
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConservation of the insert-2 motif confers Rev1 from different species with an ability to disrupt G-quadruplexes and stimulate translesion DNA synthesis†
Amit
Ketkar
a,
Reham S.
Sewilam
a,
Mason J.
McCrury
a,
Jaycelyn S.
Hall
a,
Ashtyn
Bell
a,
Bethany C.
Paxton
a,
Shreyam
Tripathi
c,
Julie E.C.
Gunderson
b and
Robert L.
Eoff
 *a
*a
aDepartment of Biochemistry and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA. E-mail: RLEoff@uams.edu; Fax: +1 501 686 8169; Tel: +1 501 686 8343
bDepartment of Physics, Hendrix College, Conway, AR 72032, USA
cArkansas School for Mathematics, Sciences, and the Arts, Hot Springs, AR 71901, USA
First published on 11th May 2023
Abstract
In some organisms, the replication of G-quadruplex (G4) structures is supported by the Rev1 DNA polymerase. We previously showed that residues in the insert-2 motif of human Rev1 (hRev1) increased the affinity of the enzyme for G4 DNA and mediated suppression of mutagenic replication near G4 motifs. We have now investigated the conservation of G4-selective properties in Rev1 from other species. We compared Rev1 from Danio rerio (zRev1), Saccharomyces cerevisiae (yRev1), and Leishmania donovani (lRev1) with hRev1, including an insert-2 mutant form of hRev1 (E466A/Y470A or EY). We found that zRev1 retained all of the G4-selective prowess of the human enzyme, but there was a marked attenuation of G4 binding affinity for the EY hRev1 mutant and the two Rev1 proteins lacking insert-2 (yRev1 and lRev1). Perhaps most strikingly, we found that insert-2 was important for disruption of the G4 structure and optimal stimulation of processive DNA synthesis across the guanine-rich motif by DNA polymerase kappa (pol κ). Our findings have implications for how Rev1 might contribute to G4 replication in different species spanning the evolutionary tree – signaling the importance of selection for enzymes with robust G4-selective properties in organisms where these non-B DNA structures may fulfill taxa-specific physiological functions.
Introduction
The accurate propagation of genomes requires the successful manipulation of DNA sequences that are prone to adopt non-B-form structures.1,2 It is well established that organisms across the spectrum of life have retained specific motifs capable of forming secondary structures to elicit and control many cellular functions. One of the best-studied examples of non-B-form structures with multi-faceted roles in the cell is the guanine-rich G-quadruplex (G4) motif.3,4 Bioinformatics analysis predicts that there are hundreds of thousands of putative quadruplex sequences (PQSs) in the human genome and that these motifs are enriched in functionally important regions, such as in the promoters of genes, at telomeres, in mitochondrial DNA, and in rDNA sequences.5,6 Targeted empirical approaches have gone on to determine that G4 structures form inside cells and that they serve as regulatory agents for a variety of cellular functions.6–8At the heart of the G4 structure is a quartet (or tetrad) of Hoogsteen-bonded guanines. The tetrad of guanines helps stabilize the overall G4 structure by coordinating monovalent cations, such as K+ and Na+, and through base stacking interactions between multiple, stacked tetrads. The thermal stability of quadruplexes can be quite high, with melting temperatures >90 °C for some G4-containing oligonucleotides. The stability of these non-canonical four-stranded structures, combined with an increased propensity to form during S-phase, makes them a formidable block to DNA replication.9 G4 structures are also susceptible to oxidation by reactive oxygen species (ROS), resulting in 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG), hydantoins, and other modifications.10 The modification of tetrad guanines by ROS may serve as a sensor, recruiting excision repair factors to sites of damage, governing epigenetic marks that control chromatin structure, and producing transcriptional changes that facilitate cellular responses to redox imbalance.11,12
Given the myriad of biological functions attributed to G4 structures and potential challenges during replication, it is not too surprising that natural selection has resulted in proteins and enzymes with attributes that are amenable to interactions with quadruplexes.13 Many G4 studies have focused on the contributions from helicases, such as DHX36, FANCJ, Pif1, and others.14–17 There are proteins and enzymes, such as DNA repair factors and specialized polymerases (pols), that coordinate with helicases to maintain the integrity of G4 motifs and other difficult-to-replicate-sequences.9,18 A number of insightful studies published over the past decade have helped define the contributions of the Rev1 translesion synthesis (TLS) pol to G4 maintenance.19–22
Rev1 is a well-studied member of the Y-family of DNA pols with a very unusual protein-template mechanism of nucleotide selection.23 Rev1 promotes TLS by (i) preferentially catalyzing dCMP insertion regardless of the identity of the template base, and (ii) through recruitment of other pols that are able to perform processive DNA synthesis across the lesions.24–27 In avian cells, Rev1 has been found to coordinate with the FANCJ helicase to promote effective histone recycling and propagation of epigenetic marks near G4 motifs.19–21 The protein-interaction motif at the extreme C-terminus of Rev1 is necessary for optimal G4 replication in avian cells, but the exact reason for this remains unclear since triple-deletion of three known Rev1-interactors – pols η, κ, and ζ – largely fails to interrupt propagation histone marks near G4 motifs, although deletion of the Rev3 subunit of pol ζ slightly elevated the epigenetic instability near the guanine-rich locus.28 It is also unclear why a catalytically inactive form of Rev1 only partially rescues G4 replication defects.19 Therefore, studies investigating the molecular features and cellular roles for the G4-related activities of Rev1 and other TLS enzymes are ongoing.
We previously reported that human Rev1 (hRev1) preferentially binds to G4 DNA substrates. We observed greater affinity for G4 substrates when the equilibrium dissociation constant (KD,DNA) values of parallel-stranded G4 structures were compared to those obtained with non-G4 substrates.29 This difference was more than an order of magnitude for several parallel-stranded G4 substrates and could be diminished by mutating residues in the insert-2 motif (e.g. E466, Y470), with mutation of L358 in the N-digit also decreasing G4 affinity, albeit the effect was less pronounced than the insert-2 mutations.30 Binding of hRev1 was found to disrupt the G4 structure to some degree, a property that did not require pol activity.29 Loss of Rev1 in human cells resulted in an elevated mutation frequency when the G4 motif was positioned in the lagging strand, as measured by the supF mutagenesis assay, and addition of the G4 stabilizing compound pyridostatin (PDS) exacerbated this effect. Complementation with either wild-type (WT) or E466K mutant hRev1 could partially suppress the increased mutation frequency of PDS-stabilized G4 motifs, but expression of hRev1 with mutations that ablated G4-selective binding were not able produce the same anti-mutagenic effect.30 These results led us to conclude that hRev1 activity helped suppress mutagenic replication near G4 sites and that G4-selective contacts were especially important for when the replisome encountered structures stabilized by PDS.
In spite of the results demonstrating G4-specific activities for Rev1 during G4 replication in avian and human cells, the enzyme seems to be dispensable for replication of the G4-containing CEB-1 minisatellite in Saccharomyces cerevisiae.22 Likewise, a genome-wide study found that loss of rev-1 in Caenorhabditis elegans did not enrich for mutations in specific sequence motifs, nor did it synergize with loss of the FANCJ homolog dog-1 to produce G4 instability.31 Thus, conservation of Rev1 function as a primary means of enacting G4 replication may not be universal in eukaryotes, but additional investigation is needed to understand the molecular basis of the differences between species.
The distinct requirements for Rev1 involvement during G4 replication in vertebrates, nematodes, and fungi raises a broader question related to how genomic/epigenomic changes influence the selection of proteins capable of maintaining those adaptive responses. A recent study used a G4-sequencing method (G4-seq) to compare the sequence composition and genomic distribution of quadruplex-forming motifs across twelve genomes covering a wide swath of the tree of life.4 What has emerged from this genome-level study is a picture of how the composition and positioning of G4 forming sequences have evolved, and in some ways, diverged across species. For example, G4-forming sequences were found to be globally less abundant in prokaryotes and yeast. Species-specific differences in the location of G4 motifs were also identified, including the enrichment of G4 motifs in the promoters of mammalian oncogenes. A reasonable supposition from these findings is that differences in the genomic landscape tied to taxa-specific phenotypes will select for varied degrees of G4-selective properties in proteins that help maintain these non-B-form DNA sequences.
With this premise as a guidepost, we have studied the G4-related biochemical properties of Rev1 orthologs from four species – human, zebrafish, yeast, and leishmania – in an effort to determine if G4-selective properties were maintained in spite of potential differences in biological function. We used oligonucleotides adopting parallel, anti-parallel, and hybrid G4 structures to mimic the breadth of G4 structural diversity in the genome. In addition to DNA binding and polymerase activity, we have also investigated the G4 unfolding properties of Rev1 in order to pinpoint molecular features that confer this enzyme with an ability to selectively bind and disrupt quadruplex structures. Finally, we provide evidence to support the notion that the G4 disruptive capacity of Rev1 is necessary for stimulation of TLS across the entirety of a G4 motif. Our findings provide some intriguing new insights into how vertebrate Rev1 may have evolved to act upon G4 DNA in a manner that separates it from more ancient versions of this TLS pol.
Results
Selection of Rev1 proteins for study
We have previously reported on the biochemical and cellular features of hRev1 activity on G4 DNA substrates.29,30 Results implicating insert-2 in G4-selective activities led us to examine conservation of this motif in organisms from across the evolutionary tree of life. The REV1 gene is conserved in eukaryotes but has not been firmly documented in either archaea or bacteria. Searching GenBank, we found that, within the domain of eukaryotes, genes encoding the Rev1 TLS pol have been found in Archaeplastida (plantae), Excavata (e.g., trypanosomes), Amoebozoa, Opisthokonta (Metazoans), and the Stramenopiles, Alveolates, and Rhizaria (SAR) clade.32 Examination of amino acid sequence alignments revealed that residues comprising insert-2 are conserved in vertebrates and could have been present during speciation events dating back to the emergence of arthropods more than 500 million years ago, with elements of insert-2 perhaps being found in some platyhelminthes (Fig. 1A).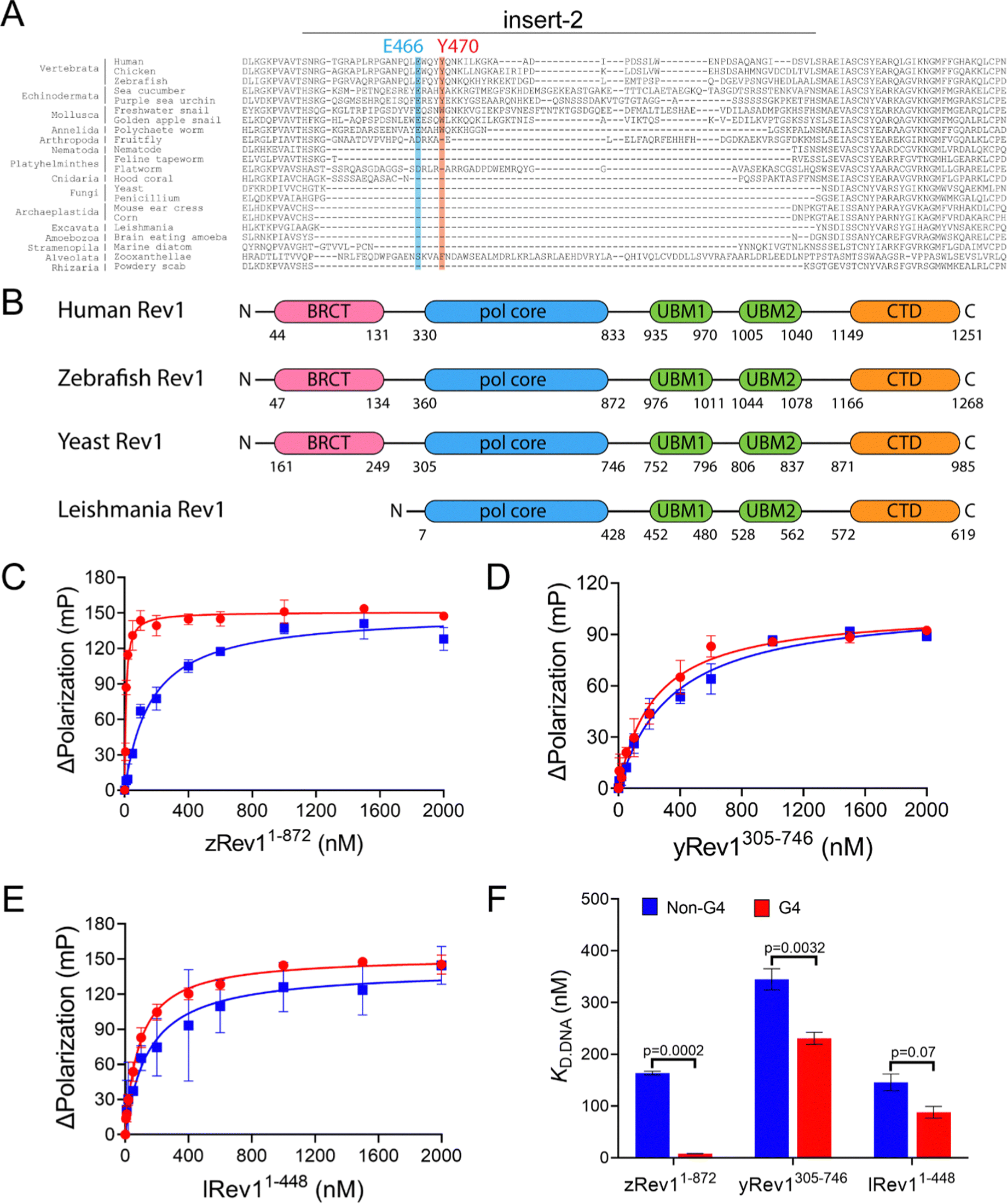 | ||
| Fig. 1 Comparison of G4 binding properties for Rev1 proteins from different species. (A) The sequence alignment of the insert-2 region for Rev1 proteins ranging from the SAR supergroup through vertebrata is shown. (B) A schematic illustration of domain arrangement and conservation is shown for the four Rev1 proteins used in this study. Binding curves for (C) zRev11-872 (D) yRev1305-746, and (E) lRev11-448 proteins with either the Myc 14/23 G4 DNA substrate (red circles) or non-G4 DNA control (blue squares) are shown. Protein was titrated into a solution containing either single-stranded (ss)-G4 DNA or ss-non-G4 DNA substrates at a concentration of 2 nM. The range of concentrations for the protein is indicated on the X-axis. The change in fluorescence polarization at each concentration was measured and plotted as a function of the protein concentration. (F) The changes in fluorescence polarization were fit to a quadratic equation to yield binding dissociation constants. The actual KD,DNA estimates are reported in Table 2. Values reported here represent the mean (± std. dev.) for three independent replicates. The reported p-values were calculated by using an unpaired Student's t-test with Welch's correction. | ||
Prompted by differences in G4 replication dynamics reported for vertebrates and other eukaryotes, we chose to study three non-human Rev1 proteins in order to better understand the influence of this motif on G4-selective binding, disruption, and catalysis. The three proteins selected for our study included: Saccharomyces cerevisiae (yeast) Rev1 (yRev1), Leishmania donovani (leishmania) Rev1 (lRev1), and Danio rerio (zebrafish) Rev1 (zRev1). We expressed and purified each of the Rev1 proteins using a bacterial expression system (Fig. S1, ESI†). In each case, the best expression was observed for constructs that encode the polymerase core domains – amino acids (a.a.) 1–872 for zRev1, a.a. 305–746 for yRev1, and a.a. 1–448 for lRev1. The only major difference in domain composition between the three enzymes was the retention of the BRCA C-terminal (BRCT) domain at the N-terminus of zRev1 (Fig. 1B). In a previous study, we obtained similar G4-selective properties when comparing the hRev1 pol core (a.a. 330–833) and full-length hRev1 (a.a. 1–1251), the latter of which possesses an intact N-terminal BRCT domain.30 Since the BRCT domain did not seem to greatly contribute to the G4-selective properties of hRev1, we had some confidence that the purified zRev1 protein would be useful for comparisons with yRev1 and lRev1. With the purified proteins in hand, we set out to determine the DNA binding and catalytic properties on both G4 and non-G4 DNA substrates.
Rev1 proteins lacking insert-2 exhibited less selective binding to G4 DNA
To determine if the G4-selective properties observed for hRev1 were conserved in Rev1 from different species, we first compared the DNA binding affinity of each of the three non-human Rev1 proteins using both G4 and non-G4 DNA substrates. We monitored the change in fluorescence polarization for FAM-labeled DNA substrates as a function of protein concentration and fit the resulting data to a quadratic equation to obtain an estimate of the equilibrium dissociation constants (KD,DNA) defining binary complex formation. We used three G4 motifs (i) the parallel-stranded Myc 14/23, which replaces guanine with thymidine at positions 14 and 23 in the Pu27-mer WT sequence from the c-MYC promoter, (ii) the anti-parallel thrombin binding aptamer (TBA), and (iii) a hybrid-forming Telo4 motif derived from the human telomeric sequence (Table 1). For each G4 substrate, we used a non-G4 sequence of identical length to evaluate the relative binding preferences for the Rev1 proteins. Both single-stranded (ss)-DNA and primer-template (p/t)-DNA substrates were prepared in either potassium-containing buffer or lithium-containing buffer. Since lithium does not effectively stabilize G4 structures, we were able to identify changes in binding equilibrium likely attributable to stable quadruplex formation.| Name | Sequence | Type of G4 fold |
|---|---|---|
| a The guanine bases involved in the formation of G4-tetrads are shown in bold. The underlined guanine in the G4 sequences was changed to cytosine in the corresponding non-G4 oligonucleotide. For DNA binding assays, both G4 and non-G4 DNA substrates were labeled at the 5′ end with 6-carboxyfluorescein (FAM). The label was on the longer template strand for binding assays with p/t-DNA. b The 42-mer template oligonucleotides were labeled at the 5′ end with FAM for DNA binding assays. These same oligonucleotide sequences were unlabeled for polymerase assays where the primer was FAM-labeled. They were also unlabeled for G4 Hemin DNAzyme experiments. c The 13-mer and 23-mer primers were labeled at the 5′ end with FAM for the polymerase assays with the p/t-DNA substrates. These same oligonucleotide sequences unlabeled for the DNA binding assays where the template was FAM-labeled. They were also unlabeled for G4 Hemin DNAzyme experiments. | ||
| Myc 14/23a | 5′-AG![[G with combining low line]](https://www.rsc.org/images/entities/b_char_0047_0332.gif) GTGGTAGGTGGTTATGAGATGAT-3′ GTGGTAGGTGGTTATGAGATGAT-3′ |
Parallel |
| Non-G4 Myc control | 5′-AGCGTGCGTAGCGTGCGTTATGAGATGAT-3′ | — |
| KRAS 22RT | 5′-AGGCGTGTGGAAGAGGAATATGAGATGAT-3′ |
Parallel |
| Non-G4 KRAS control | 5′-AGCGCGCTGTGCGAAGAGCGAATATGAGATGAT-3′ | — |
| TBA | 5′-TGTTGTGTGTTGTATGAGATGAT-3′ |
Antiparallel |
| Non-G4 TBA control | 5′-TGCTTGCTGTGCTTGCTATGAGATGAT-3′ | — |
| hTelo-4 | 5′-TTGGTTAGGTTAGGTTAGGATATGAGATGAT-3′ |
Hybrid |
| Non-G4 Telo-4 control | 5′-TTGCGTTAGCGTTAGCGTTAGCGATATGAGATGAT-3′ | — |
| 11-mer primer | 5′-ATCATCTCATA-3′ | — |
| 42-mer G4-templateb | 5′-TGAGGGTGGGTAGGGTGGGTGCGTCTGCGGCTGGCTCGAGGC-3′ | Parallel |
| 42-mer non G4-templateb | 5′-GTGAGATGTTGACCATGGGTGCGTCTGCGGCTGGCTCGAGGC-3′ | — |
| 23-mer primerc | 5′-TTTGCCTCGAGCCAGCCGCAGACGCA-3′ | — |
| 18-mer primer | 5′-GCCTCGAGCCAGCCGCAG-3′ | — |
| 13-mer primerc | 5′-TTTGCCTCGAGCCAGC-3′ | — |
Binding experiments with the G4 DNA substrates were performed, and we observed clear differences between the non-human forms of Rev1. For example, zRev1 exhibited a marked increase in affinity for two of the three G4 substrates relative to the non-G4 controls (Table 2). The increased affinity was observed for both ss-G4 DNA (Fig. 1C and Table 2) and p/t-G4 DNA (Table 3) substrates. The largest difference in KD,DNA was observed for the parallel-stranded Myc sequence where zRev1 bound the G4 substrate ∼15–20-fold more tightly than the non-G4 control (Fig. 1C and Tables 2 and 3). The affinity of zRev1 for the anti-parallel TBA G4 substrate was ∼7-fold greater than the non-G4 control (Tables 2 and 3), which differed from the more modest 3-fold increase in affinity we previously reported for hRev1.30 zRev1 binding to the hybrid Telo4 G4 DNA substrate was only moderately tighter than that of the non-G4 substrate (Tables 2 and 3). As expected, the difference between G4 and non-G4 substrates observed for zRev1 was lost for both ss- and p/t-DNA when binding was measured in the presence of lithium chloride (Tables S1 and S2, ESI†). Overall, zRev1 exhibited G4 binding properties that were very similar to hRev1.
| K D.DNA | Fold preference for G4 DNA | ||
|---|---|---|---|
| Non-G4 (nM) | G4 (nM) | (KD,NonG4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) DNA/KD,G4DNA) DNA/KD,G4DNA) |
|
| a Data represent the mean ± std. dev. (n = 3). | |||
| yRev1 (a.a. 305–746) | |||
| Myc 14/23 | 340 ± 60 | 230 ± 50 | 1.5 |
| TBA | 450 ± 70 | 320 ± 30 | 1.4 |
| hTelo-4 | 310 ± 40 | 290 ± 40 | 1.1 |
| lRev1 (a.a. 1–448) | |||
| Myc 14/23 | 150 ± 40 | 87 ± 10 | 1.7 |
| TBA | 330 ± 60 | 234 ± 34 | 1.4 |
| hTelo-4 | 410 ± 90 | 320 ± 30 | 1.3 |
| zRev1 (a.a. 1–872) | |||
| Myc 14/23 | 160 ± 30 | 8 ± 1 | 21 |
| TBA | 120 ± 40 | 14 ± 2 | 8 |
| hTelo-4 | 110 ± 30 | 34 ± 6 | 3 |
| K D.DNA | Fold preference for G4 DNA | ||
|---|---|---|---|
| Non-G4 (nM) | G4 (nM) | (KD,NonG4DNA/KD,G4DNA) |
|
| a Data represent the mean ± std. dev. (n = 3). Template strands were annealed to the 11-mer primer to form the p/t-DNA substrates. | |||
| yRev1 (a.a. 305–746) | |||
| Myc 14/23 | 480 ± 40 | 310 ± 60 | 1.5 |
| TBA | 450 ± 30 | 350 ± 30 | 1.3 |
| hTelo-4 | 380 ± 50 | 240 ± 40 | 1.6 |
| lRev1 (a.a. 1–448) | |||
| Myc 14/23 | 320 ± 20 | 210 ± 30 | 1.5 |
| TBA | 550 ± 50 | 370 ± 50 | 1.5 |
| hTelo-4 | 460 ± 50 | 320 ± 30 | 1.4 |
| zRev1 (a.a. 1–872) | |||
| Myc 14/23 | 190 ± 40 | 13 ± 4 | 15 |
| TBA | 160 ± 30 | 22 ± 5 | 7 |
| hTelo-4 | 130 ± 40 | 40 ± 8 | 3 |
By way of comparison, yRev1 exhibited a slight affinity for parallel-stranded G4 DNA relative to the non-G4 control substrates but the difference was less pronounced than that observed for either hRev1 or zRev1. We found that yRev1 bound G4 and non-G4 DNA substrates with Kd,DNA values in the 200–500 nM range irrespective of whether the primer was present or not (Fig. 1D and Tables 2 and 3). Of the three enzymes tested, lRev1 bound to the parallel-stranded G4 DNA substrates with KD,DNA values that were most similar to non-G4 control DNA (Fig. 1E and Tables 2 and 3), indicative of little preference for one substrate over the other. Likewise, neither yRev1 nor lRev1 exhibited much preference for binding the anti-parallel (TBA) or hybrid (Telo4) G4 DNA substrates more tightly than the non-G4 control substrates (Tables 2 and 3). Binding constant for yRev1 and lRev1 were not noticeably impacted by substitution of lithium for potassium (Tables S1 and S2, ESI†). Overall, comparison of the measured KD,DNA values confirmed that, of the three proteins tested, only zRev1 bound G4 substrates with an affinity that was >2-fold stronger than that of non-G4 substrates (Fig. 1F).
Moving the primer terminus closer to G4 DNA decreased the affinity of Rev1
We were curious to learn whether changing the position of the primer relative to the G4 structure impacted the observed KD,DNA values. We measured DNA binding affinity for a series of DNA substrates with a 42 nucleotide (nt) template strand, which was longer than the 29 nt template used in the first set of binding assays and allowed us to vary the position of the 3′-OH primer terminus relative to the G4 motif (Table 1). The 3′-OH of the primer was positioned 10 nts from the first tetrad-associated guanine on the 13/42-mer substrate, while the primer terminus was positioned directly adjacent to the G4 motif on the 23/42-mer substrate. Comparing the measured KD,DNA values, it was again apparent that of the three Rev1 proteins tested, only zRev1 exhibited any kind of preferential binding to G4 DNA substrates (Table 4). Similar to the shorter, 29 nt DNA substrates, zRev1 bound the ss-42-mer G4 DNA substrate with an affinity that was ∼21-fold greater than that measured for the non-G4 control (Table 4). The preference for binding to G4-containing substrates was ∼8-fold and ∼3-fold for the 13/42-mer and 23/42-mer p/t-DNA substrates, respectively. In this regard, the G4-selective binding of zRev1 decreased as the primer moved closer to the tetrad guanines. In contrast to the results for zRev1, yRev1 and lRev1 bound to the ss-42-mer, 13/42-mer, and 23/42-mer DNA without much difference between the KD,DNA values for G4 and non-G4 substrates (Table 4), indicative of the fact that the quadruplex does not impact the affinity of these enzymes for DNA.| K D.DNA | Fold preference for G4 DNA | ||
|---|---|---|---|
| Non-G4 (nM) | G4 (nM) | (KD,Non-G4DNA/KD,G4DNA) |
|
| a Data represent the mean ± std. dev. (n = 3). | |||
| yRev1 (a.a. 305–746) | |||
| ss-42-mer | 340 ± 60 | 230 ± 50 | 1.5 |
| 13/42-mer | 440 ± 80 | 300 ± 40 | 1.5 |
| 23/42-mer | 460 ± 50 | 320 ± 50 | 1.5 |
| lRev1 (a.a. 1–448) | |||
| ss-42-mer | 146 ± 83 | 87 ± 10 | 1.7 |
| 13/42-mer | 153 ± 34 | 148 ± 30 | 1 |
| 23/42-mer | 216 ± 67 | 177 ± 43 | 1.2 |
| zRev1 (a.a. 1–872) | |||
| ss-42-mer | 160 ± 30 | 8 ± 2 | 21 |
| 13/42-mer | 120 ± 40 | 15 ± 2 | 8 |
| 23/42-mer | 130 ± 30 | 42 ± 7 | 3 |
The hRev1 EY insert-2 double mutant has attenuated G4 binding affinity
In order to have a suitable negative control for comparing G4 properties and to gain greater perspective on experiments with non-human versions of Rev1, we sought to identify mutations in the human enzyme that could ablate G4-selective properties. We had previously shown that mutation of either E466A or Y470A in the insert-2 motif of hRev1 reduced the binding preference for ss-Myc-14/23 G4 DNA to around 4- to 5-fold over non-G4 DNA compared to the approximate 20-fold preference observed for WT hRev1.30 Mutation of the L358 in the N-digit to alanine reduced the preference of hRev1 for G4 substrates to around 9-fold over non-G4 DNA. However, none of the single-mutations seemed to have much of a G4-specific impact on polymerase activity. We reasoned that mutating multiple residues might exert a more pronounced effect on the ability of hRev1 to act on G4 substrates.In an attempt to improve upon the separation of G4 and non-G4 functions, we expressed and purified the E466A/Y470A (EY) hRev1 double mutant (Fig. S1, ESI†). We then measured the KD,DNA values for the EY mutant with G4 and non-G4 DNA substrates (Table 5 and Table S3, ESI†). There was a 2.7-fold and 1.3-fold difference in binding affinity for the Myc-derived ss- and p/t-G4 DNA substrates relative to the non-G4 control substrate (Table 5), suggestive of a more pronounced attenuation of the G4 binding properties when compared with the single mutants. Similar to WT enzyme, the EY mutant bound anti-parallel (TBA) G4 substrates (both ss- and p/t-) with an affinity that was around 2- to 3-fold greater than the non-G4 substrates (Table 5). Likewise, the EY mutant did not exhibit preferential binding to either the ss- or p/t-Telo4 G4 substrates relative to the non-G4 controls (Table 5).
| K D.DNA | Fold preference for G4 DNA | ||
|---|---|---|---|
| Non-G4 (nM) | G4 (nM) | (KD,NonG4DNA/KD,G4DNA) |
|
| a Data represent the mean ± std. dev. (n = 3). Template strands were annealed to the 11-mer primer to form the p/t-DNA substrates. | |||
| ss-Myc-14/23 | 650 ± 130 | 240 ± 20 | 2.7 |
| p/t-Myc-14/23 | 830 ± 120 | 630 ± 100 | 1.3 |
| ss-TBA | 150 ± 20 | 54 ± 5 | 2.7 |
| p/t-TBA | 230 ± 30 | 100 ± 20 | 2.3 |
| ss-Telo-4 | 610 ± 90 | 770 ± 100 | 0.8 |
| p/t-Telo-4 | 540 ± 70 | 310 ± 30 | 1.7 |
We then compared the binding properties of WT and EY hRev1 using the longer 42 nt DNA substrates. Similar to the shorter substrates, WT hRev1 bound the ss-42-mer G4 DNA substrate around 20-fold more tightly than the non-G4 control (Table 6). The EY mutant exhibited a 3-fold preference for the ss-42-mer G4 DNA, which was much reduced from the WT enzyme (Table 6). The difference between WT and the EY mutant was maintained for the 13/42-mer DNA substrates (Table 6). The WT enzyme bound the 13/42-mer G4 substrate ∼7-fold more tightly than the non-G4 control, while the EY mutant only maintained a meager 3-fold preference for the G4 substrate. The 23/42-mer DNA substrates were compared and there was no meaningful difference between the WT and EY mutants, with both enzymes binding the G4 substrate around 3-fold tighter than the non-G4 control. A puzzling feature of these results was the apparent lack of G4-selective binding for WT hRev1 on the 23/42-mer substrates, which is in contrast to what we have repeatedly observed with shorter p/t-DNA substrates that positioned the primer terminus adjacent to tetrad guanines.29,30 Nevertheless, we concluded that the combination of two mutations in the insert-2 motif further diminished the preferential binding of hRev1 to G4 substrates beyond that observed for the single mutants. With the EY mutant in hand as a negative control for G4-selective binding, we set out to compare the catalytic properties of the different forms of Rev1.
| K D.DNA | Fold preference for G4 DNA | ||
|---|---|---|---|
| Non-G4 (nM) | G4 (nM) | (KD,Non-G4DNA/KD,G4DNA) |
|
| a Data represent the mean ± std. dev. (n = 3). | |||
| hRev1 (a.a. 330-833) WT | |||
| ss-42-mer | 85 ± 11 | 4 ± 1 | 21 |
| 13/42-mer | 230 ± 30 | 31 ± 6 | 7 |
| 23/42-mer | 210 ± 30 | 88 ± 12 | 2.4 |
| E466A/Y470A | |||
| ss-42-mer | 270 ± 50 | 87 ± 10 | 3 |
| 13/42-mer | 250 ± 30 | 86 ± 10 | 2.9 |
| 23/42-mer | 300 ± 40 | 120 ± 20 | 2.4 |
Insert-2 impacts Rev1 catalysis by sensing G4 positioned downstream of the primer-template junction
We measured the catalytic power of the Rev1 proteins on G4 and non-G4 p/t-DNA. We first compared WT hRev1 with the EY insert-2 mutant using the 13/42-mer p/t-DNA substrates. WT hRev1 catalyzed dCMP insertion on the G4 substrate at a rate that was ∼70% as fast as the rate measured for the non-G4 13/42-mer (Fig. 2A; see Fig. S2, ESI† for gels), which was indicative of fairly robust catalysis on the G4 substrate. We then measured dCMP insertion rates for the EY mutant. On non-G4 DNA substrates, the initial velocity of dCMP insertion by the EY mutant was ∼70% as fast as WT enzyme (Fig. 2A; see Fig. S2, ESI† for gels), indicative of the fact that the double mutant largely retained WT activity on non-G4 substrates. What was most notable about the EY mutant was the sharp drop in the dCMP insertion rate on the G4 substrate, which was only around 20% of the rate measured for the non-G4 13/42-mer (Fig. 2A). In this respect, the two mutations in insert-2 seemed to have a strong effect on hRev1-catalyzed dCMP insertion when the primer was positioned 10 nts away from the G4 structure.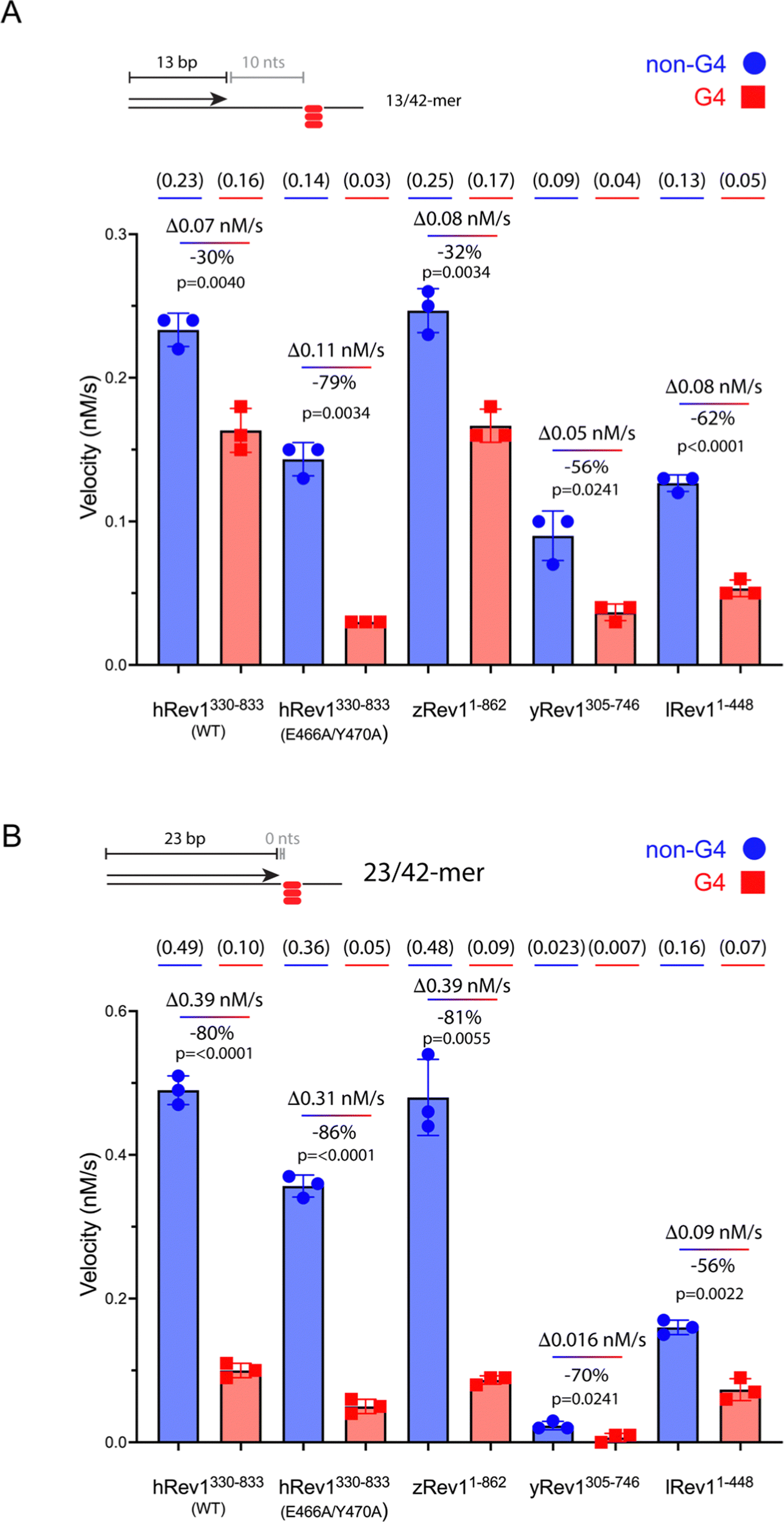 | ||
| Fig. 2 Rev1 catalysis is sensitive to G4 positioning relative to the primer terminus. (A) Single-nucleotide insertion experiments were performed with hRev1 (WT and EY mutant), zRev1, yRev1, and lRev1. A 13-mer primer was annealed to a 42-mer template strand containing either a non-G4 control sequence or the Myc 14/23 G4 motif. The initial rate of dCMP insertion was measured (nM s−1) and plotted for each enzyme. The absolute value for the rate of product formation is shown in parentheses above each data column. The absolute difference between the non-G4 and G4 DNA substrates, along with normalized change in activity for the G4 substrate, is noted for each enzyme. (B) Single-nucleotide insertion experiments were performed with hRev1 (WT and EY mutant), zRev1, yRev1, and lRev1. A 23-mer primer was annealed to a 42-mer template strand containing either a non-G4 control sequence or the Myc 14/23 G4 motif. The initial rate of dCMP insertion was measured (nM s−1) and plotted for each enzyme. The absolute value for the rate of product formation is shown in parentheses above each data column. The absolute difference between the non-G4 and G4 DNA substrates, along with normalized change in activity for the G4 substrate, is noted for each enzyme. Results shown represent the mean (± std. dev.) for three independent replicates. In both panels, results are shown for non-G4 control DNA substrates (blue circles) and G4 DNA substrates (red squares). The percent decrease in activity was calculated for each enzyme by considering the non-G4 control to be 100% active compared to the G4 substrate. The reported p-values were calculated by using an unpaired Student's t-test with Welch's correction. | ||
We then measured dCMP insertion by zRev1, yRev1, and lRev1 on the 13/42-mer DNA substrates. As we observed with the DNA binding assays, zRev1 exhibited catalytic properties that were very similar to WT hRev1, with only a modest drop in dCMP insertion rate on the G4 substrate compared to the non-G4 DNA (Fig. 2A; see Fig. S2, ESI† for gels). Both yRev1 and lRev1 had noticeably slower rates of dCMP insertion on non-G4 substrates compared to hRev1 and zRev1. The dCMP insertion rate for yRev1 and lRev1 dropped 56% and 62%, respectively, on the G4 substrate (Fig. 2A), which closely resembled the 79% drop in activity observed for the EY mutant. Thus, the presence of an intact insert-2 gave zRev1 a better ability to perform nucleotide insertion when the primer was positioned 10 nts from the G4 structure.
The rate of dCMP insertion by the different Rev1 proteins was then measured using the 23/42-mer substrates. In all cases, Rev1 exhibited strongly diminished nucleotide insertion on the 23/42-mer G4 DNA substrates (Fig. 2B; see Fig. S3, ESI† for gels). Of all the proteins tested, lRev1 was least affected by the presence of G4 in the template strand with only a 56% drop in the dCMP insertion rate. From these results and those reported previously,29,30 we concluded that Rev1 is not well adapted for performing catalysis when the primer was placed near the G4 motif. However, it was apparent that the presence of insert-2 did impart a catalytic advantage for WT hRev1 and zRev1 when the primer terminus was positioned a few nts away from the four-stranded structure.
Insert-2 and template base ejection assist in the disruption G4 integrity by Rev1
We previously observed that Rev1 has the ability to disrupt or partially unfold G4 structures without performing DNA synthesis.29 However, the molecular features involved in Rev1-mediated G4 unfolding remain undefined. We reasoned that insert-2 could contribute to the G4 disruptive capacity of Rev1. To monitor G4 folding, we used an assay that relies on the peroxidase-mimicking properties of the G4-hemin DNAzyme.33 Incubating the reaction components with a 29-mer ss-Myc 14/23 G4 DNA substrate bound by hemin resulted in an increase in absorbance near 420 nm (Fig. 3A). Performing the same experiment with non-G4 ss-DNA substrate did not elicit any colorimetric change (Fig. 3A). It should also be noted that the Myc 14/23 G4 sequence provided a better DNAzyme signal than several other G4 DNA substrates (Fig. S4, ESI†). With an assay in hand to monitor G4 integrity, we set out to test the impact of insert-2 on the G4 disruptive capacity of Rev1. | ||
| Fig. 3 Mutations in insert-2 alter the ability of hRev1 to disrupt G4 DNA. (A) A cartoon schematic is shown depicting the DNAzyme-based assay monitoring G4 integrity. Briefly, binding of hemin to an intact G4 structure catalyzes oxidation of ABTS to a peroxidation product with an absorption maximum near 420 nm. Incubating the reaction mixture with a G4-forming 29-mer Myc 14/23 ss-DNA (1 μM) oligonucleotide produces a strong absorbance peak near 420 nm (red circles). Incubating the reaction mixture with a non-G4 ss-DNA (1 μM) oligonucleotide did not produce a detectable change in absorbance near 420 nm (blue squares). (B) The maximum absorbance at 420 nm was plotted as a function of protein concentration for reactions where G4-forming 29-mer Myc 14/23 ss-DNA (1 μM) oligonucleotide was incubated with either BSA (black squares) or WT hRev1330-833 (blue circles). (C) The maximum absorbance at 420 nm was measured for reactions where G4-forming 29-mer Myc 14/23 ss-DNA (1 μM) oligonucleotide was incubated with 5 μM of the indicated proteins. (D) Cartoon schematics are shown to depict the different primer-template DNA substrates used for G4 hemin assay results shown in panel (E–G). Please note that the 3′-OH is positioned 10, 5, or 0 nts away from the first tetrad-associated guanine for the 13-mer, 18-mer, and 23-mer primers, respectively. (E) The maximum absorbance at 420 nm was measured for reactions where 13/42-mer DNA with the Myc 14/23 G4 motif in the template strand (1 μM) was incubated with 5 μM of the indicated proteins. (F) The maximum absorbance at 420 nm was measured for reactions where 18/42-mer DNA with the Myc 14/23 G4 motif in the template strand (1 μM) was incubated with 5 μM of the indicated proteins. (G) The maximum absorbance at 420 nm was measured for reactions where 23/42-mer DNA with the Myc 14/23 G4 motif in the template strand (1 μM) was incubated with 5 μM of the indicated proteins. Results shown in all panels represent the mean (± std. dev.) for three replicates. p-Values in panels (C and E–G) were calculated using an ordinary one-way ANOVA with a Dunnett's multiple comparisons test where ** = p-value < 0.01, *** = p-value < 0.001, **** = p-value < 0.0001, and ns = not significant. | ||
We first added increasing amounts of WT hRev1 into a solution containing the 29-mer ss-Myc 14/23 G4 DNA and measured DNAzyme activity. There was a concentration-dependent decrease in DNAzyme activity when WT hRev1 was incubated with G4 DNA (Fig. 3B). Incubation with the negative control BSA did not alter the DNAzyme activity of the ss-Myc 14/23 G4 substrate (Fig. 3B). These results reinforced the notion that WT hRev1 was capable of disrupting G4 structure. These experiments were performed in the absence of dNTPs, which precluded a role for pol activity during G4 disruption by WT hRev1 and was consistent with our previous findings.29
To further investigate the molecular basis of G4 disruption by hRev1, we measured DNAzyme activity for ss-Myc 14/23 G4 DNA in the presence of five hRev1 mutants. In each case, the enzyme was incubated in 5-fold molar excess of DNA substrate. Under these conditions, only WT and E466K hRev1 were able to suppress the DNAzyme activity, indicating their ability to disrupt the G4 structure (Fig. 3C). This was notable because the E466K mutant was previously found to retain many of the G4-selective properties of WT hRev1, including preferential binding and suppression of mutagenic replication past PDS-stabilized G4 DNA.30 Mutating L358, the residue responsible for ejection of the template base, abrogated the ability of hRev1 to disrupt the G4 structure (Fig. 3C). Likewise, mutating either E466 or Y470 to alanine also eliminated the ability of hRev1 to suppress the DNAzyme activity of ss-Myc 14/23 G4 DNA (Fig. 3C). Adding the EY double mutant slightly increased the G4 DNAzyme signal (Fig. 3C), indicative of a failure to disrupt the G4 structure and perhaps hinting at an ability to stabilize the G4 structure.
To examine hRev1 G4 disruptive capacity on p/t-DNA, we measured DNAzyme activity for a set of three p/t-DNA substrates. The 13/42-mer, 18/42-mer, and 23/42-mer DNA substrates positioned the primer 10, 5, and 0 nts from the first tetrad-guanine, respectively (Fig. 3D). For these experiments, we compared signal from a sample with no protein to that obtained when p/t-DNA was incubated with BSA, WT hRev1, or the EY hRev1 mutant. For all three substrates, the only protein that suppressed G4 DNAzyme activity was WT hRev1 (Fig. 3E–G). Similar to the ss-G4 DNA results (Fig. 3C), the EY mutant did not disrupt the G4 DNAzyme activity for the p/t-DNA substrates (Fig. 3E and F). In this respect, the EY mutant, which exhibited the least selective binding to G4 substrates, was unable to disrupt G4 structures while the WT enzyme could disrupt G4 stability no matter where we positioned the primer terminus.
zRev1 exhibited the strongest G4 disruptive capacity of the three non-human proteins
We repeated the G4 hemin DNAzyme assay with each of the three non-human Rev1 proteins. As with binding and catalysis, zRev1 followed a very similar trend to that observed for WT hRev1. There was a robust decrease in G4 DNAzyme activity with the ss-29-mer G4 substrate even at concentrations of zRev1 that were sub-stoichiometric to DNA (Fig. 4A). An intermediate effect on G4 DNAzyme activity was observed for yRev1, while lRev1 did not affect G4 stability even when incubated at 5-fold excess of the DNA substrate (Fig. 4A). Thus, the yRev1 and lRev1 polymerases both displayed a diminished ability to destabilize the G4 structure on ss-G4 DNA, while zRev1 had G4 properties more akin to those of hRev1. | ||
| Fig. 4 The presence of insert-2 increases the G4 disruptive capacity of Rev1. (A) The maximum absorbance at 420 nm was plotted as a function of protein concentration for reactions where G4-forming Myc 14/23 ss-DNA (1 μM) oligonucleotide was incubated with either BSA (black inverted triangles), zRev11-862 (blue circles), yRev1305-746 (red triangles), or lRev11-448 (gray squares). (B) The maximum absorbance at 420 nm was measured for reactions where 13/42-mer DNA with the Myc 14/23 G4 motif in the template strand (1 μM) was incubated with 5 μM of the indicated proteins. (C) The maximum absorbance at 420 nm was measured for reactions where 18/42-mer DNA with the Myc 14/23 G4 motif in the template strand (1 μM) was incubated with 5 μM of the indicated proteins. (D) The maximum absorbance at 420 nm was measured for reactions where 23/42-mer DNA with the Myc 14/23 G4 motif in the template strand (1 μM) was incubated with 5 μM of the indicated proteins. (E) Based on the results of the G4 hemin DNAzyme assay, the relative G4 disruptive capacity increased when moving across the tree of life from excavata through fungi to vertebrates. Results shown in panels (A–D) represent the mean (± std. dev.) for three replicates. p-Values in panels (B–D) were calculated using an ordinary one-way ANOVA with a Dunnett's multiple comparisons test where ** = p-value < 0.01, *** = p-value < 0.001, and **** = p-value < 0.0001. | ||
The G4 hemin DNAzyme assay was then performed with the non-human Rev1 proteins using the same three p/t-DNA substrates used with human enzyme (Fig. 3D). All three Rev1 proteins suppressed G4 DNAzyme activity on all three p/t-DNA substrates (Fig. 4B–D). The strongest G4 disruptive ability came from zRev1, with yRev1 producing a slightly stronger effect than lRev1 (Fig. 4B–D). In summary, for both ss- and p/t-DNA substrates, the G4 disruptive capacity was strongest with vertebrate forms of Rev1, with yeast and leishmania Rev1 providing diminishing levels of G4 disruption (Fig. 4E).
The G4 disruptive capacity of Rev1 is necessary for stimulation of DNA synthesis by pol κ
Rev1 is a very non-processive enzyme because of its almost exclusive incorporation of dCMP no matter the identity of the template base. Models of Rev1 function typically partner the enzyme with another, more processive DNA pol.20,25,27 We were curious to know if the apparent G4 disruptive ability of Rev1 could assist in DNA synthesis across the entirety of a G4 motif. To examine this possibility, we measured extension by human DNA pol kappa (pol κ) in the presence and absence of Rev1.For the pol extension assays, we used 13/42-mer DNA substrates with either a G4 or non-G4 template strand. The p/t-DNA was pre-incubated with 10 nM pol κ before initiating the reaction with the dNTP mixture (±50 nM Rev1). The total product formation was then quantified and plotted as a function of time to obtain an estimate of pol extension rates. Importantly, neither WT nor EY hRev1 had a significant impact on product formation by pol κ on non-G4 DNA substrates (Fig. 5A). However, very different results were obtained with the G4 substrates. The addition of WT hRev1 strongly increased full-length extension by pol κ on G4 substrates, whereas the EY mutant had no impact (Fig. 5B). These results support a role for quadruplex disruption by hRev1 in stimulation of TLS across the G4 motif.
 | ||
| Fig. 5 Insert-2 is important for stimulation of processive DNA synthesis across a G4 motif by pol κ. Polymerase extension assays were performed to measure the impact of Rev1 on DNA synthesis by a more processive TLS pol. Briefly, human pol κ (10 nM) was incubated with 13/42-mer primer-template DNA (200 nM) for 15 minutes before the reaction was initiated by the addition of solution that contained Rev1 (50 nM), MgCl2 (5 mM), and dNTP solution (0.2 mM total; 50 μM each for dATP, dCTP, dGTP, and dTTP). Pol extension was allowed to proceed at 37 °C before quenching at the indicated timepoints. Substrate and products were separated by PAGE using 14% (w/v) polyacrylamide gels with 7 M urea. (A) Pol extension assay results are shown for 13/42-mer non-G4 DNA where pol κ was incubated alone or in the presence of either WT hRev1 or the EY mutant. (B) Pol extension assay results are shown for 13/42-mer G4 DNA where pol κ was incubated alone or in the presence of either WT hRev1 or the EY mutant. (C) Pol extension assay results are shown for 13/42-mer non-G4 DNA where pol κ was incubated alone or in the presence of zRev1, yRev1, or lRev1. (D) Pol extension assay results are shown for 13/42-mer G4 DNA where pol κ was incubated alone or in the presence of zRev1, yRev1, or lRev1. Product formation for each reaction was quantified and normalized against the amount of product formed in the reaction with pol κ alone. The results are shown to the right of each set of gels and represent the mean (± std. dev.) of three independent replicates. Quantification of product formation by pol κ alone is re-plotted in each panel to allow more direct comparison with the reactions containing Rev1. p-Values were calculated using an ordinary one-way ANOVA with a Dunnett's multiple comparisons test where *** = p-value = 0.0008, **** = p-value < 0.0001, and ns = not significant. | ||
The pol κ extension experiments were then performed with non-human Rev1 proteins. Similar to the results with hRev1, none of the non-human Rev1 proteins greatly impacted pol κ extension on the non-G4 DNA substrate (Fig. 5C). Of the three non-human enzymes, zRev1 had the strongest stimulatory effect on pol κ extension across the G4 motif (Fig. 5D). However, we note that the addition of yRev1 did produce a band representing full-length extension, indicative of some power to stimulate pol κ-mediated synthesis past the G4 structure (Fig. 5D, middle gel). By way of comparison, lRev1 failed to stimulate the appearance of a fully extended primer (Fig. 5D, gel on right). Thus, processive DNA synthesis across the G4 motif by pol κ was most clearly stimulated by forms of Rev1 that were also adept at disrupting quadruplex structures.
Discussion
The relationships that exist between dynamic DNA structures and proteins that guard genomic integrity are multifaceted.1,2 Certain nucleic acid sequences adopt structures with distinctive chemical and biophysical properties that are connected to biological function and impact normal physiological processes.6,7,34–36 The mechanical properties of nucleic acids that allow deformation and bending ultimately govern processes like DNA replication and repair and transcription initiation and elongation, leading to the postulation of a “mechanical code” for the genome and epigenome.37 The need to retain the physiological functions of motifs with distinctive mechanical properties has undoubtedly helped shape the selection of proteins and enzymes over evolutionary time in ways that were driven by species-specific cues. Yet, much remains unknown regarding how proteins and enzymes from different species interact with non-canonical nucleic acid structures like G-quadruplexes.The last decade has seen a veritable explosion of studies focused on the chemistry and biology of G4 nucleic acids.8,12,38 From neurobiology and cancer to virology and nanotechnology applications, G4 nucleic acids have been the subject of intense investigation.39–43 The dynamic topology of the four-stranded structures and the planar nature of the tetrads each present unique molecular docking sites for proteins, peptides, and small-molecules that specifically interact with G4 DNA and RNA.44–49 Naturally occurring G4 recognition motifs have been identified, with one of the best-studied G4 recognition motifs being comprised of a series of Arg-Gly-Gly (RGG or RG) repeats.50,51 Previously referred to as Gly-Arg-rich (GAR) domains or RGG “boxes”, the RGG repeat domain is a flexible peptide motif found conserved across species in thousands of proteins that have functional roles in many cellular processes, with mutation and/or dysregulation of RGG/RG-containing proteins having been associated with diseases, such as neurological conditions, neuromuscular pathologies, and cancer.52,53 Often partnered with other nucleic acid binding domains, RGG repeats are not exclusive G4 binding domains and may help by fine-tuning binding interactions in ways that may involve biomolecular condensates through modulation of phase separation.54–56 In the case of the Fragile X Mental Retardation protein, interactions between RGG peptides and a quadruplex-duplex junction involve an intricate network of hydrogen bonds that are facilitated by a type I β-turn in the RNA-bound RGG box.56 For the G4-binding hnRNPA1 protein, the RGG-G4 interactions are thought to contribute to cooperative binding and G4 disruption through interactions with the UP1 domain of hnRNPA1.57–60
Other G4-selective proteins rely on molecular interactions that fall outside of the classic RGG motif. For example, activation-induced cytidine deaminase (AID) has conserved residues (R24, G133) that contribute to G4 recognition, but there is no clear RGG domain.61,62 The Werner syndrome protein (WRN) has been implicated in G4 processing at both the biochemical and cellular level.20,63–65 Residues in the conserved RecQ C-terminal domain contribute to the G4 unwinding activity of WRN,66 apparently without an RGG domain. Likewise, a recent structural study reported on a dual-function DNA recognition helix in the yeast Rap1 transcriptional regulator that had no obvious RGG domain but still assisted in G4 binding.67 In contrast to the electrostatic forces that dominate interactions between RGG motifs and quadruplexes, the Rap1 G4 interface primarily involved hydrophobic interactions with the planar tetrad. Similarly, a co-crystal structure of DHX36 and a Myc-derived G4 DNA substrate was solved, revealing a complex set of interactions that include a hydrophobic core of residues in the DHX36-specific domain and recognition of the single-stranded nucleic acid backbone on both faces of a partially unfolded parallel-stranded quadruplex DNA substrate.68 DHX36 is a DEAH/RHA helicase that binds and unwinds both RNA and DNA quadruplexes and has been implicated in a variety of cellular processes, ranging from DNA replication and telomere maintenance to RNA metabolism.69 Like Rev1, DHX36 appears to preferentially act on parallel-stranded G4 substrates70–72 to the point where short DHX36-derived peptides have been developed for specific recognition of parallel G4s.46,73 For DHX36, the selectivity for parallel-stranded G4 may be due to the fact that diagonal lateral loops in hybrid and anti-parallel G4 folds were predicted to interfere with binding of the hydrophobic core residues to the tetrad face. Similar to our results with hRev1 Y470A, mutation of a residue with an aromatic side-chain (Y69) greatly weakened the affinity of DHX36 for G4 DNA.68 So, while it is difficult to make direct comparisons between these studies because of differences in the G4 motifs used and other technical factors, it does seem as if the nature of G4 recognition is a malleable feature of proteins implicated in maintenance of these non-canonical structures.
Results accumulated thus far point towards insert-2 as a key part of the G4-related properties of Rev1. We previously observed that binding of hRev1 to G4 DNA selectively protected R458 from reaction with p-hydroxyphenylglyoxal.30 Interestingly, the region containing R458 (a.a. 447–461) and another region on the C-terminal side of E466 and Y470 (a.a. 473–498) are both disordered in the crystal structure of hRev1 bound to p/t-DNA.23 It is possible that, analogous to the DHX36-specific motif, binding to G4 DNA induces more structure in the region that arches over the αE helix, which contains E466, W467, Y470, and helps to form the G-loop (Fig. 6A). The AlphaFold (AF) predicted structures of hRev1 and zRev1 both possess an arch-like structure that emerges on the C-terminal side of the residues forming the G-loop (Fig. 6A). By way of comparison, both of the AF-predicted yRev1 and lRev1 completely lack the insert-2 extension (Fig. 6A), which corresponds with previously solved structures of yRev1.74,75
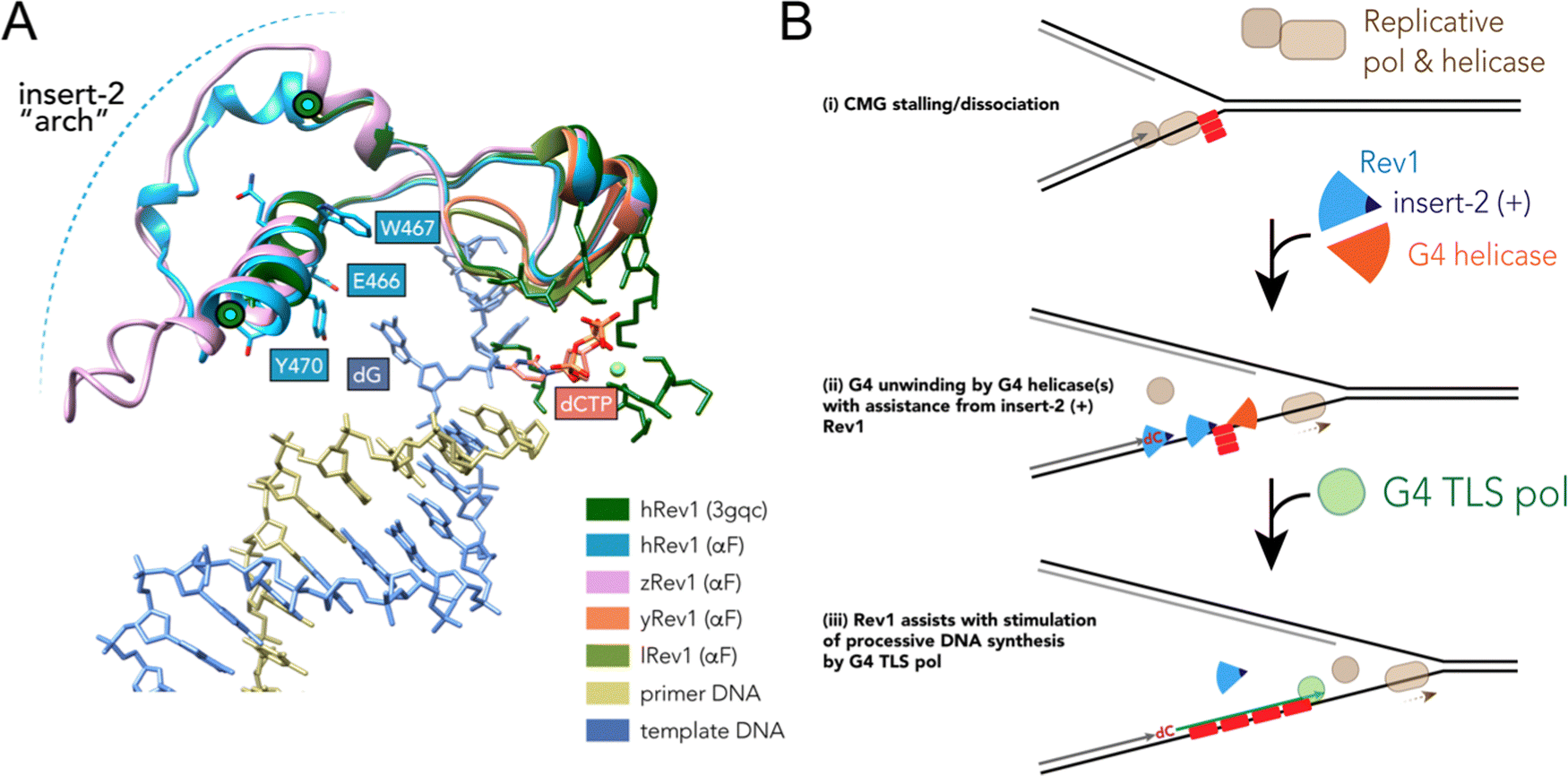 | ||
| Fig. 6 Model of selective structural features and activities important for Rev1 action during G4 replication. (A) The ternary structure of hRev1 (PDB ID 3GQC) was used as a reference structure for superimposition of models predicted by alphaFold103,104 using the Matchmaker function in UCSF Chimera version 1.14. The region of each protein near insert-2 (or the corresponding region from yRev1 and lRev1, which lack insert-2) are depicted in cartoon ribbon form. Insert-2 residues from the 3GQC structure that are in the vicinity of the ejected template base (dG) are noted and shown in ball and stick form, as is the position of the incoming dCTP. (B) A cartoon model is shown depicting Rev1-coupled G4 replication. The cartoon model was adapted in part from ref. 79, incorporating our findings related to Rev1 activity on G4 DNA. Evidence to support the model and additional details are provided in the Discussion. | ||
While insert-2 certainly seemed to provide some element of G4 selectivity, there are other features of Rev1 that still bear exploring with regard to G4 interactions. One of the more intriguing differences between the current study and previous work was with zRev1 and the anti-parallel TBA G4 substrate (Tables 2 and 3). Of all the enzymes tested in this study, only zRev1 still retained the N-terminal BRCT domain. As noted earlier, we have reported that full-length hRev1 and the pol core (a.a. 330–833) both bound parallel-stranded G4 DNA with much greater affinity than non-G4 DNA, which led us to assume that the pol core possessed all the features needed for G4 selective interactions. This was perhaps an overly simplistic assumption and does not account for cell-based results implicating Rev1 in maintenance of G4 motifs capable of forming multiple types of quadruplex folds.21 Other studies have implicated the extreme N-terminal region of murine Rev1 (mRev1) as being important for binding to DNA substrates possessing a 5′-phosphorylated terminus located on a recessed primer-template junction.76 Upon reflection, it is possible that the extreme N-terminal region of Rev1, which happens to have the only RGG motif in either the human or the mouse enzyme (but not zebrafish), could also influence binding to anti-parallel substrates. Such an interaction could conceivably provide Rev1 will greater versatility on structured templates or alter the binding mode of the enzyme. Future studies investigating these features are warranted and could shed light on these possibilities.
The potential influence of the G4-interacting insert-2 motif on DNA synthesis was very apparent when we measured pol activity using so-called “running-start” primer extension. As shown in Fig. 5, Rev1 can stimulate processive DNA synthesis across the G4 motif by pol κ, another TLS enzyme. The stimulation only occurred on substrates with a G4 motif in the template and seemed to parallel the G4 disruptive capacity measured with the G4 hemin assay (Fig. 3 and 5). To our knowledge, this is the first time such an effect has been reported. It is conceivable that a vertebrate-specific protein–protein interaction produced the stimulatory effect on pol κ observed for hRev1 and zRev1. However, we consider this scenario to be very unlikely given that the recombinant pol κ used in our pol extension assays lacked the Rev1-interaction region, and the Rev1 constructs lacked the C-terminal scaffolding region known to facilitate protein–protein interactions with TLS pols. Also, there was a slight stimulation of pol κ-catalyzed full-length extension by yRev1, which mirrored the G4 hemin results. Instead, we favor a model where Rev1 disrupts some aspect of G4 structure so that pol κ (and likely other pols) can then extend the primer across a less structured template strand. Such an interpretation expands the proposed roles for Rev1 during bypass of G4 DNA.
TLS across the entire motif presumably occurs on a completely unwound substrate, but the exact impact of Rev1 on G4 structure remains something of a mystery. The results reported here and elsewhere indicative of G4 disruption by Rev1 alone could result from changes that do not elicit unfolding of the entire four-stranded structure. Instead, the changes in the G4 hemin assay might be the result of more subtle alterations in G4 integrity (e.g., slight changes in tetrad geometry, movement of loop orientations, changes in cation stability). Indeed, G4 structures are dynamic and are known to refold spontaneously. A previous study provided evidence to support the idea that the FANCJ helicase catalyzed repeated rounds of partial G4 unfolding and refolding and that this process kept the substrate intact for interactions with Rev1.77 Given that Rev1 and FANCJ function together to help the replisome copy G4 DNA,20,78 it seems plausible that the combined G4 disruptive activities of Rev1 and FANCJ are required for persistent disruption of the G4 structure, which then allows facile DNA synthesis across the guanine rich motif.
The stepwise contribution of Rev1 to fork progression past structured or damaged DNA remains an active topic of investigation. Based on the results presented here, vertebrate Rev1 could be doing more than inserting a nucleotide or two and then recruiting other TLS pols to sites of replication stress. A recent study used the Xenopus model system to report on the multi-step mechanism of G4 replication, especially the role played by DHX36.79 In their model, the authors proposed that DHX36 remains bound to a folded G4 structure until a replication fork approaches the site. G4-induced dissociation of the replicative cdc45-MCM-GINS (CMG) helicase necessitates creation of a ss-DNA loading zone by DHX36 to allow continued unwinding.79 Dissociation of the CMG helicase left a 13–26 nt gap between the stalled polymerase and the G4 motif. This space between the primer terminus and the G4 structure may be an optimal length for recruitment of TLS machinery to assist with closure of the gap. In this respect, the ss-DNA binding and G4 disruptive properties of Rev1 might prove especially important for maintaining forward progress of the replisome (Fig. 6B). We previously reported that loss of Rev1 resulted in the accumulation of mutations near the Myc G4 motif, as measured by the supF mutagenesis assay, with mutations in the 3′ flanking region being enriched upon G4 stabilization with PDS.30 It seems possible that an outcome of Rev1-mediated G4 disruption at these gaps could be to further reduce the likelihood of mutations that ablate G4 structure and function by stimulating processive and accurate DNA synthesis. This would almost certainly occur in conjunction with the actions of other proteins and enzymes (e.g., FANCJ, RPA, and other G4 helicases), especially when one considers that loss of the Rev1-interacting FANCJ helicase is critical for suppression of G4-induced strand breaks in C. elegans.80 While not directly shown to occur at G4 sites, multiple studies have highlighted the importance of Rev1 in the suppression of ss-DNA gaps.24,25,81 An important future goal will be to determine whether the G4-selective properties we have identified for the purified Rev1 enzyme help mediate ss-DNA gap suppression in cells burdened with a more rugged G4 landscape, such as that observed for multiple cancer types.7,42,82,83
The exact enzymes and sequence of interactions that govern DNA synthesis activity during G4 replication have remained subjects of intense scrutiny.9 Although multiple TLS pols have been implicated in G4 replication,19,28,84,85 the order of addition and division of labor between DNA pols is still obscure. While we artificially combined Rev1 and pol κ in our extension assays, it is not clear what circumstances might bring the activities of these two enzymes together to copy G4 motifs in cells.
Pol switching models sometimes invoke mono- and poly-ubiquitination of PCNA as the primary or sole means of stabilizing Y-family pols at sites of replication stress during normal S-phase as well as during post-replication repair of unfilled ss-DNA gaps.86–88 Yet, Rev1 can operate outside of DNA damage tolerance mechanisms involving PCNA ubiquitination,89 and the C-terminal domain of Rev1 interacts with multiple TLS enzymes.90,91 Moreover, increasing the stable ubiquitination of PCNA requires activation of the replication stress response and may not be essential for “on-the-fly” TLS promoted by Rev1.89,92 This is not to say that Rev1 does not engage with pathways involving PCNA-ubiquitination, but if we assume that Rev1 acts as a first-responder to G4-induced replication stress, then it seems reasonable to propose that the tetrad-disruptive properties of Rev1 could combine with the G4 helicase activity of FANCJ to obviate the need to recruit additional TLS pols and fully activate the replication stress response unless widespread fork-stalling or damaged quadruplexes are encountered. Key aspects of such a model were originally proposed several years ago by the Sale laboratory.19,20 When G4 abundance is elevated or some other factor impairs fork progression, then cells may rely on Rev1 and activation of PCNA-ubiquitination pathways to further stabilize localization of other replication factors, including additional Y-family pols, to assist with G4 bypass. The C-terminal domain of Rev1 serves as a landing platform for enzymes like pol ζ and the other Y-family pols and is relevant to Rev1 function during G4 bypass.19 Given that pols η and κ possess distinct nucleotide insertion properties on G4 substrates,84,93,94 it is possible that once recruited by Rev1 and ubiquitinated PCNA, the intrinsic features of these Y-family pols could then guide distinct replication outcomes in cells experiencing high levels of G4-induced replication stress. Discerning when and how TLS pols are coordinated to assist with G4 bypass remains a challenge.
The distinct G4 properties observed for the non-human Rev1 proteins provides us with a singular opportunity to consider how the selection of proteins with an insert-2 motif could be tied to the biological functions of this TLS enzyme. Based on sequence alignment, the appearance of organisms encoding the insert-2 motif in the REV1 gene might have coincided with the emergence of the adaptive immune response in jawed vertebrates around 500 million years ago.95 This is interesting given the demonstrated role for Rev1 during somatic hypermutation (SHM) and B-cell diversification, where it may be critical for transversion mutations at G/C bases.96 Given that Ig loci have high G4 forming potential97 and AID localization is determined in-part through interactions with non-canonical DNA structures, such as G4 DNA,61,83 it is possible that the G4 properties imparted by insert-2 somehow affect the function of Rev1 during SHM. Previous work revealed that the catalytic activity of Rev1 is dispensable for recruitment of the uracil DNA glycosylase UNG that follows AID-catalyzed deoxycytosine deamination,98 which begs the question: do the G4-selective properties of Rev1 influence its function at immunoglobin loci during antibody diversification? Is the act of localizing Rev1 to G4 sites sufficient for recruitment of UNG or does the tetrad disruptive capacity of Rev1 help regulate targeted hypermutation by the AID/UNG pathway? The need for robust G4 recognition may have driven the selection for mutations in the REV1 gene leading to the emergence of insert-2, a process likely tied to taxa-specific functions for non-B DNA. It will be interesting to pursue these questions in future studies.
The G4-related properties of Rev1 could also be important in the context of cancer, where precise regulation of G4 formation and dissolution is thought to be a critical barrier to oncogenic and pro-malignant processes. This notion is supported by a general increase in the overall abundance of G4 DNA in cancer-derived cells and specimens.6,7,42 In a more recent study, Tet2 and Tet3-deficient mice that spontaneously develop germinal center-derived B cell lymphomas exhibit massively elevated genomic G4 DNA and R-loops at immunoglobin switch regions in tumor cells.82 The Tet-deficient B cells were found to be dependent upon G4 helicases like ATRX, WRN, and the Bloom syndrome helicase for survival,82 illustrating the importance of effective G4 resolution in the control of tumor growth. REV1 transcripts were not found to be differentially expressed in response to Tet-deficiency while BACH1 (FANCJ) transcripts were downregulated.82 Since Rev1 and FANCJ are known to ensure proper fork progression past quadruplex-forming sequences,20 selection against upregulation of these two G4 enzymes might be part of the mechanism driving pro-oncogenic translocations. Such a scenario fits with the increased formation of DNA double-strand breaks near G4 DNA sites observed in Tet-deficient B cells.82
A very broad view of Rev1 evolution would consider the physical and chemical properties that existed at the time when the insert-2 motif appeared. The emergence of organisms capable of shallow burrowing during the Cambrian period could have contributed to increased surface temperatures on earth,99 and this may have happened around the same time as the insert-2 motif began to appear in the Rev1 protein from some species. Combined with other biogeochemical changes, including those related to dynamic alterations in the redox state of the ocean,100 one could envision a scenario where factors influencing G4 quadruplex folding and stability (i.e. temperature and reactive oxygen species) were changing on a global scale in a way that might have necessitated selection of G4 motifs with greater intrinsic stability. It is interesting to consider the possibility that these environmental changes might have impacted the selection for proteins and enzymes, like Rev1, with features suited to handle the dynamic G4 landscape of a changing world, but these ideas exist purely in the realm of speculation. The evidence provided by our study is, however, firmly supportive of the idea that insert-2 provides Rev1 from different species with an expanded arsenal of G4-selective properties.
Material and methods
Reagents and services
All chemicals and reagents used in this study were molecular biology grade or better. The dNTP solutions were purchased from Thermo-Fisher Scientific (Waltham, MA, USA). All synthetic oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA, USA), desalted and validated by mass spectrometry. DNA sequencing was performed at the DNA Sequencing Core facility at UAMS to confirm mutations in plasmids used for expression of recombinant hRev1 mutants and non-human Rev1 proteins.DNA substrate preparation
Oligonucleotides were resuspended in 50 mM HEPES buffer (pH 7.5). For experiments with ss-DNA substrates, stock solutions of the 6-carboxyfluoroscein (FAM)-labeled G4 and non-G4 oligonucleotides were added to a solution of 50 mM HEPES buffer (pH 7.5) containing either 100 mM KCl or 100 mM LiCl. P/t-DNA substrates for the fluorescence polarization experiments were prepared by annealing each of the FAM-labeled G4 and non-G4 oligos shown in Table 1 with the 11-mer primer (1:2 molar ratio of template:primer) in a 50 mM HEPES buffer (pH 7.5) containing either 100 mM KCl or 100 mM LiCl. The p/t-DNA substrates used for enzyme activity assays were prepared similarly by mixing the 42-mer G4 or non-G4 template oligonucleotides with the 5′-FAM-labeled 23-mer or 13-mer primer (1:1.5 molar ratio of template:primer) in 40 mM HEPES buffer (pH 7.5) containing 40 mM KCl. In all cases, the mixture was heated at 95 °C for 5 min, followed by slow-cooling to room temperature. The annealed substrates were stored in amber-colored tubes at room temperature and kept in the dark.
Cloning, expression, and purification of Rev1 proteins
Expression and purification of WT hRev1330-833 (the polymerase core) from Escherichia coli was performed as previously described.29 Preparation of the L358A, E466A, E466K, and Y470A mutant forms of hRev1 were also described in our previous publication.30 The expression and purification of human pol κ19-526 was performed as described previously.101 The hRev1330-833 E466A/Y470A (EY) construct was generated as follows: initially, the E466A point mutation was generated using the pBG101 plasmid encoding WT hRev1330-833 as template, and the primer pair – (a) E466A: forward primer-5′-CCCAGCTGGCGTGGCAGTATTAC-3′; and reverse primer-5′-GTAATACTGCCACGCCAGCTGGG-3′. After confirming the mutation in the resulting PCR-amplified plasmid, the E466A mutated plasmid was used as a template to generate the second point mutation Y470A, using (b) Y470A: forward primer 5′-GGCAGTATGCCCAGAATAAAATC-3′; reverse primer-5′-GATTTTATTCTGGGCATACTGCC-3′. Mutations were confirmed by DNA sequencing. The mutant hRev1330-833 E466A/Y470A protein was expressed and purified using the same protocol as WT enzyme.Expression plasmids for the Rev1 proteins from S. cerevisiae (yRev1), L. donovani (lRev1) and D. rerio (zRev1) were purchased from Genscript Biotech Corp (Piscataway, NJ) and were designed as follows: the ORFs corresponding to the full-length lRev1 (amino acids 1–619) and zRev1 (amino acids 1–1268), and residues corresponding to the pol core (amino acids 305–746) for yRev1 were cloned into the pET28b vector. In the case of the yRev1 and lRev1 constructs, a tandem N-terminal hexa-histidine and glutathione-S-transferase (His6-GST) affinity tag was added. A sequence corresponding to the cleavage site for the HRV3C protease was inserted between the affinity tag and the Rev1 ORF, similar to the hRev1 construct. For zRev1, only a N-terminal His6-tag was included to generate a fusion construct. Subsequently, tandem stop codons were introduced to produce C-terminally truncated constructs for lRev1 (amino acid residue 448) and zRev1 (amino acid residue 862) through site-directed mutagenesis PCR. These truncations were designed to generate Rev1 proteins matching closely with the polymerase core domain of hRev1 (amino acid residues 330–833) based on primary sequence alignments.
For protein expression, each of the three plasmid constructs (lRev11-448, yRev1305-746 and zRev11-862) was transformed into E. coli BL21(DE3) competent cells and colonies were grown at 37 °C on LB-agar medium containing 50 μg mL−1 kanamycin for selection. Large-scale cultures (between 4–6 liters) were grown in LB medium containing 50 μg mL−1 kanamycin on a shaker by incubation at 37 °C until an optical density of OD600nm ∼ 0.5–0.6 was reached. At this stage, protein expression was induced by the addition of 1 mM isopropyl β-D-thiogalactopyranoside. The cultures were then incubated at 18 °C for an additional 15 hours before harvesting.
Cells were harvested by centrifugation, and the cell pellets were washed with phosphate buffered saline, flash-frozen in liquid nitrogen, and stored at −80 °C until further use. For purification, frozen cell pellets were thawed on ice and resuspended in a 50 mM Tris–HCl (pH 7.4) buffer containing 0.5 M NaCl, 10% (v/v) glycerol, 5 mM 2-mercaptoethanol (β-ME), lysozyme (1 mg mL−1) and a protease inhibitor cocktail (Fisher Scientific) was added to the harvested pellet. The suspension was sonicated and supernatant recovered by ultracentrifugation (35 000 × g, 1 h, 4 °C). Each protein was purified by affinity chromatography using Ni Sepharose (GE Healthcare Life Sciences). Briefly, the protein was bound to a nickel-chelating column. The column was then subjected to sequential wash steps with increasing imidazole in each wash buffer, and fractions containing protein as measured by in-line UV absorbance were collected and analyzed by SDS-PAGE to confirm which UV peak contained the purified Rev1. All three non-human Rev1 proteins eluted between 60–100 mM imidazole from the Ni Sepharose column. In the case of zRev1, the purified protein was dialyzed to remove imidazole and then loaded on a Superdex-75 size exclusion column (GE Healthcare Life Sciences) for a further polishing step. This two-step purification resulted in a zRev1 protein that was ∼95% pure, as confirmed by SDS-PAGE analysis (Fig. S1, ESI†). The purified protein was concentrated to ∼40–50 μM in a 25 mM HEPES (pH 7.5) buffer containing 0.2 M NaCl, 5 mM β-ME and 30% (v/v) glycerol and aliquots were frozen at −80 °C until further use. In the case of yRev1 and lRev1, the affinity-purified proteins from the first step were dialyzed to remove imidazole and were purified further by an additional affinity step using a glutathione sepharose column (GE Healthcare Life Sciences). The bound proteins were treated with HRV3C protease (Fisher Scientific) on the column as recommended by the manufacturer. The untagged Rev1 proteins were eluted in 25 mM HEPES pH 7.5 buffer containing 0.2 M NaCl, 10% (v/v) glycerol and 5 mM β-ME. The protein was found to be ∼80–90% pure as determined by SDS-PAGE, and was concentrated to ∼30–50 μM. Glycerol was added to the concentrated proteins to a final concentration of 30% (v/v), and aliquots were stored frozen at −80 °C.
DNA binding by fluorescence polarization
The affinity of the Rev1 proteins towards 5′-FAM labeled non-G4 and G4 DNA substrates described in this study was determined using fluorescence polarization on a plate-reader (Biotek SynergyH4), similar to experiments described previously.29 Briefly, titrations of various concentrations of each protein with a fixed concentration of the ss- and p/t-DNA substrates (2 nM) were performed at room temperature (approximately 25 °C) in 40 mM HEPES (pH 7.5) buffer containing 5 mM MgCl2, 0.1 mM EDTA, 2 mM β-ME, 0.1 mg mL−1 BSA and either 100 mM KCl or 100 mM LiCl. The change in fluorescence polarization at every concentration of protein was measured, plotted as a function of protein concentration, and was fit to a quadratic equation, as described previously,29 to determine the values of equilibrium dissociation constants. Three independent experiments were performed for all assays and results were expressed as mean ± std. dev. (Fig. S5–S12, ESI†). A two-tailed unpaired Student's t-test with Welch's correction was used to determine if there was a statistically significant difference in the measured binding constants for G4 and non-G4 DNA substrates.Measurement of Rev1 cytidyl transferase activity
The cytidyl transferase activity of the Rev1 proteins was determined with non-G4 and G4 DNA template-primer substrates of varying primer length. All assays were performed in 40 mM HEPES buffer (pH 7.5) containing 100 mM KCl, 0.1 mg mL−1 BSA, and 5 mM dithiothreitol at 37 °C using 50 nM protein and 200 nM DNA. Reactions were initiated by adding a mixture of 5 mM MgCl2 and 1 mM dCTP. Aliquots of 15 μL were withdrawn from each reaction mixture at intervals of 0, 10, 30, 60, 120, 300 and 600 seconds and added to tubes containing 85 μL of quench solution (95% v/v formamide, 20 mM EDTA, 0.1% w/v bromophenol blue), and heated at 95 °C for 5 min. Aliquots of these quenched reactions were loaded on to 7 M Urea/14% (w/v) polyacrylamide gels and electrophoresed at a constant power of 35 W for 2–3 hours to separate the products (Fig. S2 and S3, ESI†). Gels were scanned on a Typhoon Imager (GE Life Sciences, Marlborough, MA, USA), and the bands corresponding to substrate and products in each lane were quantified using the ImageJ software.102 Total product formation for each protein with both the G4 and non-G4 DNA substrates was plotted as a function of time. Product formation was calculated by dividing the sum of intensities of all product bands in a lane by the sum of intensities of all bands in that lane and then multiplying by the substrate concentration. The initial portion of the velocity curve was fit to a linear equation to estimate the rate of product formation. Three independent experiments were performed for all assays and results were expressed as mean ± std. dev. A two-tailed unpaired Student's t-test with Welch's correction was used to determine if there was a statistically significant difference in polymerase activity for G4 and non-G4 DNA substrates.Polymerase extension assay
Polymerase extension assays were performed to measure the enzyme activity of human pol κ19-526 on G4 and non-G4 13/42 p/t-DNA substrates. The extension experiments were performed in the presence and absence of Rev1, including WT and mutant hRev1, as well as zRev1, yRev1, and lRev1. The assay was performed in the same buffer used for the cytidyl transferase assays, using 10 nM pol κ enzyme, 200 nM 13/42 G4/non-G4 DNA, 5 mM MgCl2, and 0.2 mM dNTP solution (50 μM each for dATP, dCTP, dGTP and dTTP). Prior to each reaction, the pol κ enzyme was incubated with DNA for 15 min, thereby giving pol κ access to the primer before Rev1. Reactions were initiated by adding a cocktail containing Rev1 (50 nM) and the Mg-dNTP mixture. All reactions were performed at 37 °C, and aliquots were withdrawn after 0, 0.5, 1, 2, 5, 10, 30 and 60 minutes from each reaction into a quench solution. Separation of products, visualization and quantification was done as described above for the cytidyl transferase reactions. Product formation for the pol extension assay was calculated by measuring the substrate band and all of the product bands. The ratio of product over total DNA (i.e. all product bands/[substrate + all product bands]) was multiplied by the concentration of DNA used in the assay (200 nM) to obtain an estimate of the concentration of product formed at each time point. Re-plotting product formation as a function of time allowed us to calculate a rate of pol extension. The rate of pol extension for each condition was normalized to the rate of pol extension by the pol κ enzyme alone for that DNA substrate, which was taken as 100%, and reported as percent activity. Three independent experiments were performed for all assays and results were expressed as mean ± std. dev. Product formation was quantified using Fiji.102 All the results were plotted using the GraphPad Prism software (San Diego, CA). A two-tailed unpaired Student's t-test with Welch's correction was used to determine if there was a statistically significant difference in polymerase activity for G4 and non-G4 DNA substrates.G4 Hemin DNAzyme assay
We used a DNAzyme-based assay to monitor the integrity of G4 structures. Our protocol largely followed that used recently to study the G4-related activities of the DHX36 helicase.33 DNA substrates were prepared in 100 mM Tris–HCl buffer (pH 7.5) containing 100 mM KCl. The G4-Hemin reactions were performed in 25 mM HEPES-NaOH buffer (pH 7.4) containing 100 mM KCl, 5 mM MgCl2, 0.05% (v/v) Triton-X 100 and 1% (v/v) dimethyl sulfoxide (DMSO). The reactions were performed in 96 well plates and followed an exact order of addition where DNA was added to reaction buffer before the addition of protein. Following addition of protein, the reaction was incubated for 15 minutes at room temperature. Hemin was added next at a concentration that was twice the final concentration and the reaction was again allowed to incubate for 15 minutes at room temperature. After the 15 minute incubation with hemin, the DNAzyme reaction was initiated by addition of an equal volume of solution containing H2O2, 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid (ABTS) resuspended in the reaction buffer. Reactions were performed at 25 °C. The final concentrations in the reaction mixture were 1 μM DNA, 2.5 μM hemin, 0.4 mM H2O2, 2.5 mM ABTS and variable amounts of protein. We compared multiple G4-forming sequences and found that the Myc 14/23 motif provided the most robust DNAzyme activity (Fig. S4A and B, ESI†). All experiments reported in the main text used a derivative of the Myc 14/23 sequence or the non-G4 control. For experiments investigating ss-DNA, a 29-mer Myc 14/23 oligonucleotide was used. For experiments with p/t-DNA, a 42-mer Myc 14/23 template oligonucleotide was used with primers of varying lengths. In all cases, unlabeled oligonucleotides were used (i.e. no FAM-label). To ensure that any carryover of β-ME from the protein storage buffer did not interfere in the oxidation reaction, mock protein dilution experiments were performed (Fig. S4C and D, ESI†). Absorbance readings were taken from 400–460 nm over a period of 2–3 hours since there was a time-dependent increase in the colorimetric change that plateaued at slightly different times depending on the presence or absence of protein. The maximum absorbance at 420 nm was used as an estimate of G4 integrity. Three replicates were performed for each condition and the results were plotted in GraphPad Prism. Where indicated, p-values were calculated using an ordinary one-way ANOVA with a Dunnett's correction for multiple comparisons.Data availability
Unaltered raw data that pertains to figures, graphs, and tables in the manuscript are available through the Open Science Foundation at https://osf.io/vbw5z.Conflicts of interest
The authors have no conflicts of interest to report.Acknowledgements
This work was supported in part by a National Science Foundation grant (NSF-MCB 1903357 to R. L. E. and J. E. C. G.) with additional support from the Arkansas Breast Cancer Research Program (AWD00053282 to R. L. E.), a Barton Bridging Award from the UAMS College of Medicine (to R. L. E.), and the UAMS Translational Research Institute grant (UL1 TR003107) through the National Center for Advancing Translational Sciences of the NIH. The authors thank Alicia Byrd, PhD, and Samantha Kendrick, PhD, for critically reading their manuscript and for helpful suggestions related to the study.References
- G. Wang and K. M. Vasquez, Mutat. Res., 2006, 598, 103–119 CrossRef CAS PubMed.
- R. D. Wells, Trends Biochem. Sci., 2007, 32, 271–278 CrossRef CAS PubMed.
- J. A. Capra, K. Paeschke, M. Singh and V. A. Zakian, PLoS Comput. Biol., 2010, 6, e1000861 CrossRef PubMed.
- G. Marsico, V. S. Chambers, A. B. Sahakyan, P. McCauley, J. M. Boutell, M. D. Antonio and S. Balasubramanian, Nucleic Acids Res., 2019, 47, 3862–3874 CrossRef CAS PubMed.
- V. S. Chambers, G. Marsico, J. M. Boutell, M. Di Antonio, G. P. Smith and S. Balasubramanian, Nat. Biotechnol., 2015, 33, 877–881 CrossRef CAS PubMed.
- R. Hänsel-Hertsch, D. Beraldi, S. V. Lensing, G. Marsico, K. Zyner, A. Parry, M. Di Antonio, J. Pike, H. Kimura, M. Narita, D. Tannahill and S. Balasubramanian, Nat. Genet., 2016, 48, 1267–1272 CrossRef PubMed.
- G. Biffi, D. Tannahill, J. Miller, W. J. Howat and S. Balasubramanian, PLoS One, 2014, 9, e102711 CrossRef PubMed.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- L. K. Lerner and J. E. Sale, Genes, 2019, 10, E95 CrossRef PubMed.
- A. M. Fleming and C. J. Burrows, Chem. Soc. Rev., 2020, 49, 6524–6528 RSC.
- M. Stein and K. A. Eckert, Genes, 2021, 12, 1779 CrossRef CAS PubMed.
- A. M. Fleming and C. J. Burrows, NAR Cancer, 2021, 3, zcab038 CrossRef PubMed.
- T. M. Bryan, Mol. Basel Switz., 2019, 24, 3439 CAS.
- J. P. Vaughn, S. D. Creacy, E. D. Routh, C. Joyner-Butt, G. S. Jenkins, S. Pauli, Y. Nagamine and S. A. Akman, J. Biol. Chem., 2005, 280, 38117–38120 CrossRef CAS PubMed.
- K. Paeschke, M. L. Bochman, P. D. Garcia, P. Cejka, K. L. Friedman, S. C. Kowalczykowski and V. A. Zakian, Nature, 2013, 497, 458–462 CrossRef CAS PubMed.
- T. B. C. London, L. J. Barber, G. Mosedale, G. P. Kelly, S. Balasubramanian, I. D. Hickson, S. J. Boulton and K. Hiom, J. Biol. Chem., 2008, 283, 36132–36139 CrossRef CAS PubMed.
- C. G. Wu and M. Spies, Nucleic Acids Res., 2016, 44, 8742–8753 CrossRef CAS PubMed.
- W.-C. Tsao and K. A. Eckert, Int. J. Mol. Sci., 2018, 19, E3255 CrossRef PubMed.
- P. Sarkies, C. Reams, L. J. Simpson and J. E. Sale, Mol. Cell, 2010, 40, 703–713 CrossRef CAS PubMed.
- P. Sarkies, P. Murat, L. G. Phillips, K. J. Patel, S. Balasubramanian and J. E. Sale, Nucleic Acids Res., 2012, 40, 1485–1498 CrossRef CAS PubMed.
- D. Schiavone, G. Guilbaud, P. Murat, C. Papadopoulou, P. Sarkies, M.-N. Prioleau, S. Balasubramanian and J. E. Sale, EMBO J., 2014, 33, 2507–2520 CrossRef CAS PubMed.
- J. Lopes, A. Piazza, R. Bermejo, B. Kriegsman, A. Colosio, M.-P. Teulade-Fichou, M. Foiani and A. Nicolas, EMBO J., 2011, 30, 4033–4046 CrossRef CAS PubMed.
- M. K. Swan, R. E. Johnson, L. Prakash, S. Prakash and A. K. Aggarwal, J. Mol. Biol., 2009, 390, 699–709 CrossRef CAS PubMed.
- S. Nayak, J. A. Calvo, K. Cong, M. Peng, E. Berthiaume, J. Jackson, A. M. Zaino, A. Vindigni, M. K. Hadden and S. B. Cantor, Sci. Adv., 2020, 6, eaaz7808 CrossRef CAS PubMed.
- S. Tirman, A. Quinet, M. Wood, A. Meroni, E. Cybulla, J. Jackson, S. Pegoraro, A. Simoneau, L. Zou and A. Vindigni, Mol. Cell, 2021, 81, 4026–4040 CrossRef CAS PubMed.
- J. K. Hicks, C. L. Chute, M. T. Paulsen, R. L. Ragland, N. G. Howlett, Q. Guéranger, T. W. Glover and C. E. Canman, Mol. Cell. Biol., 2010, 30, 1217–1230 CrossRef CAS PubMed.
- S. Sharma and C. E. Canman, Environ. Mol. Mutagen., 2012, 53, 725–740 CrossRef CAS PubMed.
- C. M. Wickramasinghe, H. Arzouk, A. Frey, A. Maiter and J. E. Sale, DNA Repair, 2015, 29, 83–90 CrossRef CAS PubMed.
- S. Eddy, A. Ketkar, M. K. Zafar, L. Maddukuri, J.-Y. Choi and R. L. Eoff, Nucleic Acids Res., 2014, 42, 3272–3285 CrossRef CAS PubMed.
- A. Ketkar, L. Smith, C. Johnson, A. Richey, M. Berry, J. H. Hartman, L. Maddukuri, M. R. Reed, J. E. C. Gunderson, J. W. C. Leung and R. L. Eoff, Nucleic Acids Res., 2021, 49, 2065–2084 CrossRef CAS PubMed.
- I. van Bostelen, R. van Schendel, R. Romeijn and M. Tijsterman, PLoS Genet., 2020, 16, e1008759 CrossRef PubMed.
- K. Clark, I. Karsch-Mizrachi, D. J. Lipman, J. Ostell and E. W. Sayers, Nucleic Acids Res., 2016, 44, D67–D72 CrossRef CAS PubMed.
- H. Liu, Y.-N. Lu, T. Paul, G. Periz, M. T. Banco, A. R. Ferré-D’Amaré, J. D. Rothstein, L. R. Hayes, S. Myong and J. Wang, J. Am. Chem. Soc., 2021, 143, 7368–7379 CrossRef CAS PubMed.
- J. Robinson, F. Raguseo, S. P. Nuccio, D. Liano and M. Di Antonio, Nucleic Acids Res., 2021, 49, 8419–8431 CrossRef CAS PubMed.
- H. Tateishi-Karimata and N. Sugimoto, Nucleic Acids Res., 2021, 49, 7839–7855 CrossRef CAS PubMed.
- N. Pandya, S. R. Bhagwat and A. Kumar, Biochim. Biophys. Acta, Rev. Cancer, 2021, 1876, 188594 CrossRef CAS PubMed.
- A. Basu, D. G. Bobrovnikov, B. Cieza, J. P. Arcon, Z. Qureshi, M. Orozco and T. Ha, Nat. Struct. Mol. Biol., 2022, 29, 1178–1187 CrossRef CAS PubMed.
- R. Hänsel-Hertsch, M. Di Antonio and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2017, 18, 279–284 CrossRef PubMed.
- J. Dong, M. P. O’Hagan and I. Willner, Chem. Soc. Rev., 2022, 51, 7631–7661 RSC.
- J. Liu, L. Yan, S. He and J. Hu, Nano Res., 2022, 15, 3504–3513 CrossRef CAS PubMed.
- E. Ruggiero and S. N. Richter, Nucleic Acids Res., 2018, 46, 3270–3283 CrossRef CAS PubMed.
- R. Hänsel-Hertsch, A. Simeone, A. Shea, W. W. I. Hui, K. G. Zyner, G. Marsico, O. M. Rueda, A. Bruna, A. Martin, X. Zhang, S. Adhikari, D. Tannahill, C. Caldas and S. Balasubramanian, Nat. Genet., 2020, 52, 878–883 CrossRef PubMed.
- E. Wang, R. Thombre, Y. Shah, R. Latanich and J. Wang, Nucleic Acids Res., 2021, 49, 4816–4830 CrossRef CAS PubMed.
- D. D. Le, M. Di Antonio, L. K. M. Chan and S. Balasubramanian, Chem. Commun. Camb. Engl., 2015, 51, 8048–8050 RSC.
- S. Haldar, Y. Zhang, Y. Xia, B. Islam, S. Liu, F. L. Gervasio, A. J. Mulholland, Z. A. E. Waller, D. Wei and S. Haider, J. Am. Chem. Soc., 2022, 144, 935–950 CrossRef CAS PubMed.
- K.-W. Zheng, J.-Y. Zhang, Y. He, J.-Y. Gong, C.-J. Wen, J.-N. Chen, Y.-H. Hao, Y. Zhao and Z. Tan, Nucleic Acids Res., 2020, 48, 11706–11720 CrossRef CAS PubMed.
- K. M. Patil, D. Chin, H. L. Seah, Q. Shi, K. W. Lim and A. T. Phan, Chem. Commun. Camb. Engl., 2021, 57, 12816–12819 RSC.
- A. Ambrus, D. Chen, J. Dai, R. A. Jones and D. Yang, Biochemistry, 2005, 44, 2048–2058 CrossRef CAS PubMed.
- J. Dickerhoff, B. Onel, L. Chen, Y. Chen and D. Yang, ACS Omega, 2019, 4, 2533–2539 CrossRef CAS PubMed.
- M. N. Chowdhury and H. Jin, Wiley Interdiscip. Rev.: RNA, 2022, e1748 Search PubMed.
- P. Thandapani, T. R. O’Connor, T. L. Bailey and S. Richard, Mol. Cell, 2013, 50, 613–623 CrossRef CAS PubMed.
- P. Rajyaguru and R. Parker, Cell Cycle Georget. Tex, 2012, 11, 2594–2599 CrossRef CAS PubMed.
- F. Gebauer, T. Schwarzl, J. Valcárcel and M. W. Hentze, Nat. Rev. Genet., 2021, 22, 185–198 CrossRef CAS PubMed.
- M. Kiledjian and G. Dreyfuss, EMBO J., 1992, 11, 2655–2664 CrossRef CAS PubMed.
- E. L. Spaulding, A. M. Feidler, L. A. Cook and D. L. Updike, Nat. Commun., 2022, 13, 6585 CrossRef CAS PubMed.
- N. Vasilyev, A. Polonskaia, J. C. Darnell, R. B. Darnell, D. J. Patel and A. Serganov, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E5391–E5400 CrossRef CAS PubMed.
- M. Ghosh and M. Singh, Nucleic Acids Res., 2018, 46, 10246–10261 CrossRef CAS PubMed.
- A. Kumar and S. H. Wilson, Biochemistry, 1990, 29, 10717–10722 CrossRef CAS PubMed.
- A. Kumar, J. R. Casas-Finet, C. J. Luneau, R. L. Karpel, B. M. Merrill, K. R. Williams and S. H. Wilson, J. Biol. Chem., 1990, 265, 17094–17100 CrossRef CAS PubMed.
- H. Idriss, A. Kumar, J. R. Casas-Finet, H. Guo, Z. Damuni and S. H. Wilson, Biochemistry, 1994, 33, 11382–11390 CrossRef CAS PubMed.
- Q. Qiao, L. Wang, F.-L. Meng, J. K. Hwang, F. W. Alt and H. Wu, Mol. Cell, 2017, 67, 361–373 CrossRef CAS PubMed.
- W. T. Yewdell, Y. Kim, P. Chowdhury, C. M. Lau, R. M. Smolkin, K. T. Belcheva, K. C. Fernandez, M. Cols, W.-F. Yen, B. Vaidyanathan, D. Angeletti, A. B. McDermott, J. W. Yewdell, J. C. Sun and J. Chaudhuri, Immunity, 2020, 53, 952–970 CrossRef CAS PubMed.
- M. Fry and L. A. Loeb, J. Biol. Chem., 1999, 274, 12797–12802 CrossRef CAS PubMed.
- R. R. Damerla, K. E. Knickelbein, S. Strutt, F.-J. Liu, H. Wang and P. L. Opresko, Cell Cycle, 2012, 11, 3036–3044 CrossRef CAS PubMed.
- P. Mohaghegh, J. K. Karow, R. M. Brosh, V. A. Bohr and I. D. Hickson, Nucleic Acids Res., 2001, 29, 2843–2849 CrossRef CAS PubMed.
- A. Ketkar, M. Voehler, T. Mukiza and R. L. Eoff, J. Biol. Chem., 2017, 292, 3154–3163 CrossRef CAS PubMed.
- A. Traczyk, C. W. Liew, D. J. Gill and D. Rhodes, Nucleic Acids Res., 2020, 48, 4562–4571 CrossRef CAS PubMed.
- M. C. Chen, R. Tippana, N. A. Demeshkina, P. Murat, S. Balasubramanian, S. Myong and A. R. Ferré-D’Amaré, Nature, 2018, 558, 465–469 CrossRef CAS PubMed.
- A. Antcliff, L. D. McCullough and A. S. Tsvetkov, Aging, 2021, 13, 25578–25587 CrossRef CAS PubMed.
- P. M. Yangyuoru, D. A. Bradburn, Z. Liu, T. S. Xiao and R. Russell, J. Biol. Chem., 2018, 293, 1924–1932 CrossRef CAS PubMed.
- P. J. Smaldino, E. D. Routh, J. H. Kim, B. Giri, S. D. Creacy, R. R. Hantgan, S. A. Akman and J. P. Vaughn, PLoS One, 2015, 10, e0132668 CrossRef PubMed.
- R. Tippana, H. Hwang, P. L. Opresko, V. A. Bohr and S. Myong, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8448–8453 CrossRef CAS PubMed.
- L. T. A. Nguyen and D. T. Dang, Mol. Biotechnol., 2023, 65, 291–299 CrossRef CAS PubMed.
- D. T. Nair, R. E. Johnson, L. Prakash, S. Prakash and A. K. Aggarwal, Science, 2005, 309, 2219–2222 CrossRef CAS PubMed.
- T. M. Weaver, L. M. Cortez, T. H. Khoang, M. T. Washington, P. K. Agarwal and B. D. Freudenthal, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25494–25504 CrossRef CAS PubMed.
- F. H. de Groote, J. G. Jansen, Y. Masuda, D. M. Shah, K. Kamiya, N. de Wind and G. Siegal, DNA Repair, 2011, 10, 915–925 CrossRef CAS PubMed.
- C. G. Wu and M. Spies, Nucleic Acids Res., 2016, 44, 8742–8753 CrossRef CAS PubMed.
- K. Lowran, L. Campbell, P. Popp and C. G. Wu, Genes, 2019, 11, 5 CrossRef PubMed.
- K. Sato, N. Martin-Pintado, H. Post, M. Altelaar and P. Knipscheer, Sci. Adv., 2021, 7, eabf8653 CrossRef CAS PubMed.
- W. Koole, R. van Schendel, A. E. Karambelas, J. T. van Heteren, K. L. Okihara and M. Tijsterman, Nat. Commun., 2014, 5, 3216 CrossRef PubMed.
- A. Taglialatela, G. Leuzzi, V. Sannino, R. Cuella-Martin, J.-W. Huang, F. Wu-Baer, R. Baer, V. Costanzo and A. Ciccia, Mol. Cell, 2021, 81, 4008–4025 CrossRef CAS PubMed.
- V. Shukla, D. Samaniego-Castruita, Z. Dong, E. González-Avalos, Q. Yan, K. Sarma and A. Rao, Nat. Immunol., 2022, 23, 99–108 CrossRef CAS PubMed.
- Y.-Z. Xu, P. Jenjaroenpun, T. Wongsurawat, S. D. Byrum, V. Shponka, D. Tannahill, E. A. Chavez, S. S. Hung, C. Steidl, S. Balasubramanian, L. M. Rimsza and S. Kendrick, NAR Cancer, 2020, 2, zcaa029 CrossRef PubMed.
- M. Stein, S. E. Hile, M. H. Weissensteiner, M. Lee, S. Zhang, E. Kejnovský, I. Kejnovská, K. D. Makova and K. A. Eckert, DNA Repair, 2022, 119, 103402 CrossRef CAS PubMed.
- R. Bétous, L. Rey, G. Wang, M.-J. Pillaire, N. Puget, J. Selves, D. S. F. Biard, K. Shin-ya, K. M. Vasquez, C. Cazaux and J.-S. Hoffmann, Mol. Carcinog., 2009, 48, 369–378 CrossRef PubMed.
- M. Bienko, C. M. Green, N. Crosetto, F. Rudolf, G. Zapart, B. Coull, P. Kannouche, G. Wider, M. Peter, A. R. Lehmann, K. Hofmann and I. Dikic, Science, 2005, 310, 1821–1824 CrossRef CAS PubMed.
- M. Bienko, C. M. Green, S. Sabbioneda, N. Crosetto, I. Matic, R. G. Hibbert, T. Begovic, A. Niimi, M. Mann, A. R. Lehmann and I. Dikic, Mol. Cell, 2010, 37, 396–407 CrossRef CAS PubMed.
- J.-R. Lin, M. K. Zeman, J.-Y. Chen, M.-C. Yee and K. A. Cimprich, Mol. Cell, 2011, 42, 237–249 CrossRef CAS PubMed.
- C. E. Edmunds, L. J. Simpson and J. E. Sale, Mol. Cell, 2008, 30, 519–529 CrossRef CAS PubMed.
- S. D’Souza and G. C. Walker, Mol. Cell. Biol., 2006, 26, 8173–8182 CrossRef PubMed.
- Y. Pustovalova, I. Bezsonova and D. M. Korzhnev, FEBS Lett., 2012, 586, 3051–3056 CrossRef CAS PubMed.
- C. Tellier-Lebegue, E. Dizet, E. Ma, X. Veaute, E. Coïc, J.-B. Charbonnier and L. Maloisel, PLoS Genet., 2017, 13, e1007119 CrossRef PubMed.
- S. Eddy, L. Maddukuri, A. Ketkar, M. K. Zafar, E. E. Henninger, Z. F. Pursell and R. L. Eoff, Biochemistry, 2015, 54, 3218–3230 CrossRef CAS PubMed.
- S. Eddy, M. Tillman, L. Maddukuri, A. Ketkar, M. K. Zafar and R. L. Eoff, Biochemistry, 2016, 55, 5218–5229 CrossRef CAS PubMed.
- M. M. de Camargo and L. A. Nahum, Hum. Genomics, 2005, 2, 132–137 CrossRef PubMed.
- J.-C. Weill and C.-A. Reynaud, Nat. Rev. Immunol., 2008, 8, 302–312 CrossRef CAS PubMed.
- C. Tang and T. MacCarthy, Front. Immunol., 2021, 12, 671944 CrossRef CAS PubMed.
- H. Zan, C. A. White, L. M. Thomas, T. Mai, G. Li, Z. Xu, J. Zhang and P. Casali, Cell Rep., 2012, 2, 1220–1232 CrossRef CAS PubMed.
- S. van de Velde, B. J. W. Mills, F. J. R. Meysman, T. M. Lenton and S. W. Poulton, Nat. Commun., 2018, 9, 2554 CrossRef PubMed.
- S. J. van de Velde, C. T. Reinhard, A. Ridgwell and F. J. R. Meysman, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 33043–33050 CrossRef CAS PubMed.
- G. E. Coggins, L. Maddukuri, N. R. Penthala, J. H. Hartman, S. Eddy, A. Ketkar, P. A. Crooks and R. L. Eoff, ACS Chem. Biol., 2013, 8, 1722–1729 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Varadi, S. Anyango, M. Deshpande, S. Nair, C. Natassia, G. Yordanova, D. Yuan, O. Stroe, G. Wood, A. Laydon, A. Žídek, T. Green, K. Tunyasuvunakool, S. Petersen, J. Jumper, E. Clancy, R. Green, A. Vora, M. Lutfi, M. Figurnov, A. Cowie, N. Hobbs, P. Kohli, G. Kleywegt, E. Birney, D. Hassabis and S. Velankar, Nucleic Acids Res., 2022, 50, D439–D444 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00027c |
| This journal is © The Royal Society of Chemistry 2023 |