig. S2, ESI†).
| ASO | Sequence | Sugar composition | Length |
|---|---|---|---|
| 1 | TGCTACAAAACCCA | LNA gapmer 3-8-3 | 14 |
| 2 | TGCTACAAAACCCA | OMe gapmer 3-8-3 | 14 |
| 3 | UGCUACAAAACCCA | Full 2′OMe | 14 |
| 4 | TGCT*ACAAAACCCA | LNA gapmer 3-8-3 | 14 |
| 5 | TGCTA*CAAAACCCA | LNA gapmer 3-8-3 | 14 |
| 6 | AUUAAUAAAAAUGC | LNA gapmer 3-8-3 | 14 |
| 7 | AACCCAGAAUAAAU | LNA gapmer 3-8-3 | 14 |
| 8 | TGATACAAAACCCA | LNA gapmer 3-8-3 | 14 |
| 9 | TGATACAAAACCCA | MOE gapmer 3-8-3 | 14 |
| 10 | TGCTAAAAAACCCA | LNA gapmer 3-8-3 | 14 |
| 11 | TGCTAAAAAACCCA | MOE gapmer 3-8-3 | 14 |
| 12 | GTCTGTGGAAGCG | LNA gapmer 2-8-3 | 13 |
| 13 | CGTCAGTATGCGAATC | LNA gapmer 3-8-3-1 | 16 |
 | ||
| Fig. 2 (A) ASO/RNA duplex dissociation mechanism; (B) comparison of denaturation melting temperatures measured via DSF, UV and CD; (C) denaturation melting temperatures measured via DSF for 24-mer RNA target and 48-mer RNA target; (D) denaturation melting temperature of ASO-mismatched sequences (wing and DNA) for LNA and MOE ASO gapmers measured via DSF. | ||
To evaluate the contribution of the dye on the stabilization of the duplex, the temperature values obtained by DSF were then compared to Tm values measured using UV absorbance (Fig. 2B and Fig. S4, ESI†). The difference between Tmax and Tm was about ±3 °C, confirming the potential of the DSF assay to deliver accurate data for several ASOs and RNAs while increasing the throughput significantly. The flexibility of the assay was also validated by measuring Tmax values for duplexes with the longer version of the RNA target. For our purpose, the 48-mer RNA was designed as an extension on both 3′ and 5′ of the 24-mer probe (Fig. 1B) and the Tmax values remained unchanged.
To validate the results obtained by DSF, we also performed circular dichroism (CD) spectroscopy. CD can be used to study the chirality of macromolecules in solution, influenced by secondary, tertiary, and quaternary structures.20–22 Increasing the temperature while observing changes in the CD spectra makes it possible to quantify temperature-dependent unfolding or dissociation of macromolecules without the need for dyes. CD spectra were recorded for ASO 1–3 and ASO 13 in the absence and presence of RNA. Temperature ramps were performed from 40 °C to 90 °C and melting temperatures were determined (Fig. S5, ESI†), showing consistent results with those obtained from UV-Vis spectroscopy and DSF. Additionally, the CD of the individual ASOs indicates some differences in the secondary structure. ASO 1 and ASO 2 share characteristic peaks at 220 and 280 nm (Fig. S5, ESI†), while the 280 nm peak has shifted to 270 nm for ASO 3. Introducing RNA to each of the ASOs resulted in a significant change in the CD spectrum for ASO 1–3 but not for ASO 13, confirming the latter's inability to bind RNA.
Kinetics of ASO–RNA duplex formation through high through-put surface plasmon resonance (SPR)
Although very informative for the in silico development of an oligonucleotide therapy pipeline, DSF does not give any information on the kinetics stability of the ASO/RNA complex.To date, in fact the main empirical characterization of ASOs revolves around their Tm. This evaluation allows the determination and comparison between the affinities of the ASOs and their mRNA targets at their respective melting temperatures but provides no information on the binding kinetics. For oligonucleotides, the off-rates are directly correlated to their length and chemistry, highlighting the relationship between off-rates and affinity. Therefore, improving the koff of an mRNA targeting oligonucleotide drug is relatively easy and could be modulated by adjusting these two components. In contrast, the optimization of on-rates, kon, is largely independent of oligonucleotide length.23 However, the value of these changes is questionable as mRNAs have short half-lives, and dissociation from cleaved targets needs to be fast for their catalytic mechanism.
A limited number of attempts have been made to study RNA and ASO binding by SPR.24 Using the same tool ASO sequences along with the 5′-biotinylated version of the 24-mer RNA, we developed a high through-put SPR assay which would allow for the analysis of several sequences. The SPR kinetic studies are conducted close to or at physiological temperatures (25 °C or 37 °C), which are below the melting temperature of the complex. One of the most arduous steps while developing the protocol has been finding regeneration conditions that are strong enough to release ASOs from the RNA, without degrading the RNA probe on the chip. This is particularly true while working with LNA gapmers due to their stronger affinity for the RNA target, which makes them less prone to separation without the help of high temperature, or strong ionic buffers. Although the DSF showed a substantial affinity difference between ASO 1 and ASO 2, the SPR assay at 25 °C showed Kd in the similar range (picomolar) (Table S2, ESI†), but the gap between the modifications becomes larger when the assay is run at 37 °C (Table S3, ESI†), where MOE and 2′OMe ASOs jump to nanomolar Kd. As observed previously through DSF (Fig. 2C and D), the introduction of a mismatch in the DNA stretch of the gapmer seems to be less tolerated than in the wings (ASO 8 and ASO 10, Fig. 3B and C). The koff was in this case considerably higher at 37 °C (Table S3, ESI†) than at 25 °C (Table S2, ESI†). At 37 °C, one mismatch was enough to dramatically reduce the affinity of the MOE gapmers (ASO 9 and ASO 11, Fig. 3B and C), confirming the decrease of the Tm values.
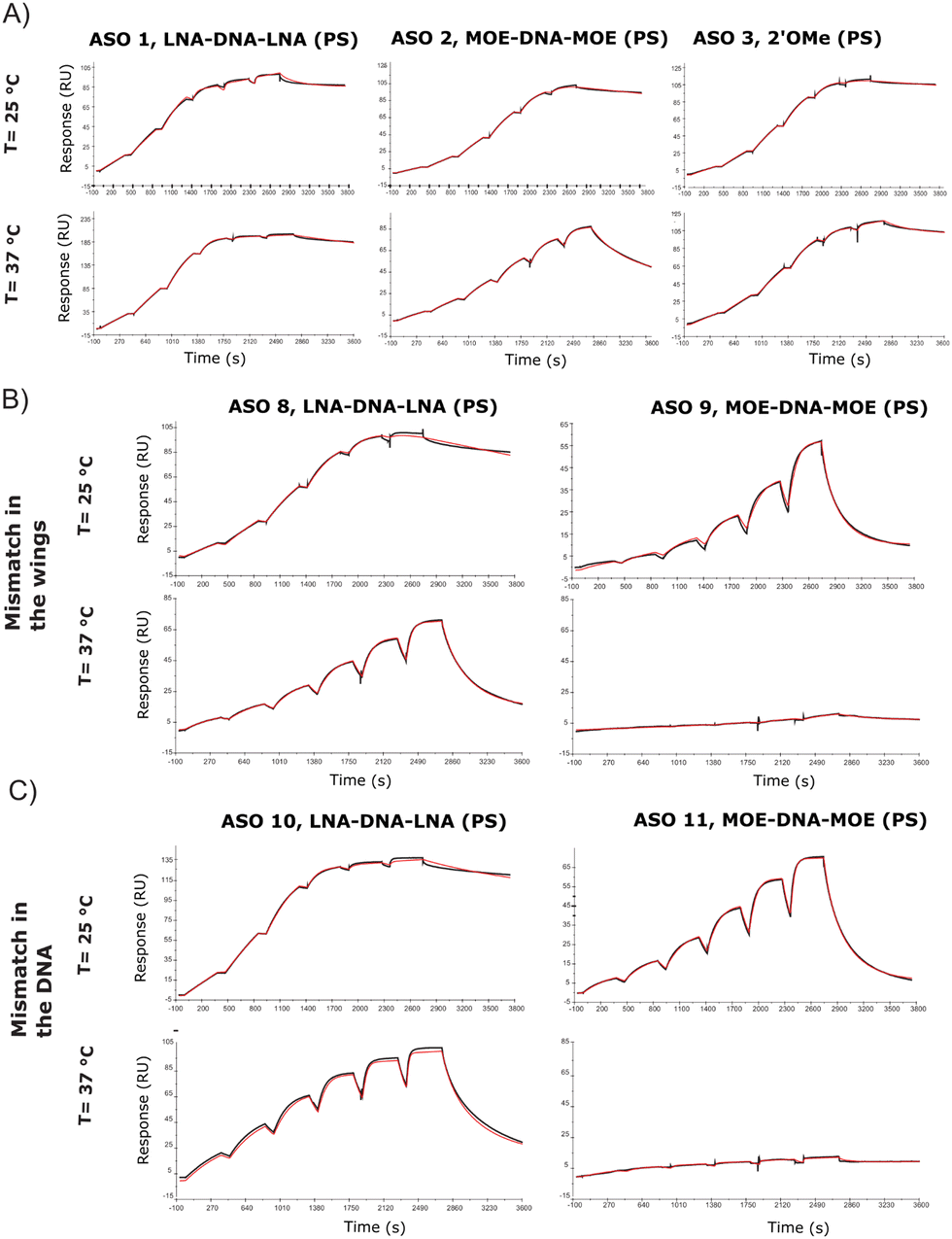 | ||
| Fig. 3 Surface plasmon resonance. (A) SPR sensorgrams and the corresponding 1:1 binding model at 25 °C and 37 °C for ASO 1, 2 and 3. (B) SPR sensorgrams and the corresponding 1:1 binding model at 25 °C and 37 °C for ASO 8 and 9, carrying a mismatch in the gapmer wing; (C) SPR sensorgrams and the corresponding 1:1 binding model at 25 °C and 37 °C for ASO 10 and 11, carrying a mismatch in the DNA sequence. | ||
Thermodynamic parameters of the ASO–RNA duplex through isothermal calorimetry (ITC)
ITC is used to measure the exothermic heat of the reaction when one strand interacts with its complementary counterpart, as the duplex gets formed. The technique has rarely been applied to oligonucleotides, but the titration assay provides a more accurate thermodynamic characterization of the ASO–RNA duplex formation, compared to UV concentration-dependent measurements. ITC reports the dissociation constant Kd, which is a measurement of the affinity of the two strands at equilibrium. According to the equation, ΔG = ΔH − TΔS, changes in enthalpy and entropy account for the Gibbs free energy of ligand binding. It is preferred to improve the enthalpy and the entropy simultaneously but this is hard to achieve because the gain in enthalpy is always accompanied with the loss of entropy. This is the pronounced enthalpy–entropy compensation, a phenomenon frequently observed in ligand–protein binding. The ITC experiments were run at 37 °C and at 45 °C, where the self-interactions and homodimers are supposed to be reduced for both RNA and ASOs (Fig. 4 and Table S4, Fig. S6, ESI†). For most of the experiments, the stoichiometry value was lower than 1.0, indicating that one of the strands was not fully available for the reaction. This suggests that at lower temperatures, a fraction of strands is unavailable to form duplexes during the injection due to self-interaction or dimerization. It was previously shown in the literature that in the case of RNA duplexes, the stoichiometry value decreased as the temperature was increased from 25 °C to 37 °C, even when the same stock solutions were used for both temperature conditions.25 This, together with an increased favourable enthalpy, suggests that all the strands tend to form a duplex under the described experimental conditions (within 90 s), but there is a small enthalpic penalty for removing the structure from the isolated oligonucleotide solutions before forming the enthalpic-favoured duplex.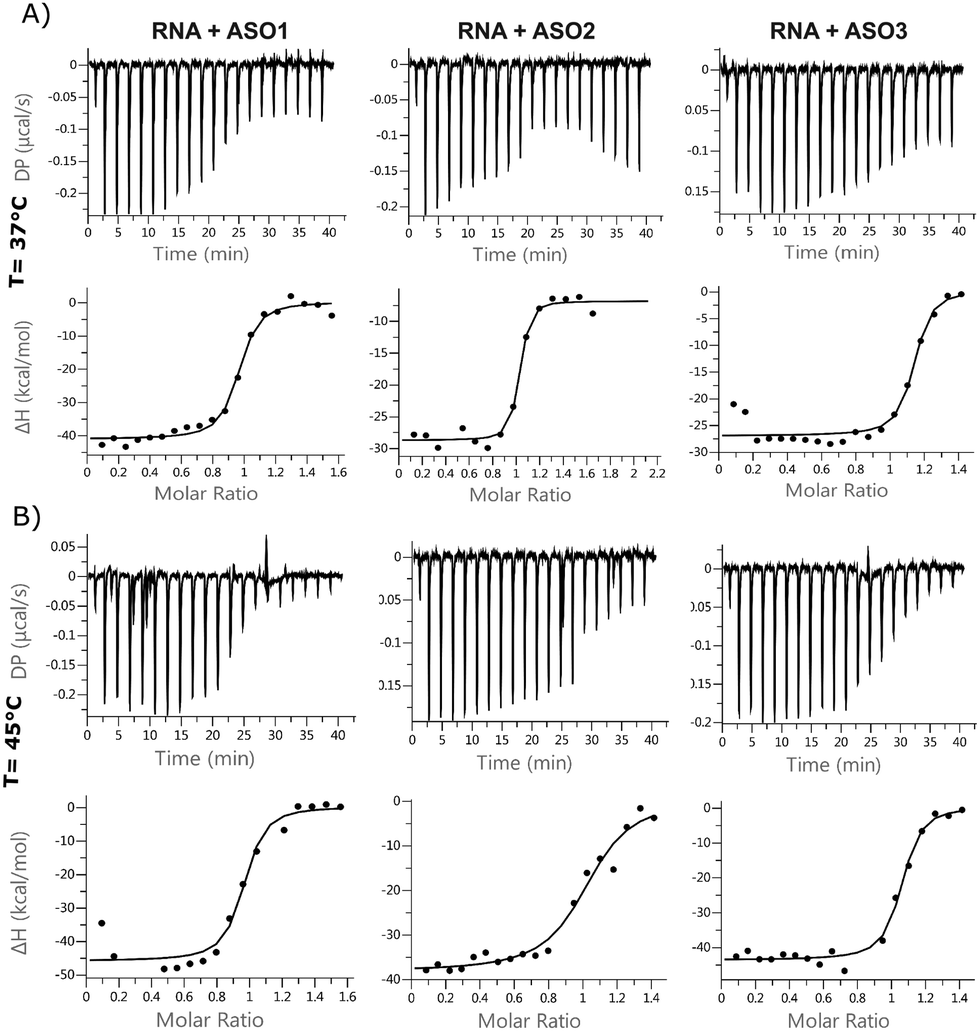 | ||
| Fig. 4 Isothermal calorimetry. Binding isotherms for a 1:1 system of ASO 1, 2, 3 and 24nt RNA at (A) 37 °C and (B) 45 °C. | ||
The entropic component was particularly favourable for the LNA gapmer at 37 °C compared to the other modifications, which is in line with the DSF data and can be attributed to an increased pre-organization of the single-stranded LNA oligonucleotide towards double strand RNA helices. Minor differences were observed for the MOE gapmer and the fully modified 2′OMe sequence compared to the LNA gapmers. All ASOs control injections showed a slight decrease in enthalpy while increasing the concentration of oligonucleotides in the cell, probably due to a change of the buffer conditions or due to some nonspecific aggregation of the PS oligonucleotides. Interestingly, the ΔG of different sequences was almost the same. Also, ΔG at different temperatures was almost identical; the enthalpic factor decreased substantially with temperature and compensated the increase of the entropic component.
Small angle X-ray scattering (SAXS) provides structural information on ASOs
Single-stranded RNA structures are essential to fully characterise interactions with complementary sequences and proteins. Due to high flexibility, only a small number of high-resolution structures while bound to proteins is currently available, and no single stranded RNA or oligonucleotide structure has been reported in the literature yet. In the case of single stranded oligonucleotides, the complexity is increased due to phosphorothioate linkage which adds variability to the sample due to its chirality. Small angle X-ray scattering (SAXS) provides structural information at low resolution. It reports the comprehensive ensemble of all the conformations, and by avoiding perturbation of dyes or chemical labelling, it captures the variability of flexible systems. Previously, SAXS/EOM studies reported a striking difference between the conformations of dT30 and dA30 homopolymer sequences at low salt concentrations (20 mM NaCl):26 dA30 has a strong propensity for base-stacking, which forces the backbone in a tortuous wind, juxtaposed to dT30 which presents straighter conformations. Unfortunately, there has not been any reported SAXS characterization of ASO sequences yet. We investigated ASO 1–3 sequences via SAXS, both in the absence and presence of the 24-mer target RNA (Fig. 5A and C). The ASO sequences all showed a peculiar shape, where the central part and the terminal monomers seem to have a higher flexibility and freedom of rotation. At the resolution measured, no significant structural differences between the ASOs could be observed, indicating that the overall radius and maximum dimensions of the ASO ensembles remain constant between the different ASO folds. To characterize and validate the ASO–RNA interaction, we measured ASO 1–3 in the presence of RNA. While rationalizing the differences observed with the previous assays, we could hypothesize that their binding properties can partially be explained by the different conformations the single stranded sequence assumes before reaching the target RNA. The addition of RNA to the ASOs showed a clear shift in scattering intensity and we could observe an increase in distance parameters (Fig. 5A, B and Table S5, ESI†), consistent with ASO–RNA complex formation.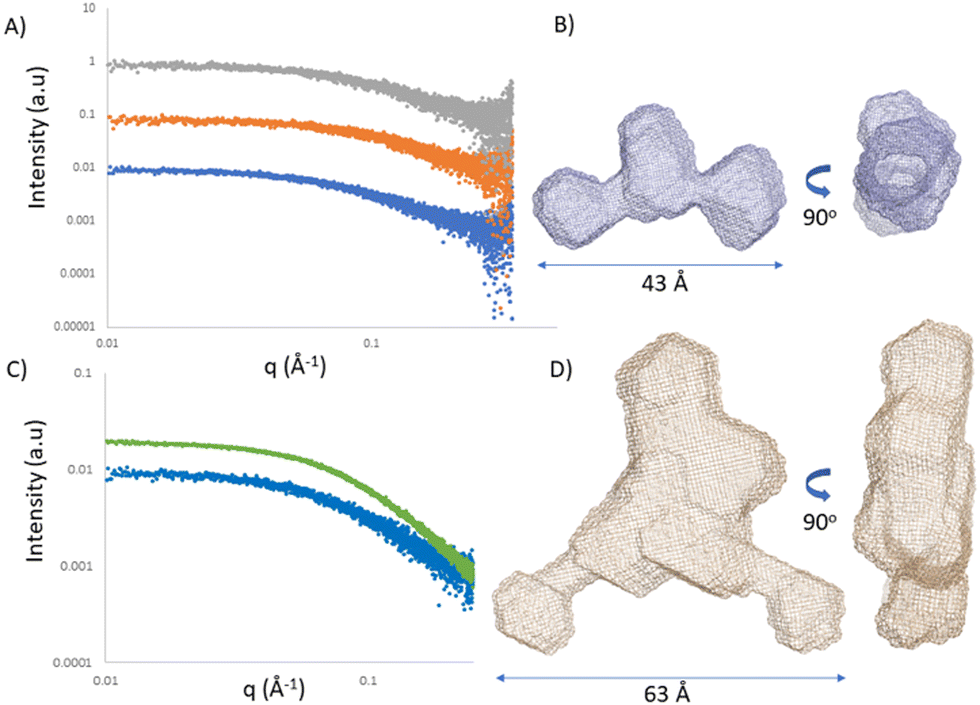 | ||
| Fig. 5 Small angle X-ray scattering. (A) Solution scattering profiles of ASO 1 (blue) ASO 2 (orange) and ASO 3 (grey) displaced by 1 logarithmic unit. (B) Ab initio model of ASO 2. (C) Solution scattering profile of ASO 2 in the absence (blue) and presence (green) of RNA. (D) Ab initio model of ASO 2 in the presence of RNA. | ||
Conclusions
We measured thermodynamics of duplex formation using differential scanning fluorimetry, comparing it to the traditional UV-Vis and CD spectroscopies and showed that we could reach a sufficient high throughput applicable for ASOs’ therapeutic studies. We then evaluated thermodynamic and kinetic values through isothermal titration calorimetry and surface plasmon resonance. The advantages of a full in vitro characterization of an ASO library are numerous, starting from empirical data integration with the current AI prediction models.The nature of the DSF assay allows the analysis of longer RNA target strands, adding flexibility to the protocol and a more accurate comparison between ASOs, resulting from the same RNA walk. The DSF throughput can be substantial in multi-well thermal cycling machines, thereby allowing the sensitive and simultaneous measurement of 384 samples in a single experiment. Compared to UV-Vis spectroscopy, the throughput is increased by a factor of 50, the experimental time is reduced by a factor of 5 along with the required amounts of RNA and ASO. The SPR assay here describes the distinction of sequences based on their kinetics properties in different temperature and salt conditions, allowing a better understanding of the influence of monomer and backbone modifications on the overall kinetic parameters.
A theoretical explanation of the observation that long, complementary oligonucleotides tend to be less potent than shorter versions, which consequently have a smaller affinity, has been proposed by Pedersen et al.4 However, to date, this theory is derived only from a small sample of data. They showed that the effect is due to affinity rather than length; we, therefore, designed biophysical techniques that will allow a fast generation of affinity, kinetics and structural data to accelerate an understanding on the development of ASOs.
Material and methods
Oligonucleotides’ synthesis
Oligonucleotides were synthesized on a 1 μmol scale using a K&A Laborgeräte H8 SE oligonucleotide synthesizer with commercially available 0.1 M phosphoramidites (Sigma-Aldrich) and CUTAG support. Detritylation was performed using a solution of 3% dichloroacetic acid in dichloromethane (Sigma-Aldrich). 5-(Benzylthio)-1H-tetrazole (BTT) (Sigma-Aldrich) was used as an activator. Cyanoethyl deprotection was performed using diethylamine/toluene (20% v/v) (Sigma-Aldrich). Xanthane hydride (TCI chemicals) was dissolved in pyridine immediately before use (0.2 M).After synthesis, the oligonucleotides were cleaved and deprotected for 1 hour at 65 °C using 1:1 methylamine (40%) aqueous ammonia (35%). After cleavage, the crude oligonucleotide was evaporated to dryness in a SpeedVac vacuum concentrator, purified via HPLC, and the pure fractions were freeze dried to give a white powder (see Fig. S1, ESI†).
Differential scanning fluorimetry (DSF)
The procedure was adapted from the method described by Silvers et al.14 All experiments were performed in a 384 qPCR plate (SP-1384, Thermo Scientific) using a LightCycler® 480 Instrument II (Roche) with PBS buffer (0.01 M phosphate buffer, 2.7 mM KCl, 137 mM NaCl, pH 7.4), or no salt-free version of the buffer (0.01 M phosphate buffer, pH 7.4).In a typical experiment, RNA and ASOs were dissolved in a buffer and dispensed into the plate to reach a final concentration of 4 μM in 10 μL. Then 50 nL of Quant-iT™ RiboGreen® (RIBO, Life Technologies, Ex max = ∼500 nm, Em max = ∼525 nm) dye stock solution in DMSO was added to each well. The plates were then sealed with an optically clear adhesive seal (Thermo Scientific) and centrifuged at 1000 × g for 2 min.
The fluorescence intensity was monitored using the SYBR Green I/HRM dye filter combination (465–510 nm) from 20 to 95 °C at a ramp rate of 0.03 °C s−1 with 36 acquisitions per °C. Melting profiles were obtained through Tmax analysis using the LightCycler® 480 software. To ensure that the measurements were statistically valid, which means that they are reproducible, three to six runs were performed. The replicates were averaged, and the standard deviation was used for error determination. In some measurements, the concentration of RNA or ASO was also varied to see if this could influence the resulting melting curve/temperature.
UV-Vis spectroscopy
UV melting curves were recorded on a Cary 3500 UV-Vis spectrophotometer equipped with a Peltier temperature controller. The RNA and ASO were dissolved in PBS buffer (0.01 M phosphate buffer, 2.7 mM KCl, 137 mM NaCl, pH 7.4) to a final concentration of 4 μM in an Eppendorf tube, warmed to 90 °C and slowly left to cool to room temperature. Then, they were transferred to a quartz cuvette with 1 mm path length and mineral oil was layered on top of the oligonucleotide solution to prevent evaporation. Melting curves were recorded at 260 nm with a ramp rate of 0.5 °C min−1 and absorbances were recorded every 2 minutes.Circular dichroism (CD) spectroscopy
CD measurements were conducted on a Chirascan V100 CD spectrophotometer (Applied Photophysics Limited, Leatherhead, UK) equipped with a xenon arc lamp and a Peltier temperature controller (Quantum Northwest Inc., Washington State, U.S.) at a wavelength range of 310–210 nm, a bandwidth of 1 nm and a step size of 1 nm. CD spectra were recorded for ASOs and RNA alone (50 μM of ASO 1–3, ASO 13 and RNA in PBS buffer) and for ASOs in the presence of 24nt RNA. ASO/RNA samples were prepared by mixing each 50 μM ASO sample with the 50 μM RNA sample in a 1:1 ratio. 140 μL of sample was loaded into a short pathlength cuvette and a baseline spectrum of PBS buffer was collected prior to each sample spectrum. After acquiring a baseline and sample spectrum at 20 °C, temperature was increased 0.7 °C min−1 from 40 °C to 90 °C while collecting a CD spectrum every 1 °C. The time-per-point was changed from 1 s per step in static measurements to 0.5 s per step when starting a temperature ramp. Baseline subtraction was performed using the Chirascan Data-Pro Viewer program. Melting temperatures were obtained by data analysis using the Chirascan Global 3 Analysis software.
Surface plasmon resonance (SPR)
The SPR binding experiments were performed on a Biacore S200 optical biosensor unit (GE Healthcare) at 25 and 37 °C. Sensor chips, Series S SA (research grade, GE Healthcare), were equilibrated at room temperature prior to use. The running buffer used for protein tethering and subsequent ligand binding experiments was 0.01 M phosphate buffer, 2.7 mM KCl and 137 mM NaCl, at a pH of 7.4. The SA surface was conditioned by five 60 s injections of a 50 mM NaOH solution (pH 8.3) to remove unbound streptavidin from the chip, followed by four 100 s buffer washing. This was immediately followed by an injection of 5′ biotinylated 24-mer RNA in running buffer at a concentration of 25 nM with a contact time of 20 to 100 s to achieve the desired densities of 200–400 RU. The reference surface was prepared accordingly, omitting the injection of RNA over the reference surface. The subsequent binding experiments were all performed at a flow rate of 25 μL min−1 and by employing the method of single-cycle kinetics. This approach involves the sequential injection of a series of compound concentrations without regeneration steps. A contact time of 400 s was selected, which was followed by a 17 min dissociation phase to allow for a proper estimation of the dissociation rate constant. Compounds were dissolved in water to 1 mM concentration. The tested concentrations for ASO 1–13 have been 36, 18, 9, 4.5, 2.25, and 1 nM. Prior to the analysis of compound binding, two running buffer blanks were injected to equilibrate the instrument. The data collection rate was set to 10 Hz and the experiments were repeated at least three times to allow for error estimations.This double-referenced data set has been fitted using a simple 1:1 interaction model with drift in order to extract kinetic and affinity data. All data were expressed as mean ± standard deviation and further analysed using GraphPad Prism 8.
Small angle X-ray scattering (SAXS)
SAXS measurements were conducted at the Diamond Light Source at the 12.4 keV beamline B21.27 35 μL of 100 μM ASO 1–3 was loaded in the presence and absence of 24nt RNA using the EMBL Arinax sample handling robot onto the capillary. Blank buffer samples were measured before and after each sample and used as background for subtraction. Scattering intensity was detected on a Eiger 4 M hybrid photon-counting detector (Dectris Ltd, Baden, Switzerland) in the momentum transfer (q) range of 0.0032–0.38 Å−1 [q = 4πsin(θ)/λ, where 2θ is the scattering angle] with each data set comprising 18 exposures for 180 seconds each. Data merging, averaging and subtraction were performed using the data processing tool Scatter (https://www.bioisis.net).
Author contributions
R. S., M. L., O. L., A. T., A. G. performed the experiments and analysed the data. R. S., S. G. and A. G. designed the study and A. G., R. S. wrote the main manuscript with input from all the authors for the specific experiments.Conflicts of interest
All the authors are employees of AstraZeneca at the time of writing this manuscript.Notes and references
- S. Sciabola, H. Xi, D. Cruz, Q. Cao, C. Lawrence, T. Zhang, S. Rotstein, J. D. Hughes, D. R. Caffrey and R. V. Stanton, PLoS One, 2021, 16, e0238753 CrossRef CAS PubMed.
- E. M. Straarup, N. Fisker, M. Hedtjärn, M. W. Lindholm, C. Rosenbohm, V. Aarup, H. F. Hansen, H. Ørum, J. B. R. Hansen and T. Koch, Nucleic Acids Res., 2010, 38, 7100–7111 CrossRef CAS PubMed.
- M. Frieden, S. M. Christensen, N. D. Mikkelsen, C. Rosenbohm, C. A. Thrue, M. Westergaard, H. F. Hansen, H. Ørum and T. Koch, Nucleic Acids Res., 2003, 31, 6365–6372 CrossRef CAS PubMed.
- L. Pedersen, P. H. Hagedorn, M. W. Lindholm and M. Lindow, Mol. Ther. Nucleic Acids, 2014, 3, e149 CrossRef CAS PubMed.
- M. E. Østergaard, C. L. De Hoyos, W. B. Wan, W. Shen, A. Low, A. Berdeja, G. Vasquez, S. Murray, M. T. Migawa, X. Liang, E. E. Swayze, S. T. Crooke and P. P. Seth, Nucleic Acids Res., 2020, 48, 1691–1700 CrossRef PubMed.
- W. Shen, C. L. De Hoyos, M. T. Migawa, T. A. Vickers, H. Sun, A. Low, T. A. Bell, M. Rahdar, S. Mukhopadhyay, C. E. Hart, M. Bell, S. Riney, S. F. Murray, S. Greenlee, R. M. Crooke, X. Liang, P. P. Seth and S. T. Crooke, Nat. Biotechnol., 2019, 37, 640–650 CrossRef CAS PubMed.
- T. P. Prakash, J. Yu, W. Shen, C. Li De Hoyos, A. Berdeja, H. Gaus, X. Liang, S. T. Crooke and P. P. Seth, ACS Med. Chem. Lett., 2021, 12, 922–927 CrossRef CAS PubMed.
- N. Papargyri, M. Pontoppidan, M. R. Andersen, T. Koch and P. H. Hagedorn, Mol. Ther. Nucleic Acids, 2020, 19, 706–717 CrossRef CAS PubMed.
- J.-L. Mergny and L. Lacroix, Oligonucleotides, 2003, 13, 515–537 CrossRef CAS PubMed.
- J.-L. Mergny and L. Lacroix, Curr. Protoc. Nucleic Acid Chem., 2009, 37, 17.1.1–17.1.15 Search PubMed.
- S. Boivin, S. Kozak and R. Meijers, Protein Expr. Purif., 2013, 91, 192–206 CrossRef CAS PubMed.
- K. Gao, R. Oerlemans and M. R. Groves, Biophys. Rev., 2020, 12, 85–104 CrossRef CAS PubMed.
- S. Shi, A. Semple, J. Cheung and M. Shameem, J. Pharm. Sci., 2013, 102, 2471–2483 CrossRef CAS PubMed.
- R. Silvers, H. Keller, H. Schwalbe and M. Hengesbach, ChemBioChem, 2015, 16, 1109–1114 CrossRef CAS PubMed.
- J. S. Matarlo, L. R. H. Krumpe, W. F. Heinz, D. Oh, S. R. Shenoy, C. L. Thomas, E. I. Goncharova, S. J. Lockett and B. R. O’Keefe, Cell Chem. Biol., 2019, 26, 1133–1142.e4 CrossRef CAS PubMed.
- A. Donlic, M. Zafferani, G. Padroni, M. Puri and A. E. Hargrove, Nucleic Acids Res., 2020, 48, 7653–7664 CrossRef CAS PubMed.
- A. A. Ageeli, K. R. McGovern-Gooch, M. M. Kaminska and N. J. Baird, Nucleic Acids Res., 2019, 47, 1468–1481 CrossRef CAS PubMed.
- N. Gupta, N. Fisker, M.-C. Asselin, M. Lindholm, C. Rosenbohm, H. Ørum, J. Elmén, N. G. Seidah and E. M. Straarup, PLoS One, 2010, 5, e10682 CrossRef PubMed.
- M. W. Lindholm, J. Elmén, N. Fisker, H. F. Hansen, R. Persson, M. R. Møller, C. Rosenbohm, H. Ørum, E. M. Straarup and T. Koch, Mol. Ther., 2012, 20, 376–381 CrossRef CAS PubMed.
- A. Cappannini, K. Mosca, S. Mukherjee, S. N. Moafinejad, R. R. Sinden, V. Arluison, J. Bujnicki and F. Wien, Nucleic Acids Res., 2023, 51, D226–D231 CrossRef CAS PubMed.
- J. Jaumot, R. Eritja, S. Navea and R. Gargallo, Anal. Chim. Acta, 2009, 642, 117–126 CrossRef CAS PubMed.
- G. R. Bishop and J. B. Chaires, Curr. Protoc. Nucleic Acid Chem., 2002, 11, 7.11.1–7.11.8 Search PubMed.
- D. Pörschke and M. Eigen, J. Mol. Biol., 1971, 62, 361–381 CrossRef PubMed.
- C. S. Schneider, A. G. Bhargav, J. G. Perez, A. S. Wadajkar, J. A. Winkles, G. F. Woodworth and A. J. Kim, J. Controlled Release, 2015, 219, 331 CrossRef CAS PubMed.
- K. Halvorsen and P. F. Agris, Anal. Biochem., 2014, 465, 127–133 CrossRef CAS PubMed.
- A. Plumridge, S. P. Meisburger and L. Pollack, Nucleic Acids Res., 2017, 45, e66 CrossRef CAS PubMed.
- N. P. Cowieson, C. J. C. Edwards-Gayle, K. Inoue, N. S. Khunti, J. Doutch, E. Williams, S. Daniels, G. Preece, N. A. Krumpa, J. P. Sutter, A. D. Tully, N. J. Terrill and R. P. Rambo, J. Synchrotron Radiat., 2020, 27, 1438–1446 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00072a |
| ‡ AstraZeneca is neither developing nor commercialising this PCSK9 specific LNA antisense oligonucleotide – SPC5001. |
| This journal is © The Royal Society of Chemistry 2023 |