
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards a simple in vitro surface chemistry pre-screening method for nanoparticles to be used for drug delivery to solid tumours†
Roman
Schmid
 a,
Juliane
Kaiser
b,
Ramona
Willbold
b,
Nomusa
Walther
b,
Rainer
Wittig
*b and
Mika
Lindén
*a
a,
Juliane
Kaiser
b,
Ramona
Willbold
b,
Nomusa
Walther
b,
Rainer
Wittig
*b and
Mika
Lindén
*a
aInorganic Chemistry II, Albert-Einstein-Allee 11, Ulm University, 89081 Ulm, Germany. E-mail: mlinden@uni-ulm.de
bInstitute for Laser Technologies in Medicine & Metrology (ILM) at Ulm University, Helmholtzstrasse 12, 89081 Ulm, Germany. E-mail: rainer.wittig@ilm-ulm.de
First published on 2nd August 2023
Abstract
An efficient nanoparticulate drug carrier intended for chemotherapy based on intravenous administration must exhibit a long enough blood circulation time, a good penetrability into the tumour volume, as well as an efficient uptake by cancer cells. Limiting factors for the therapeutic outcome in vivo are recognition of the nanoparticles as foreign objects, which triggers nanoparticle uptake by defence organs rich in macrophages, e.g. liver and spleen, on the time-scale of accumulation and uptake in/by the tumour. However, the development of nanomedicine towards efficient nanoparticle-based delivery to solid tumours is hampered by the lack of simple, reproducible, cheap, and predictive means for early identification of promising nanoparticle formulations. The surface chemistry of nanoparticles is known to be the most important determinant for the biological fate of nanoparticles, as it influences the extent of serum protein adsorption, and also the relative composition of the protein corona. Here we preliminarily evaluate an extremely simple screening method for nanoparticle surface chemistry pre-optimization based on nanoparticle uptake in vitro by PC-3 cancer cells and THP-1 macrophages. Only when both selectivity for the cancer cells as well as the extent of nanoparticle uptake are taken into consideration do the in vitro results mirror literature results obtained for small animal models. Furthermore, although not investigated here, the screening method does also lend itself to the study of actively targeted nanoparticles.
Introduction
Nanoparticle-based drug delivery is seen as one of the most promising means for enhancing the therapeutic outcome due to enhanced specificity, improved drug bioavailability, and biodistribution. This is of special importance in oncology, where side-effects related to non-specific drug delivery in combination with the need for high systemic drug doses are severe problems. In the case of solid tumours, the enhanced permeability and retention (EPR) effect is supposed to help nanoparticles accumulate in the tumour.1 Further enhancement of the cellular specificity can be attempted through linking of ligands to the outer surface of the nanoparticles, which addresses receptors that are overexpressed on the target cells. However, a large fraction of intravenously injected nanoparticles are typically removed from the blood circulation very soon after administration through immune response-triggered nanoparticle uptake by the mononuclear phagocyte system (MPS, also known as the reticuloendothelial system, RES) consisting of phagocytic cells, typically monocytes and macrophages.2 Organs rich in phagocytotic cells are the liver and spleen, and therefore nanoparticle accumulation in these organs with time is observed upon intravenous administration of nanoparticles. Thus, in order to enhance the portion of particles getting accumulated in the tumour area, the blood circulation time should be long enough. Here, the surface chemistry of the nanoparticles naturally plays an important role. Although there is no clear consensus about the relationship between the composition of the protein corona and the biodistribution, the commonly accepted paradigm is that the recognition of nanoparticles by the MPS system correlates with the extent of opsonin binding to the nanoparticles.3 Therefore, nanoparticles intended for intravenous administration are typically surface functionalized by a hydrophilic polymer, in most cases poly(ethylene glycol), PEG, in order to decrease the extent of serum protein adsorption.4–7 As can be expected, not only the surface polarity but also the surface charge influences the extent of protein adsorption, and also which proteins adsorb.8,9 Nanoparticles exhibiting a low effective charge typically adsorb less serum proteins, which explains why zwitterionic nanoparticle coatings in addition to non-ionic polymers can be used to minimize protein adsorption.10 Highly charged particles, on the other hand, are typically removed quickly from the blood stream by RES upon intravenous injection,11–14 which can be attributed to enhanced protein adsorption to such particles.However, escaping RES and thus prolonging the nanoparticle circulation time is not the only important aspect concerning the therapeutic outcome. The nanoparticles should also be able to invade the tumour and also be efficiently internalized by the target cells in order to serve as efficient drug carriers, as most anticancer drugs are active intracellularly. For example, it has been shown that a zwitterionic coating that reduces protein adsorption also lead to a lower nanoparticle uptake by HeLa cancer cells in vitro.15 It has also been reported that the cell-specificity (targetability) of antibody-tagged nanoparticles towards acute myeloid leukaemia stem cells depends on the exact composition of the protein corona,16 suggesting that a total suppression of protein adsorption may be counterproductive when aiming for high cell-specificity in vivo, even in the case that such nanoparticles may exhibit a prolonged blood circulation time. Cationic nanoparticles, on the other hand, are known to be internalized much more efficiently by cancer cells as compared to anionic nanoparticles,17–20 and lead to enhanced therapeutic outcomes.21,22 Thus, it is clear that this is a classic optimization problem, but the mutual influences of this multitude of parameters remain largely unknown, and any attempts to establish such links are associated with an immense experimental effort even when only a very small number of nanoparticles would be considered. From a nanoparticle development point of view, simple screening means would therefore be highly beneficial in order to speed up the surface chemistry optimization, and to allow focusing the detailed studies on the most promising particles for a given application. Although simple in vitro measurements can never fully replace in vivo characterizations, it is also clear that broad in vivo screenings are not feasible, neither from an economical nor an ethical point of view. Finding a surface chemistry that allow for a high specificity towards the target cells in combination with a fast uptake kinetics is preferable. Mesoporous silica nanoparticles (MSNs) are attracting considerable interest as drug carrier systems due to their easy functionalization, high drug loading capacities, and flexible tuning of particle size and shape. However, their biodistribution characteristics still need optimization. An increasing number of small animal in vivo studies have been published recently, using MSNs with different types of surface functionalities. This allows some conclusions concerning the relation between the MSN surface chemistry, the general biodistribution, and tumour accumulation of intravenously injected particles. In a recent study, hyaluronic acid-functionalized MSNs with a particle diameter of about 180 nm were shown to exhibit a long blood circulation time, much longer than corresponding cationic amino-functionalized particles.23 In another study, an increase on the blood circulation time and in the tumour targetability of pure hyaluronic acid-based nanoparticles via PEGylation was demonstrated in vivo, where these effects were dependent on the degree of PEGylation.24 A fast accumulation of amino-functionalized MSNs in the liver upon intravenous injection is in agreement with previous reports.14,25 All-silica MSNs as well as carboxy-silane functionalized MSNs have also been shown to accumulate fast in liver and spleen.26–28 PEGylation has been shown to decrease uptake in the liver in several cases, as compared to corresponding non-PEGylated particles.24,26,29,30 On the other hand, as discussed above, amino-functionalized MSNs have repeatedly been shown to be efficiently taken up by cancer cells in vitro and to accumulate in solid tumours, leading to efficient tumour-directed drug delivery.31–33
Inspired by these literature reports, we conducted a study in order to investigate if we could mirror these results by a simple and reproducible in vitro uptake study involving THP-1 cells as models for naïve macrophages, and PC-3 prostate cancer cells as a cancer model. Both the selectivity at a given time-point as well as the relative amount of particles taken up by the cells are evaluated, in order to allow for classification of the particles. In addition, the results are complemented by preliminary protein corona analysis using SDS-PAGE. It is shown that the simple in vitro evaluation nicely mirrors the in vivo responses that have been seen for such particles, and this approach could potentially as such or in modified form serve as a fast pre-screening method for surface chemistry optimization of nanoparticles intended for drug delivery to solid tumours.
Experimental
Chemicals and materials
Tetramethyl orthosilicate (TMOS), (3-aminopropyl) trimethoxysilane (APTMS), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC-HCl), N-hydroxysuccinimide (NHS), glycerine, hyaluronic acid sodium salt from Streptococcus equi (1.5–1.8 × 103 kDa), O-[2-(Fmoc-amino)-ethyl]-O′-(2-carboxyethyl)polyethylene glycol 3000 (Fmoc-NH-PEG3k-COOH), piperidine, dimethylformamide (DMF), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), propidium iodide (PI) and phalloidin-TRITC were purchased from Sigma-Aldrich Chemie GmbH, Schnelldorf, Germany. cetyltrimethylammonium bromide (CTAB), sodium hydroxide, acetone, ethanol, methanol and hydrochloric acid were purchased from VWR International GmbH, Darmstadt, Germany. Carboxyethylsilanetriol di-sodium salt (CES, 25 wt% in water, 1.49 M) was purchased from ABCR GmbH & Co. KG, Karlsruhe, Germany. Ammonium nitrate, sodium dodecyl sulfate (SDS) sodium dodecyl sulfate (SDS), tris(hydroxymethyl)aminomethane (TRIS) and glycine were purchased from Carl Roth GmbH & Co. KG, Karlsruhe, Germany. (4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) and Accutase were purchased from Merck KGaA, Darmstadt, Germany. N-Hydroxysulfosuccinimide (Sulfo-NHS), phosphate buffered saline (PBS), PageRuler™ Broad range unstained protein ladder and black 96-well microplates were purchased from Thermo Fisher Scientific, Karlsruhe, Germany, fetal calf serum (FCS) was purchased from Cytiva HyClone™. ATTO647N-NH2 and ATTO647N-NHS were purchased from ATTO-TEC GmbH, Siegen, Germany. THP-1 monocyte cell line was purchased from Cell Lines Service, Eppelheim, Germany. RPMI 1640 medium, streptomycin/penicillin, glutamine, sodium pyruvate and Trypsin-EDTA were purchased from Life Technologies, Darmstadt, Germany. Fetal bovine serum was purchased from Applichem, Darmstadt, Germany. Ibitreat dishes were purchased from ibidi, Munich, Germany. The Cell Titer Blue assay was purchased from Promega, Mannheim, Germany. The FCS was heat inactivated, all other chemicals were used as supplied by the manufacturer without further purification.Synthesis of mesoporous silica nanoparticles
Mesoporous silica nanoparticles (MSN) were synthesized as described by Rosenholm et al.20 In a 2 L round-bottomed flask, 7.88 g of the surfactant CTAB (7.88 g, 21.6 mmol) was dissolved in a mixture of water (922 mL), methanol (849 mL) and sodium hydroxide solution (1 M, 4.56 mL, 4.6 mmol). A mixture of the silica precursors TMOS (2.18 mL, 14.7 mmol) and APTMS (0.36 mL, 2.1 mmol) was added at a stirring rate of 500 rpm. The solution was stirred for 45 min and consecutively reduced to 300 rpm. After 18 h, the particles were precipitated with ammonium nitrate (25 g, 19.6 mmol) and separated via centrifugation. In order to yield all-silica nanoparticles, the precipitate was washed with ethanol twice and with acetone once by dispersing them for 1 h each step in an ultrasonic bath. Finally, the dried particles were calcined at 550 °C for 5.5 h (heating rate 1.5 °C min−1). These particles are denoted “MSN-c” in the following. For preservation of amino-functionalities the separated as-synthesized nanoparticles were, instead of calcination, washed with ethanol and the surfactant was extracted by dispersing the particles with acidic ethanol (4 g 32% HCl in 1 L ethanol) for 1 h. Consecutive to a final washing step in ethanol, the particles were dried at 60 °C overnight to yield amino-functionalized “MSN-NH2”. The amount of amino groups on the particles was determined by TGA in the temperature range 150 °C–700 °C.Carboxy-functionalization of mesoporous silica particles
A solution of 25 wt% CES in water (1.49 M, 336 μL, 0.5 mmol) was added to a dispersion of MSNc particles in methanol (7.5 mg mL−1, 20 mL). The mixture was agitated for 3 h at RT. Finally, the particles were centrifuged, washed with methanol and acetone in an ultrasonic bath and dried at 60 °C overnight. These particles are denoted “MSN-COOH” in the following. The amount of carboxy groups on the particles was determined by TGA in the temperature range of 250 °C to 700 °C. The contribution of silanol condensation to the mass loss was accounted for by using non-functionalized particles that had otherwise been exposed to the same conditions as reference.Fluorescence dye-attachment
Carboxy-functionalized particles were dispersed in HEPES buffer (10 mg mL−1, 10 mL). Particulate carboxy groups were activated with EDC (5.649 M, 89 μL, 0.5 mmol) and an aqueous NHS solution (0.434 M, 2.3 mL, 1 mmol). After agitation for 30 min the particles were centrifuged, briefly washed and re-dispersed in HEPES buffer (10 mg mL−1, 10 mL). The activated MSN-COOH were fluorescently labelled with a solution of ATTO647N-NH2 in DMSO (1.092 mM, 100 μL, 0.109 μmol) for 1 h at RT. The particles were centrifuged and washed thoroughly with water once and methanol twice and dried in vacuo at 60 °C overnight to yield fluorescence labelled “MSN-COOH”. To a dispersion of amino-functionalized particles in HEPES buffer (10 mg mL−1, 10 mL) a solution of ATTO647N-NHS ester in DMSO (1.186 mM, 100 μL, 0.119 μmol) was added and the mixture was agitated for 1.5 h at RT. Upon centrifugation, the particles were thoroughly washed with water once and methanol twice and dried in vacuo at 60 °C overnight. These particles are denoted “MSN-NH2” in the following.Functionalization of MSN-NH2 with PEG
EDC-HCl (16 mg, 83 μmol) and NHS (19 mg, 165 μmol) were dissolved in a HEPES-buffered aqueous solution of Fmoc-NH-PEG3k-COOH (0.83 mM, 19 mL, 15.7 μmol) and agitated for 45 min at RT. NHS-activated PEG solution (0.83 mM, 6.375 mL, 5.3 μmol) was mixed with a dispersion of amino-functionalized MSN-NH2 (8.8 mg mL−1, 9.1 mL) and reacted for 2 h at room temperature (RT). The particles were separated via centrifugation and washed with water and acetone in an ultrasonic bath. After drying the particles at 60 °C for 40 h, the PEGylated MSNs were labelled with an ATTO647N-COOH dye as described above. The cleavage of the Fmoc protection group was achieved by dispersing the particles in a 25 wt% solution of piperidine in DMF (5 mg mL−1, 5 mL) and agitation for 15 min. The particles were separated via centrifugation and the cleavage procedure was repeated once again. Finally, the particles were thoroughly washed with DMF and acetone and dried at 60 °C overnight to yield fluorescence labelled “MSN-PEG”. The amount of PEG on the particles was determined by TGA in the temperature range of 150 °C to 700 °C. The contribution of silanol condensation and aminopropyl-functions to the mass loss was accounted for by using non-functionalized particles that had otherwise been exposed to the same conditions as a reference.Functionalization of MSN-NH2 with hyaluronic acid
EDC-HCl (6.4 mg, 33 μmol) and sulfo-NHS (14.6 mg, 67 μmol) were dissolved in a HEPES-buffered (25 mM, pH = 7.2) aqueous solution of hyaluronic acid (2 mg mL−1, 1 mL) and agitated for 45 min at RT. Upon addition of a dispersion of amino-functionalized MSN-NH2 (20 mg mL−1, 1 mL) the coupling reaction proceeded for another 3.5 h. The particles were separated via centrifugation and thoroughly washed with water once and MeOH twice in an ultrasonic bath and finally dried in vacuo at 60 °C overnight yielding particles denoted “MSN-HA”. The amount of hyaluronic acid bound to the MSN-HA particles was determined by TGA in the temperature range of 150 °C to 700 °C corrected by the mass loss measured for reference particles that had not been exposed to hyaluronic acid, but otherwise had undergone the same treatment conditions as the MSN-HA particles.Nanoparticle characterization
Amounts of functionalities bound to the particles was measured via thermogravimetric analysis (TGA) using a TG209 F1 Libra (Netzsch, Germany) at a heating rate of 10 °C min−1 in nitrogen/oxygen (70%/30%) atmosphere and normalized on the silica rest mass at 1000 °C. Upon nitrogen sorption measurements of the dried particles (in vacuo, 100 °C, 22 h) on a Quadrasorb-1 (Quantachrome Instruments, Germany) at −196 °C, the specific surface areas, pore diameters and volumes were determined via BET model and equilibrium NLDFT kernel calculations (silica, cylindrical pores, relative pressure range of 0–0.9). Zeta potentials of MSNs were measured with a Zetasizer NanoZS Zen3600 (Malvern Panalytical, Germany) in an aqueous HEPES buffer (25 mM, pH 7.2, particle concentration: 0.1 mg mL−1). Particle morphology and size was examined via transmission electron microscope (TEM) with a Jeol 1200 (Jeol, Germany) using a HT voltage of 120 kV and a beam current of 65 μA. Mean fluorescence intensities (MFIs) of ATTO-dye labelled particles were determined in an aqueous 10% sucrose solution (particle concentration: 0.1 mg mL−1) using black 96-well microplates and a Spark 10M microplate reader (Tecan, Crailsheim, Germany). In order to account for different fluorescence labelling efficiencies, the MFI of each particle type was measured at a constant concentration of 100 μg mL−1 in aqueous 10% sucrose solution and normalized to the MFI of “MSN-NH2” resulting in conversion factors (“rMFIs”) which were used to calculate “rated GMFIs” (rGMFIs) in flow cytometry experiments. ATR-IR spectra were measured on a Tensor 27 (Bruker, USA) with a total of 200 scans per sample. Depending on the coupling chemistry used for surface functionalization, either a spectrum measured for MSN-c or for MSN-NH2 was used as a background spectrum.Protein adsorption onto silica nanoparticles from FCS
Formation of protein corona on the set of different functionalized MSNs was examined by dispersing the particles in PBS with a focused ultrasonic bath. The dispersions were mixed with pure, heat inactivated FCS in 2 mL Eppendorf tubes resulting in 10 vol% FCS and a silica concentration of 5 mg mL−1. Note, that the silica concentration was normalized on the rest mass at 700 °C (TGA) in order to account for different functionalization degrees. After protein adsorption for 24 h at 37 °C under agitation, the particles were centrifuged (21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 rcf, 10 min) and the supernatants were discharged. Upon briefly washing the particles with purified water thrice, the protein corona was eluted by dispersing the samples in an aqueous solution of 10 wt% SDS (0.39 M), doubling the initial silica concentration, and further incubation at 95 °C for 30 min. Finally, the particles were centrifuged again (21100 rcf, 10 min), and the SDS supernatants were stored at −80 °C until further use.
100 rcf, 10 min) and the supernatants were discharged. Upon briefly washing the particles with purified water thrice, the protein corona was eluted by dispersing the samples in an aqueous solution of 10 wt% SDS (0.39 M), doubling the initial silica concentration, and further incubation at 95 °C for 30 min. Finally, the particles were centrifuged again (21100 rcf, 10 min), and the SDS supernatants were stored at −80 °C until further use.
SDS-PAGE of SDS supernatants
Upon thawing and another centrifugation step (21100 rcf, 10 min), 40 μL of SDS supernatant was mixed with 8 μL Laemmli buffer (Tris-HCl buffer (0.5 M; pH 6.8 (adjusted with HCl); 1.2 mL), SDS (1.2 g), bromophenol blue (6 mg), glycerine (4.7 mL), water (2.9 mL) and 1,4-dithiothreitol (0.9 g). Additionally, as controls, solutions containing SDS buffer (150 μL) and Laemmli buffer (30 μL; “SDS”) as well as FCS (25 μL), SDS buffer (185 μL) and Laemmli buffer (90 μL; “FCS”) were prepared. The protein samples and controls were incubated at 95 °C for 5 min. Consecutively, 5 μL broad range unstained protein ladder, 7.5 μL of protein sample, 15 μL of SDS control and 2.5 μL of FCS control were loaded into Mini-PROTEAN TGX Stain-Free Precast Gels (Bio-Rad Laboratories GmbH, Germany). SDS-PAGE was run at a voltage of 300 V in an aqueous electrophoresis buffer (SDS (3.5 mM), Tris (49.5 mM), glycine (383.6 mM)) with a Mini-PROTEAN Tetra Vertical Electrophoresis Cell and a PowerPac Universal Power Supply. Gel documentation and analysis was performed with a Gel Doc™ EZ Imager (Bio-Rad Laboratories GmbH, Germany) and Image Lab software version 6.0.1. The lanes on every gel were divided into three protein molecular mass regions: high molecular weight (>88 kDa), medium molecular weight (88–47 kDa) and low molecular weight proteins (<47 kDa). The overall absolute grey values of each of the band regions were first normalized on the 70 kDa band of the broad range ladder to obtain staining-normalized intensities. Finally, the band region intensities were normalized on the respective intensities of the MSN-NH2 particles.
Cell culture
Cells were cultivated using standard conditions (37 °C, 5% CO2, >95% relative humidity). The THP-1 monocyte cell line was originally isolated from peripheral blood of a one year old boy suffering from acute monocytic leukemia.34 THP-1 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1% streptomycin/penicillin, 2 mM glutamine, and 1 mM sodium pyruvate. Differentiation to macrophages (M0) was conducted using Phorbol-12-myristat-13-acetate (PMA) as described previously.35 PC-3 prostate cancer (PCa) cells were cultivated in RPMI 1640 medium supplemented with 10% fetal bovine serum, and 1% streptomycin/penicillin. To retain the option of using the current dataset in a context of molecular targeting analysis in the future, experiments were conducted using a genetically engineered subclone of PC-336 (PC3L1) which was further engineered for doxycycline inducible expression of the FOLH1/PSMA targeting ligand (resulting in PC3L1-FOLH1) using a vector system described previously.37 However, since the transgene was not induced the clone was phenotypically identical to the original PC-3 cell line.Evaluation of cellular particle uptake
Nanoparticle internalization was assessed qualitatively via confocal laser scanning fluorescence microscopy, as well as quantitatively using flow cytometry. A constant particle concentration of 10 μg mL−1 was used. THP-1 cells were seeded at a density of 268 mm−2 and grown for 72 h in presence of PMA, followed by a 24 h incubation period in MSN-containing growth medium. PC-3 cells were seeded at a density of 150–200 mm−2 and grown for 24 to 48 h prior to a 24 h incubation period in MSN-containing growth medium.Flow cytometry
Flow cytometry was conducted using a CyFlow Space instrument (Sysmex-Partec, Görlitz, Germany) and FloMax 3.0 software (Sysmex-Partec). Cells were detached using Trypsin-EDTA (for THP-1) or Accutase (for PC3L1-FOLH1), washed with PBS/1% BSA and subsequently resuspended in PBS/1% BSA. For detection of dead cells, propidium iodide (PI) was added at 2 μg mL−1. A total of 30000 to 50000 events was acquired, respectively. Debris and dead cells were identified by scattering characteristics (FSC, SSC) and/or enhanced PI-mediated fluorescence and excluded from further analysis. Quantification of internalization efficiencies was performed using the geometric mean fluorescence intensity (GMFI) data for ATTO647N-mediated fluorescence of particle-treated cells vs. controls from flow cytometry (n = 3 for each cell type and particle, respectively).
Confocal laser scanning fluorescence microscopy
Confocal laser scanning fluorescence microscopy (CLSM) was conducted using a Leica TCS SP8 system (Leica Microsystems, Wetzlar, Germany). Cells were seeded in ibitreat dishes and grown in presence of nanoparticles for 24 h. At the end of the incubation period, cells were fixed using PBS-buffered formaldehyde (4%) for 10 min at RT. Cells were washed with PBS and subsequently stained using DAPI (0.1 μg mL−1 in PBS for visualization of nuclei) during 5 min at RT, followed by phalloidin-TRITC (50 nM in PBS/1% BSA for visualization of actin cytoskeleton) during 30 min at RT. Subsequent to washing, cells were kept in PBS for confocal microscopy. Here, excitation of DAPI, TRITC and ATTO647N was achieved using integrated laser emitting at 405 nm, 552 nm and 638 nm, respectively. DAPI-mediated fluorescence was detected at 410–601 nm, TRITC-mediated fluorescence was detected at 557–713 nm, and ATTO647N-mediated fluorescence was detected at 643–795 nm using a HP CL APO 63×/1.40 OIL CS2 oil immersion objective (Leica Microsystems) and a pinhole setting of 1 Airy unit.Evaluation of cell viability
For cytotoxicity studies, PC-3 cells were seeded at 200 cells per mm2 in 96-well microplates. THP-1 cells were seeded at 268 cells per mm2 to 96 well microplates and differentiated to macrophages during 72 h. Nanoparticles were added after 24 h in case of PC-3 cells and after 72 h in case of THP-1 macrophages at various concentrations, and incubated for further 24 h. Subsequently, cell viability was determined using the Cell Titer Blue assay. Resorufin-mediated fluorescence (excitation: 544 nm, detection: 590 nm), the intensity of which correlates to the number of viable cells per well, was quantified using a Fluostar Omega microplate reader (BMG Labtech, Ortenberg, Germany). Fluorescence intensity values were blank corrected and viability was calculated as a percentage of an untreated control. For each sample, four independent experiments were performed in triplicates.Results and discussion
Monodisperse MSNs with a mean particle diameter of 190 nm and a radially aligned pore system were synthesized. The surfactant was removed either by acidic extraction yielding amino-functionalized particles (MSN-NH2) or by calcination. Calcined particles were further covalently functionalized with a carboxy-silane resulting in MSN-COOH particles. Further covalent functionalization of the MSN-NH2 particles with PEG3k (MSN-PEG) or hyaluronic acid (MSN-HA) resulted in altogether four different sets of particles. Key physicochemical characteristics of the particles are summarized in Table 1, and a representative transmission electron microscopy (TEM) image of the MSN-NH2 particles is shown in Fig. 1. TEM images of MSN-COOH, MSN-PEG and MSN-HA are shown in Fig. S1.† The high specific surface areas and pore volumes of the basic particles are in agreement with what is to be expected for this kind of particles (see Fig. S2 and Table S1†). Successful surface functionalization was proven by thermogravimetric analysis, zeta-potential, and ATR-IR spectroscopy measurements. Exemplary thermogravimetric measurements of both the native and the functionalized particles are depicted in ESI Fig. S3,† and the ATR-IR spectra including peak assignments are shown in Fig. S4.† While the MSN-NH2 and MSN-PEG particles exhibited an unaltered, positive zeta potential at pH 7.2, functionalization with hyaluronic acid led to a shift towards a negative zeta potential. The MSN-COOH particles exhibited strongly negative zeta potentials as expected. | ||
| Fig. 1 Exemplary transmission electron microscopy image of the MSN-NH2 particles. | ||
| MSN-NH2 | MSN-PEG | MSN-HA | MSN-COOH | |
|---|---|---|---|---|
| Mean diameter ± SD/nm | 176 ± 16 | 176 ± 16 | 211 ± 19 | 184 ± 17 |
| Functional groups/mg g−1 (silica) | 116 | 110 | 108 | 16 |
| Functional groups/mmol g−1 (silica) | 2.00 | 1.90 | 7.19 × 10−5 | 0.22 |
| Zeta potential (HEPES) ± SD/mV | +14 ± 3.2 | +10 ± 3.6 | −27 ± 4.3 | −44 ± 6.5 |
As a simple but informative MPS model we used THP-1 monocytes,34 which were differentiated to an adherently growing naïve macrophage-like phenotype (M0) by incubation with the phorbol ester phorbol-12-myristate-13-acetate (PMA). Particularly liver macrophages may be best represented by the M0 phenotype,38 and in line with this argumentation THP-1 M0 cells were recently used as a liver-resident macrophage model in an organ-on-chip application.39 M0-differentiated THP-1 cells can be further polarized to pro-inflammatory (M1) and tissue repair (M2) phenotypes and are as such a well-accepted in vitro model for the evaluation of the effects of nanomaterials on the immune system.35,40–42 Although some researchers prefer primary ex vivo models such as (human) peripheral blood mononuclear cells (PBMC), these bear significant disadvantages such as the requirement of chemokine supplementation for prolonged growth and considerable batch-to-batch/donor variation, which particularly impedes data interpretation in repetitive screening approaches. In contrast, THP-1 cells have been demonstrated to retain a relatively stable genetic background and monocyte/macrophage-like phenotype over several passages, which facilitates the reproducibility of results from screening approaches.43
Successful nanoparticle dispersability, and dispersion stability in cell medium supplemented with 10% FCS was proven by dynamic light-scattering. Hydrodynamic diameters of about 200 nm was measured for all particles (10 μg mL−1 concentration) apart from for the MSN-HA particles, for which a hydrodynamic diameter of about 300–400 nm was measured, suggesting some slight particle aggregation in this case (see ESI S5†). No loss of viability compared to untreated controls could be observed subsequent to incubation with MSNs under investigation at doses up to 25 μg mL−1 for THP-1 cells and up to 50 μg mL−1 for PC-3 cells, respectively (ESI Fig. S6†). The internalization of the particles by THP-1 M0 macrophages and the prostate cancer cell line PC-3 was investigated subsequent to a 24 h incubation at a particle concentration of 10 μg mL−1via both confocal laser scanning fluorescence microscopy (CLSM) and flow cytometry. Intracellular localization of the particles as well as the distribution patterns observed in flow cytometry were verified by CLSM (Fig. 2). Particle uptake could be visualized in both cell lines irrespective of particle type, but variation in signal intensities were evident. However, the particle-related fluorescence signals were narrowly localized and clearly distinguishable within the cells, indicating that the fluorescence dye was still attached to the MSNs and that neither particle dissolution nor diffusion of free dye into the cytosol impacted further analysis via flow cytometry. The low contribution of particle dissolution effects can be attributed to the fact that most of the particle internalization occur during the first 1–2 hours of incubation under these conditions.44
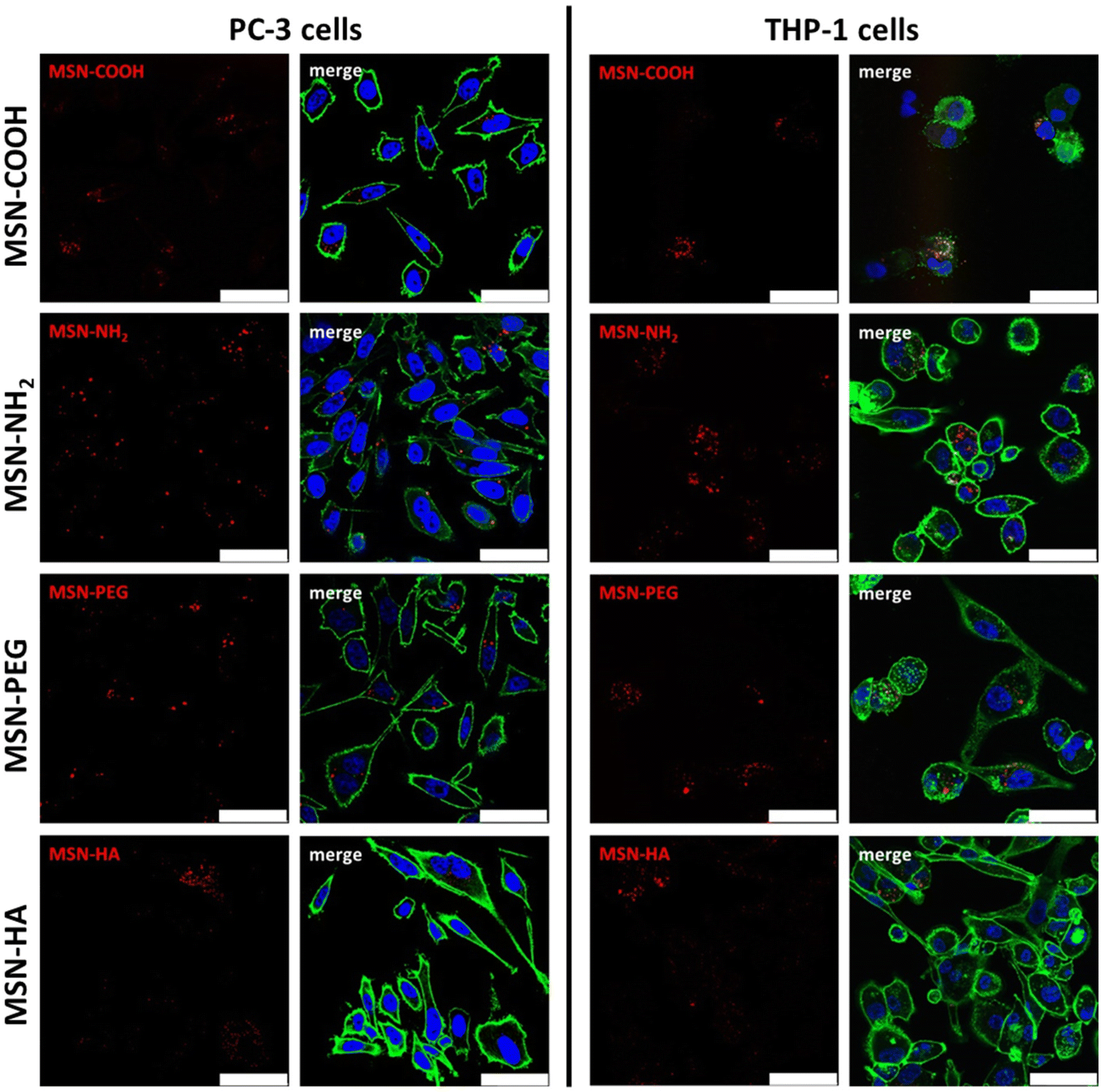 | ||
| Fig. 2 Exemplary micrographs of PC-3 (left column) and THP-1 (right column) cells treated with 10 μg ml−1 of MSNs for 24 h. Subsequent to fixation, cells were stained with fluorescent probes indicative for the actin cytoskeleton (Phalloidin-TRITC, green) and the nucleus (DAPI, blue). The nanoparticles are ATTO647N-labelled and depicted red in the images (scale bar: 50 μm). | ||
Exemplary histograms of ATTO647N-mediated fluorescence of viable cells in response to MSN treatment obtained from flow cytometry experiments are shown in Fig. 3. Gaussian-like distributions demonstrate a relatively homogeneous uptake of MSN-COOH, MSN-NH2 and MSN-HA in PC-3 cells, whereas the internalization of MSN-PEG was rather inhomogeneous, resulting in a broader distribution in the histogram (Fig. 3a). THP-1 cells revealed an overall broader distribution of fluorescence intensities in the flow cytometry measurements for all MSNs tested (Fig. 3b). The bar plots shown in Fig. 2 show the quantitation of MSN uptake, where the mean of the geometric mean relative fluorescence intensities resulting from triplicate flow cytometry measurements were weighted with a particle specific factor reflecting the ATTO647N-labelling efficiency, as recently suggested45 (see Fig. S6†).
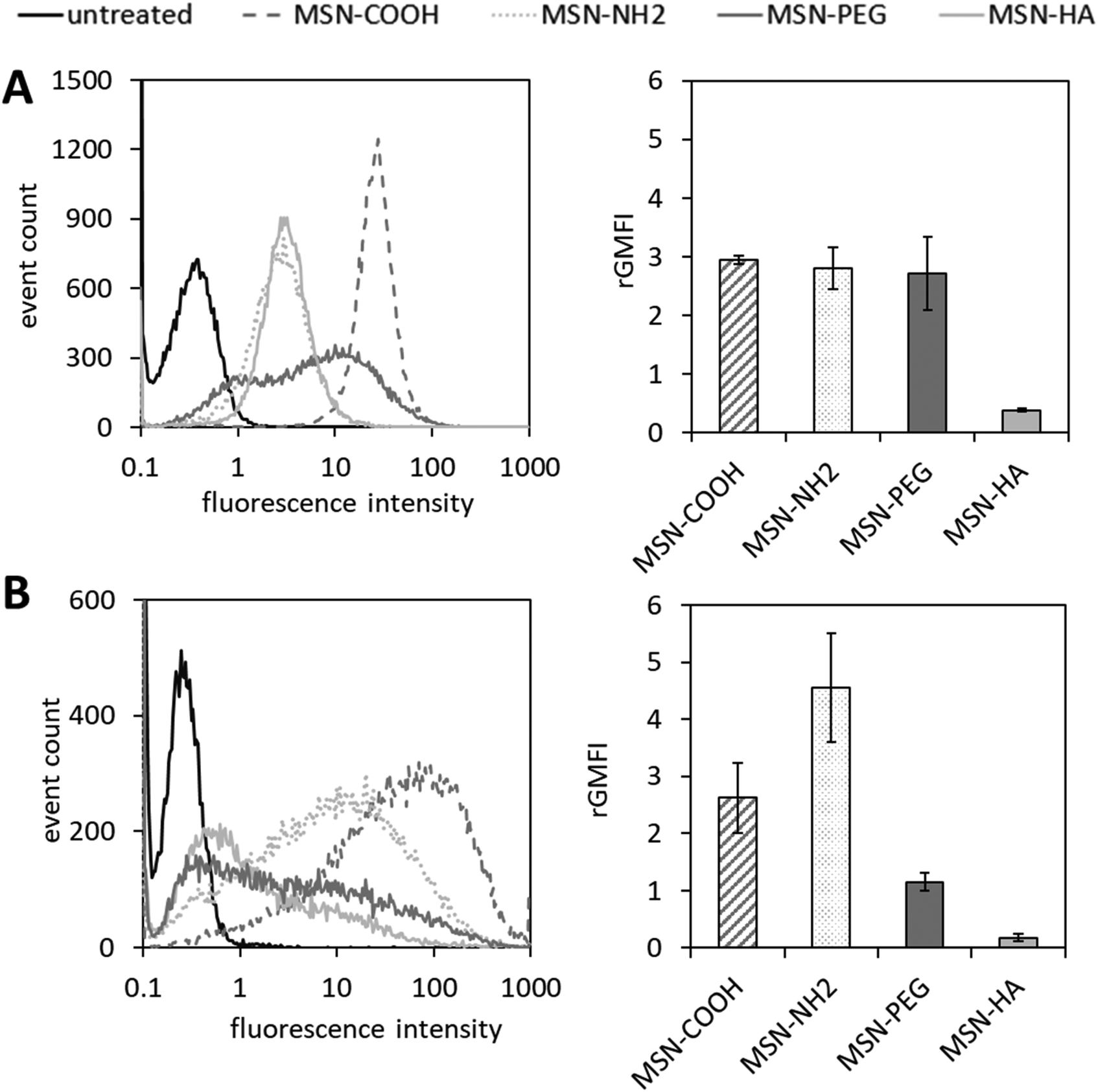 | ||
| Fig. 3 Analysis of MSN internalization by (A) PC-3 cells and (B) THP-1 cells via flow cytometry. Cell lines were treated with ATTO647N-labelled MSNs at 10 μg mL−1 for 24 h at 37 °C. Surface functionalities are color coded, respectively. Exemplary histograms of one out of three replicate measurements with similar outcomes are shown on the left side. For quantitation of internalization characteristics for each MSN species to a certain cell type, geometric mean fluorescence intensities (GMFIs) of MSN-treated cell populations were blank corrected (i.e. the geometric MFI of the corresponding untreated control populations was subtracted), rated with a particle-specific factor which reflects the ATTO647N-labelling efficiency (see Fig. S6†) and plotted as bar graphs (n = 3, error bars are SD). | ||
Rated GMFI values for particle internalization to both cell lines are shown in Fig. 4 as a function of zeta potential measured in HEPES buffer (pH = 7.2). In PC-3 cells, the positively charged MSN-NH2 and MSN-PEG particles are taken up to the same extent as the negatively charged MSN-COOH, and their cellular uptake is much higher (by a factor of ∼7) than that of the likewise negative MSN-HA particles. At first sight, this seems to contradict the expected influence of particle charge on the uptake of nanoparticles by endocytotic cells, as electrostatic attraction between positively charged particles with negatively charged phospholipids present in the outer cell membrane are assumed to be beneficial for cellular uptake efficiency.46,47 However, it was previously reported48 that considerable serum protein adsorption, as observed here for the MSN-COOH particles (see Fig. 5), can partially shield a negative surface charge resulting in elevated, endocytosis-driven nanoparticle uptake into human osteoblasts as compared to their positively charged counter-parts. With the low protein adsorption levels on MSN-HA (as discussed later) in mind, the strongly reduced uptake of these particles into PC-3 cells can be understood.
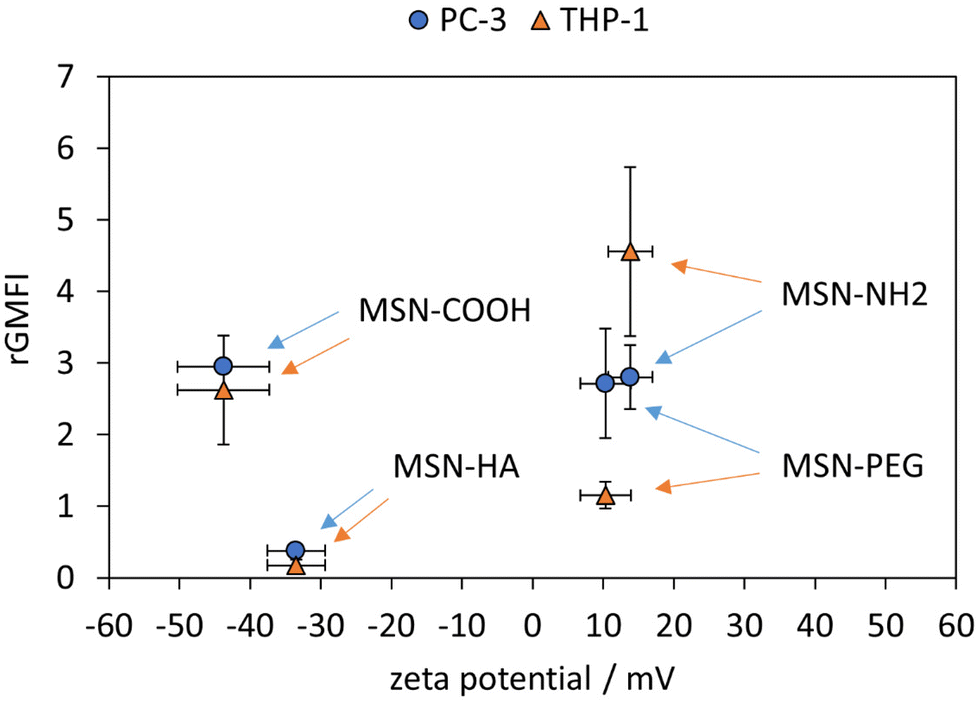 | ||
| Fig. 4 rGMFIs measured for PC-3 cells and THP-1 after incubation with 10 μg mL−1 particles for 24 h as a function of the MSN zeta potential. Zeta potentials were measured in aqueous HEPES buffer (25 mM, pH 7.2) (n = 3, error bars are SD). | ||
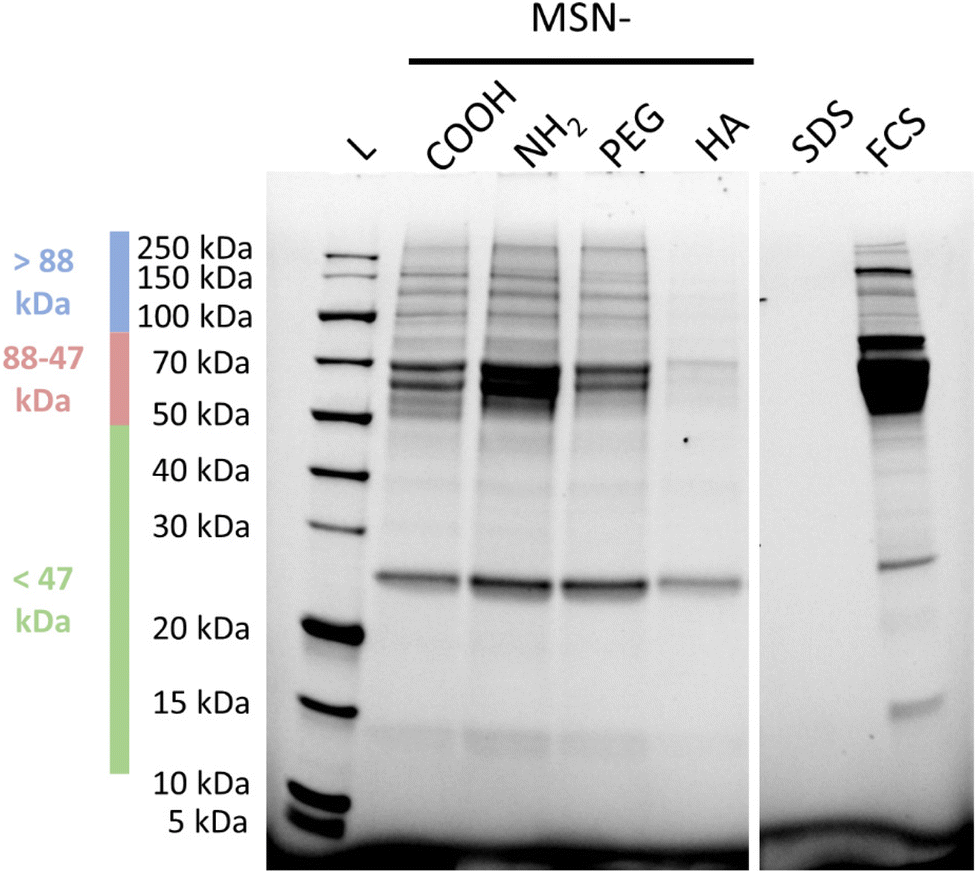 | ||
| Fig. 5 Representative SDS-PAGE of SDS supernatants after adsorption of serum proteins from 10% FCS in PBS onto MSNs at a silica concentration of 5 mg mL−1 for 24 h at 37 °C. L: broad range protein ladder, SDS: SDS control, FCS: 8% FCS control. | ||
The corresponding nanoparticle uptake by adherently growing macrophage-like THP-1 cells (M0) as determined by flow cytometry measurements are also summarized in Fig. 4. No correlation between particle charge (positive vs. negative particle charge) and uptake was observed, as highlighted by the almost 20 times higher rGMFI values measured for the anionic MSN-COOH as compared to anionic MSN-HA particles, and the approximately two-fold higher uptake of MSN-COOH vs. cationic MSN-PEG particles. The by far highest particle uptake was observed for the MSN-NH2 particles, although the MSN-PEG exhibited a virtually identical, positive zeta potential. Thus, in the case of THP-1 cells other factors than particle charge strongly influenced particle uptake. Nanoparticle uptake into macrophages have been repeatedly linked to protein corona formation. We therefore studied the composition of the protein corona of the different nanoparticles by SDS-PAGE. A representative gel is shown in Fig. 4.
Indeed, already based on visual inspection it is evident that the MSN-HA particles adsorbed by far the least amounts of serum proteins of all studied particles, and only for 26 kDa a strong band is seen, which can be ascribed to Apolipoprotein A-1 (Apo-A1),49 and a clearly fainter band in the range of 68 kDa ascribable to BSA can be seen. Albumin has previously been shown to interact weakly with HA-coated silica surfaces at neutral pH.50 Apo-A1 and serum albumin are common constituents of the protein corona of various nanoparticles subsequent to plasma exposition.51 Clearly stronger band intensities in the same molecular weight regions, and additionally in the molecular weight range >88 kDa, were seen for both MSN-COOH and MSN-PEG particles, and the band intensities were very similar for both sets of particles. By far the highest band intensities were observed for the MSN-NH2 particles, which is not surprising keeping in mind that most plasma proteins have a pI < 7.4.52 Thus, it is evident that the order of nanoparticle uptake by the THP-1 cells followed the order to serum protein adsorption to the particles.
We have previously shown that the integrated band intensities can be used for a semi-quantitative comparison of the relative amounts of protein adsorbed.53 In Fig. 6(a), integrated intensities obtained from three separate molecular weight windows (>88 kDa (high), 88–47 kDa (medium), and <47 kDa (low)), are shown, where the intensities have been normalized to those obtained for the respective molecular weight windows of the MSN-NH2 particles. As can be seen, the smaller molecular weight proteins, mainly corresponding to the strong band attributed to Apolipoprotein A-I, adsorb to all particles, while pronounced particle-dependent differences were observed for the heavier proteins, and especially so for the proteins with molecular weights in the medium range, mainly ascribed to BSA. While the correlation between the extent of adsorption of neither the heavy proteins nor of the lighter proteins showed a direct correlation with the particle uptake by the THP-1 cells, (ESI Fig. S7†) an almost perfectly linear correlation was observed between the relative amounts of medium molecular weight proteins and the cellular uptake, as shown in Fig. 6. These results suggest an active role of the protein corona, and more specifically albumin, in mediating the nanoparticle uptake by the THP-1 cells. A direct connection between serum protein adsorption,54 and specifically BSA,8,55 and nanoparticle uptake has also been demonstrated previously, and our preliminary results imply an even quantitative connection between these parameters.
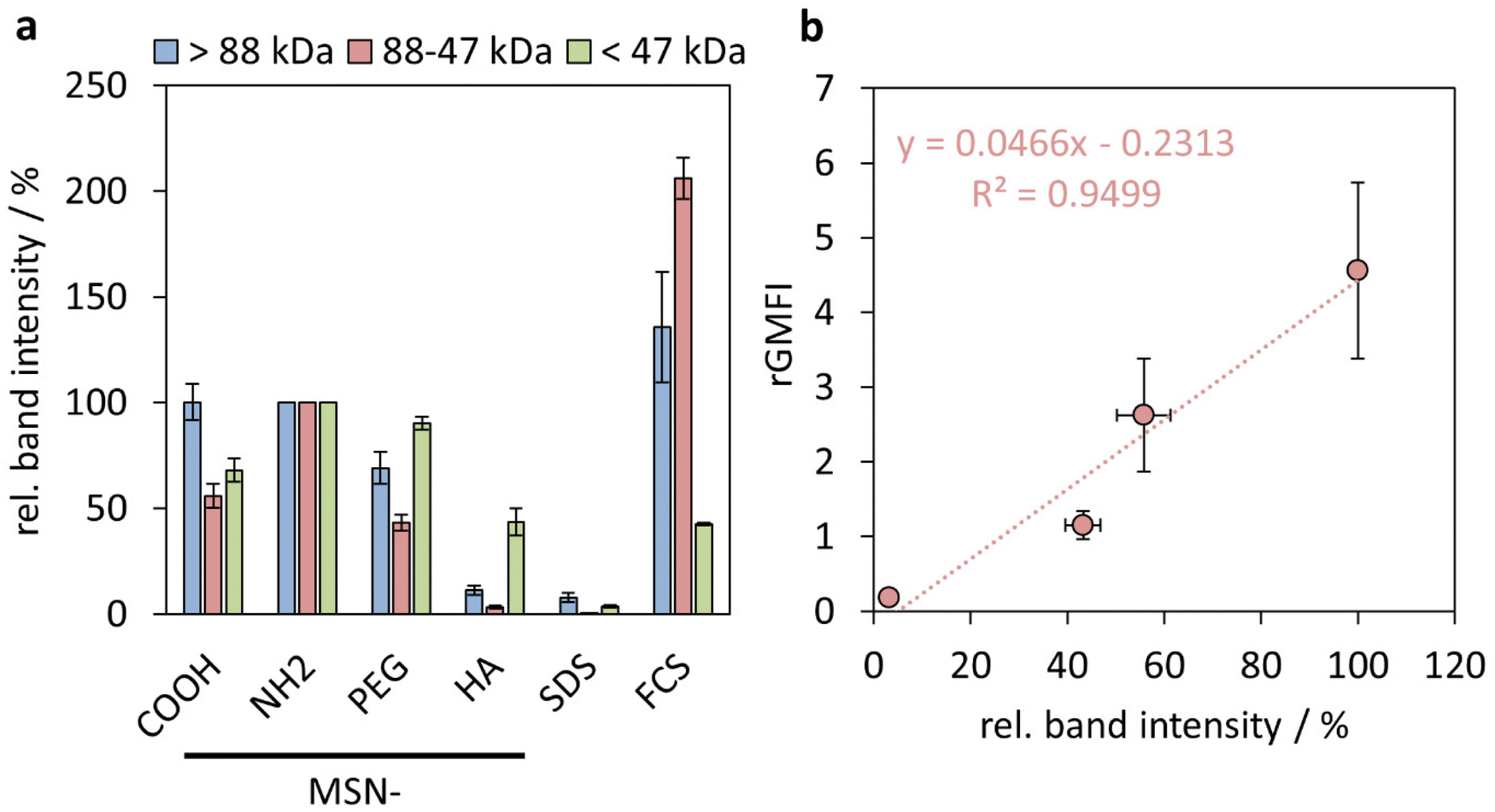 | ||
| Fig. 6 (a) Relative band intensities for the three protein mass windows: high molecular weight (>88 kDa), medium molecular weight (88–47 kDa) and low molecular weight (<47 kDa). Band intensities were normalized on the respective intensities of MSN-NH2. (b) rGMFIs measured for THP-1 cells after particle treatment for 24 h as a function of relative band intensities of medium molecular weight proteins (n = 3, error bars = SD). | ||
Having determined the nanoparticle uptake in the two model cell lines, the next step was to investigate how the uptakes can be converted into a general measure by which the nanoparticles can be classified in terms of their potential ability to most efficiently deliver drugs to tumours. The most straightforward means for grading the nanoparticles would be in terms of their selectivity towards the cancer cells. The selectivity was therefore defined simply as the ratio between the corresponding relative uptakes, uptakePC-3/uptakeTHP-1. The corresponding ratios obtained for the studied nanoparticles are shown in Fig. 7. The selectivity for the cancer cells is the highest for the MSN-PEG particles, closely followed by the MSN-HA particles.
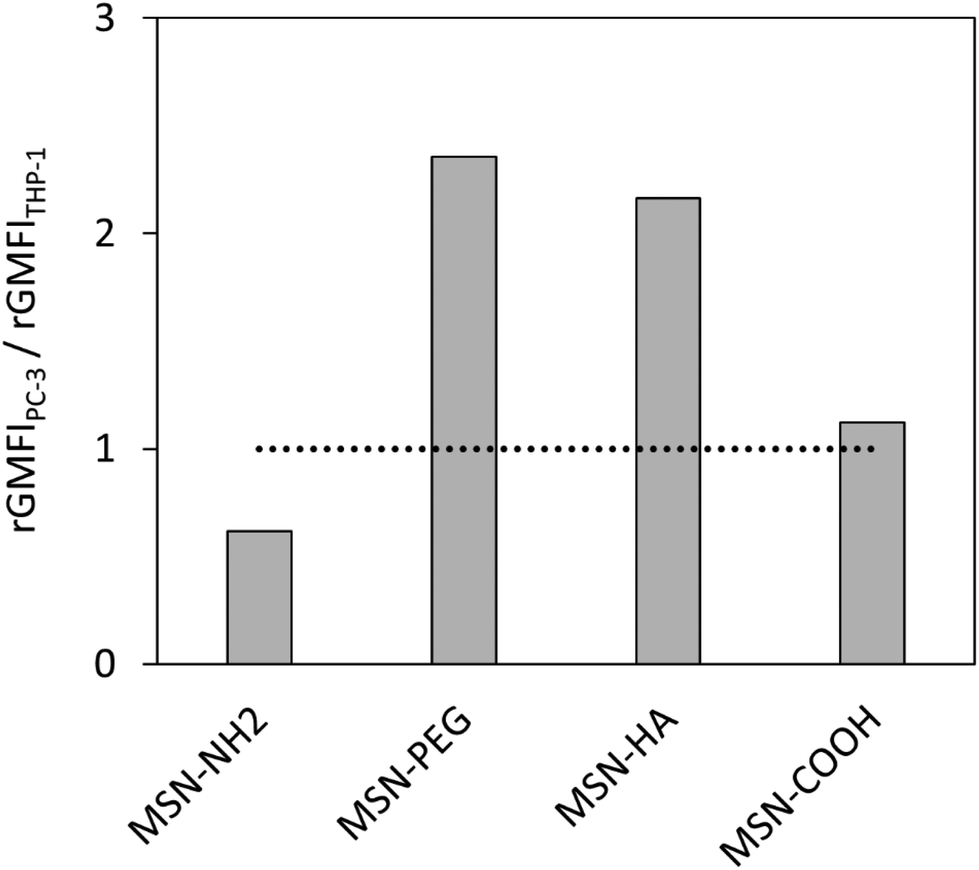 | ||
| Fig. 7 Nanoparticle uptake ratio calculated as rGMFIPC-3/rGMFITHP-1. | ||
Surface charge of nanocarriers generally plays a critical role for tumour targeting in vivo. However, findings reported in the literature are heterogeneous and suggest that a combination of surface charge extent, size and shape of the nanocarrier determines its fate in the organism.56 Different studies suggest that a modest negative surface charge appears to be favourable for circumvention of MPS clearance and improved blood compatibility, thereby extending blood circulation time and improving tumour delivery.57,58 On the other hand, cationic nanocarriers were recently used for the improvement of tumour immunosurveillance59 as well as, more specifically, the administration of cancer vaccines which preferentially target antigen presenting cells.18 These findings were reflected by our in vitro system: whereas internalization of anionic MSN-COOH particles was still slightly higher in PC-3 vs. THP-1 cells, cationic MSN-NH2 particles had a preference for THP-1 cells.
To further account for the overall amount of nanocarrier internalization, a key parameter for reaching a high intracellular drug concentration in the target cells, we define a new value for comparison, specifically focusing on this aspect, as uptakePC-3x the selectivity, i.e. (uptakePC-3)2/uptakeTHP-1. This is equivalent to an activity times selectivity-measure for catalyst classification. The so obtained values are shown in Fig. 8. As can be seen, the MSN-PEG particle is clearly the most promising one according to this analysis. This result is in full agreement with what is to be expected for the drug delivery efficiency of the studied particles to solid tumours if administered intravenously.
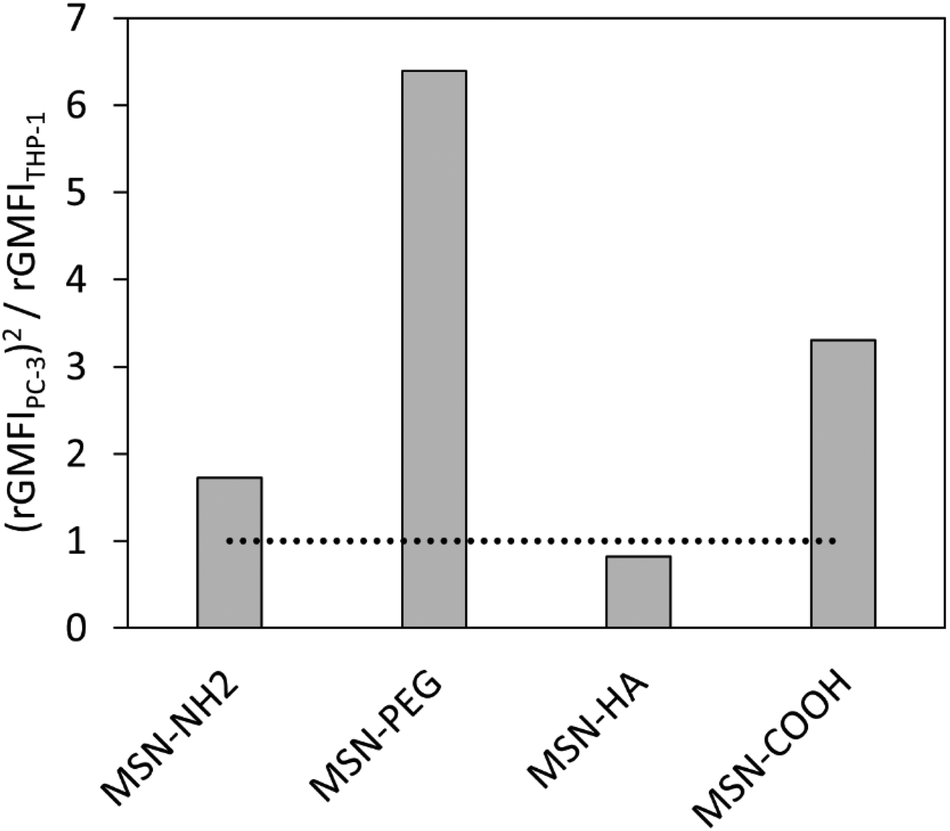 | ||
| Fig. 8 Nanoparticle uptake ratio calculated as (rGMFIPC-3)2/rGMFITHP-1. | ||
Naturally this simple means for analysis is not quantitative, but can serve as a simple preliminary tool for grading particles during in vitro particle screenings for identifying the most promising nanoparticle surface chemistries for further more detailed in vitro and in vivo evaluations. We are well aware that the model is oversimplified, but encouraged by the fact that the outcome appears to be reasonable. Experimental parameters like incubation time, particle concentration or cell lines could be varied but were chosen based on some consideration. A low particle concentration of 10 μg mL−1 was chosen in order to minimize potential contributions from particle aggregation and sedimentation on the cellular uptake, as well as to avoid potential cellular saturation effects. The used incubation time of 24 h is within relevant in vivo time-scale, and minimizes potential influences of different nanoparticle internalization kinetics. The cancer cell line can naturally be varied at will, based on the application aimed for, and particles intended for active targeting can also easily be screened with the suggested methodology. We emphasize that the main aim of the suggested screening is to identify the most promising surface chemistries for efficient drug delivery to solid tumours through intravenous (or intraperitoneal) administration, and the model is probably relatively insensitive to other important parameters for in vivo responses of nanoparticles, like particle size and shape.
Conclusions
The identification of the most promising nanoparticle formulations for drug delivery to solid tumours is a bottleneck in the field of bionanomedicine. Chemists can synthesize a battery of different particles within a time-frame of weeks and months, and testing of all those particles in vivo is expensive and time-consuming, and also not ethically acceptable. Here we propose a very simple in vitro nanoparticle surface chemistry screening methodology based on established cell-lines that ensures reproducibility and comparability of the results. As the efficiency of the drug delivery is dependent both upon the possibility of the nanoparticles to escape the RES as well as on the extent of which they are accumulating in the tumour area and internalized by the cancer cells, a macrophage model cell line, THP-1, as well as a model cancer cell line, PC-3, was used for the screening. Realistic mirroring of in vivo results obtained by intravenous administration of comparable particles was achieved only when taking into account both the selectivity of the nanoparticles for the cancer cells, as well as the extent of nanoparticle uptake by the cancer cells. The screening method can naturally easily be modified to account for other cancer cell types, and is also suitable for screening of nanoparticles where active, receptor-mediated cell targeting is aimed for. While the screening method promises to be suitable for early identification of promising nanoparticle surface chemistries, it seems less suitable for the optimization of other important nanoparticle characteristics like size and probably also shape. When increasingly more data becomes available, the predictability of the screening method can be judged, and further optimized.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the DFG funded projects WI3868/6-1 as well as SFB 1279 is gratefully acknowledged. The authors want to thank Cornelia Egger for nitrogen sorption measurements, Petra Kruse and Eva Winkler for technical assistance in cell culture experiments, as well as Kea Fedke and Lionel Kroner for TG measurements.References
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- Y. N. Zhang, W. Poon, A. J. Tavares, I. D. McGilvray and W. C. W. Chan, J. Controlled Release, 2016, 240, 332, DOI:10.1016/j.jconrel.2016.01.020.
- W. P. Caron, S. Rawal, G. Song, P. Kumar, J. C. Lay and W. C. Zamboni, J. Pharmacol. Exp. Ther., 2013, 347, 599–606, DOI:10.1124/jpet.113.208801.
- J. D. Friedl, V. Nele, G. de Rosa and A. Bernkop-Schnürch, Adv. Funct. Mater., 2021, 31, 2103347, DOI:10.1002/adfm.202103347.
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. H. Müller, Colloids Surf., B, 2000, 18, 301, DOI:10.1016/S0927-7765(99)00156-3.
- W. R. Gombotz, W. Guanghui, T. A. Horbett and A. S. Hoffman, J. Biomed. Mater. Res., 1991, 25, 1547, DOI:10.1002/jbm.820251211.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93, DOI:10.1016/j.ijpharm.2005.10.010.
- J. A. Champion, S. M. Pustulka, K. Ling and S. L. Pish, ACS Appl. Mater. Interfaces, 2020, 12, 48284, DOI:10.1021/acsami.0c12341.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, J. Am. Chem. Soc., 2012, 134, 2139, DOI:10.1021/ja2084338.
- C. Yao, P. Wang, L. Zhou, R. Wang, X. Li, D. Zhao and F. Zhang, Anal. Chem., 2014, 86, 9749, DOI:10.1021/ac5023259.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941, DOI:10.1038/nbt.3330.
- L. Kou, Y. D. Bhutia, Q. Yao, Z. He, J. Sun and V. Ganapathy, Front. Pharmacol., 2018, 9 DOI:10.3389/fphar.2018.00027.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505–15, DOI:10.1021/mp800051m.
- J. S. Souris, C. H. Lee, S. H. Cheng, C. T. Chen, C. S. Yang, J. an A. Ho, C. Y. Mou and L. W. Lo, Biomaterials, 2010, 31, 5564, DOI:10.1016/j.biomaterials.2010.03.048.
- S. Ashraf, J. Park, M. A. Bichelberger, K. Kantner, R. Hartmann, P. Maffre, A. H. Said, N. Feliu, J. Lee, D. Lee, G. U. Nienhaus, S. Kim and W. J. Parak, Nanoscale, 2016, 8, 17794, 10.1039/c6nr05805a.
- M. Beck, T. Mandal, C. Buske and M. Lindén, ACS Appl. Mater. Interfaces, 2017, 9, 18566, DOI:10.1021/acsami.7b04742.
- A. Panariti, G. Miserocchi and I. Rivolta, Nanotechnol., Sci. Appl., 2012, 7, 87, DOI:10.2147/NSA.S25515.
- J. Heuts, W. Jiskoot, F. Ossendorp and K. van der Maaden, Pharmaceutics, 2021, 13, 596, DOI:10.3390/pharmaceutics13050596.
- S. Jambhrunkar, Z. Qu, A. Popat, J. Yang, O. Noonan, L. Acauan, Y. Ahmad Nor, C. Yu and S. Karmakar, Mol. Pharm., 2014, 11, 3642, DOI:10.1021/mp500385n.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Lindén, ACS Nano, 2009, 3, 19, DOI:10.1021/nn800781r.
- H. X. Wang, Z. Q. Zuo, J. Z. Du, Y. C. Wang, R. Sun, Z. T. Cao, X. D. Ye, J. L. Wang, K. W. Leong and J. Wang, Nano Today DOI:10.1016/j.nantod.2016.04.008.
- R. Wittig, J. M. Rosenholm, E. von Haartman, J. Hemming, F. Genze, L. Bergman, T. Simmet, M. Lindén and C. Sahlgren, Nanomedicine, 2014, 9 DOI:10.2217/nnm.13.62.
- K. Song, Z. Tang, Z. Song, S. Meng, H. Guo, Y. Zhu and X. Wang, Pharmaceutics, 2022, 14, 1265, DOI:10.3390/pharmaceutics14061265.
- K. Y. Choi, K. H. Min, H. Y. Yoon, K. Kim, J. H. Park, I. C. Kwon, K. Choi and S. Y. Jeong, Biomaterials, 2011, 32, 1880, DOI:10.1016/j.biomaterials.2010.11.010.
- P. Dogra, N. L. Adolphi, Z. Wang, Y. S. Lin, K. S. Butler, P. N. Durfee, J. G. Croissant, A. Noureddine, E. N. Coker, E. L. Bearer, V. Cristini and C. J. Brinker, Nat. Commun., 2018, 9 DOI:10.1038/s41467-018-06730-z.
- X. He, H. Nie, K. Wang, W. Tan, X. Wu and P. Zhang, Anal. Chem., 2008, 80, 9597, DOI:10.1021/ac801882g.
- L. Miller, G. Winter, B. Baur, B. Witulla, C. Solbach, S. Reske and M. Lindén, Nanoscale, 2014, 6, 4928, 10.1039/c3nr06800e.
- M. Xie, Y. Xu, H. Shen, S. Shen, Y. Ge and J. Xie, Int. J. Pharm., 2014, 474, 223, DOI:10.1016/j.ijpharm.2014.08.027.
- L. Kramer, G. Winter, B. Baur, A. J. Kuntz, T. Kull, C. Solbach, A. J. Beer and M. Lindén, Nanoscale 10.1039/c7nr02050c.
- Q. He, Z. Zhang, F. Gao, Y. Li and J. Shi, Small DOI:10.1002/smll.201001459.
- Y. He, L. Luo, S. Liang, M. Long and H. Xu, J. Biomater. Appl., 2017, 32 DOI:10.1177/0885328217724638.
- V. Mamaeva, R. Niemi, M. Beck, E. Ozliseli, D. Desai, S. Landor, T. Gronroos, P. Kronqvist, I. K. N. Pettersen, E. McCormack, J. M. Rosenholm, M. Linden and C. Sahlgren, Mol. Ther., 2016, 24, 926, DOI:10.1038/mt.2016.42.
- R. Wittig, J. M. Rosenholm, E. von Haartman, J. Hemming, F. Genze, L. Bergman, T. Simmet, M. Lindén and C. Sahlgren, Nanomedicine, 2014, 9 DOI:10.2217/nnm.13.62.
- S. Tsuchiya, M. Yamabe, Y. Yamaguchi, Y. Kobayashi, T. Konno and K. Tada, Int. J. Cancer, 1980, 26, 171, DOI:10.1002/ijc.2910260208.
- Z. Zhu, C. Scalfi-Happ, A. Ryabova, S. Gräfe, A. Wiehe, R. U. Peter, V. Loschenov, R. Steiner and R. Wittig, J. Photochem. Photobiol., B, 2018, 185, 215, DOI:10.1016/j.jphotobiol.2018.06.015.
- S. M. Wittig-Blaich, L. A. Kacprzyk, T. Eismann, M. Bewerunge-Hudler, P. Kruse, E. Winkler, W. S. L. Strauss, R. Hibst, R. Steiner, M. Schrader, D. Mertens, H. Sültmann and R. Wittig, Neoplasia, 2011, 13, 579, DOI:10.1593/neo.11294.
- S. Wittig-Blaich, R. Wittig, S. Schmidt, S. Lyer, M. Bewerunge-Hudler, S. Gronert-Sum, O. Strobel-Freidekind, C. Müller, M. List, A. Jaskot, H. Christiansen, M. Hafner, D. Schadendorf, I. Block and J. Mollenhauer, Oncotarget, 2017, 8, 23760, DOI:10.18632/oncotarget.15863.
- C. Wang, C. Ma, L. Gong, Y. Guo, K. Fu, Y. Zhang, H. Zhou and Y. Li, Front. Immunol., 2021, 12 DOI:10.3389/fimmu.2021.803037.
- K. M. Bircsak, R. DeBiasio, M. Miedel, A. Alsebahi, R. Reddinger, A. Saleh, T. Shun, L. A. Vernetti and A. Gough, Toxicology, 2021, 450, 152667, DOI:10.1016/j.tox.2020.152667.
- T. Brzicova, E. Javorkova, K. Vrbova, A. Zajicova, V. Holan, D. Pinkas, V. Philimonenko, J. Sikorova, J. Klema, J. Topinka and P. Rossner, Nanomaterials, 2019, 9, 687, DOI:10.3390/nano9050687.
- Y. Matsuoka, Y. Kawauchi, K. Kawauchi, A. Takiyama, S. Kojima, Y. Kuroda and N. Kojima, Glycoconjugate J., 2019, 36, 185, DOI:10.1007/s10719-019-09870-6.
- A. D. Schoenenberger, A. Schipanski, V. Malheiro, M. Kucki, J. G. Snedeker, P. Wick and K. Maniura-Weber, ACS Biomater. Sci. Eng., 2016, 2, 908, DOI:10.1021/acsbiomaterials.6b00006.
- W. Chanput, J. J. Mes and H. J. Wichers, Int. Immunopharmacol., 2014, 23, 37, DOI:10.1016/j.intimp.2014.08.002.
- M. Fisichella, H. Dabboue, S. Bhattacharyya, G. Lelong, M. L. Saboungi, F. Warmont, P. Midoux, C. Pichon, M. Guérin, T. Hevor and J. P. Salvetat, J. Nanosci. Nanotechnol., 2010, 10 CrossRef CAS PubMed.
- B. Baumann, R. Wittig and M. Lindén, Nanoscale, 2017, 9, 12379, 10.1039/c7nr02015e.
- O. Harush-Frenkel, N. Debotton, S. Benita and Y. Altschuler, Biochem. Biophys. Res. Commun., 2007, 353, 26, DOI:10.1016/j.bbrc.2006.11.135.
- A. Verma and F. Stellacci, Small, 2010, 6, 12, DOI:10.1002/smll.200901158.
- S. Shahabi, L. Treccani, R. Dringen and K. Rezwan, ACS Appl. Mater. Interfaces, 2015, 7, 13821, DOI:10.1021/acsami.5b01900.
- A. Pochert, I. Vernikouskaya, F. Pascher, V. Rasche and M. Lindén, J. Colloid Interface Sci., 2017, 488, 1, DOI:10.1016/j.jcis.2016.10.085.
- I. Berts, G. Fragneto, L. Porcar, M. S. Hellsing and A. R. Rennie, J. Colloid Interface Sci., 2017, 504, 315, DOI:10.1016/j.jcis.2017.05.037.
- S. Winzen, S. Schoettler, G. Baier, C. Rosenauer, V. Mailaender, K. Landfester and K. Mohr, Nanoscale, 2015, 7, 2992, 10.1039/c4nr05982d.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780, 10.1039/c1cs15233e.
- C.-Y. Lin, C.-M. Yang and M. Lindén, RSC Adv., 2019, 9, 33912, 10.1039/c9ra05585a.
- O. Lunov, T. Syrovets, C. Loos, J. Beil, M. Delacher, K. Tron, G. U. Nienhaus, A. Musyanovych, V. Mailänder, K. Landfester and T. Simmet, ACS Nano, 2011, 5, 1657, DOI:10.1021/nn2000756.
- Y. Yan, K. T. Gause, M. M. J. Kamphuis, C. S. Ang, N. M. O'Brien-Simpson, J. C. Lenzo, E. C. Reynolds, E. C. Nice and F. Caruso, ACS Nano, 2013, 7, 10960, DOI:10.1021/nn404481f.
- X. Duan and Y. Li, Small, 2013, 9, 1521, DOI:10.1002/smll.201201390.
- Y. Yamamoto, Y. Nagasaki, Y. Kato, Y. Sugiyama and K. Kataoka, J. Controlled Release, 2001, 77, 27–38, DOI:10.1016/s0168-3659(01)00451-5.
- K. Xiao, Y. Li, J. Luo, J. S. Lee, W. Xiao, A. M. Gonik, R. G. Agarwal and K. S. Lam, Biomaterials, 2011, 32, 3435, DOI:10.1016/j.biomaterials.2011.01.021.
- R. Smith, E. I. Wafa, S. M. Geary, S. H. Alhaj-Suliman and A. K. Salem, Sci. Adv., 2022, 8 DOI:10.1126/sciadv.abk3150.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm00966a |
| This journal is © The Royal Society of Chemistry 2023 |