
An advanced biphasic porous and injectable scaffold displays a fine balance between mechanical strength and remodeling capabilities essential for cartilage regeneration†
Gabriela
Zavala
abc,
Sergio M.
Viafara-García
abc,
Javier
Novoa
abc,
Carmen
Hidalgo
abc,
Ingrid
Contardo
ad,
Paulo
Díaz-Calderón
ad,
Wilfredo
Alejandro González-Arriagada
ae,
Maroun
Khoury
abc and
Juan Pablo
Acevedo
 *abc
*abc
aCentro de Investigación e Innovación Biomédica (CIIB), Universidad de los Andes, Chile. E-mail: jpacevedo@uandes.cl
bCells for Cells and REGENERO, The Chilean Consortium for Regenerative Medicine, Santiago, Chile
cIMPACT, Center of Interventional Medicine for Precision and Advanced Cellular Therapy, Santiago, Chile
dFacultad de Medicina, Escuela de Nutrición y Dietética, Biopolymer Research & Engineering Laboratory (BiopREL), Universidad de los Andes, Chile
eFacultad de Odontología, Universidad de los Andes, Chile
First published on 25th August 2023
Abstract
An important challenge in tissue engineering is the regeneration of functional articular cartilage (AC). In the field, biomimetic hydrogels are being extensively studied as scaffolds that recapitulate microenvironmental features or as mechanical supports for transplanted cells. New advanced hydrogel formulations based on salmon methacrylate gelatin (sGelMA), a cold-adapted biomaterial, are presented in this work. The psychrophilic nature of this biomaterial provides rheological advantages allowing the fabrication of scaffolds with high concentrations of the biopolymer and high mechanical strength, suitable for formulating injectable hydrogels with high mechanical strength for cartilage regeneration. However, highly intricate cell-laden scaffolds derived from highly concentrated sGelMA solutions could be deleterious for cells and scaffold remodeling. On this account, the current study proposes the use of sGelMA supplemented with a mesophilic sacrificial porogenic component. The cytocompatibility of different sGelMA-based formulations is tested through the encapsulation of osteoarthritic chondrocytes (OACs) and stimulated to synthesize extracellular matrix (ECM) components in vitro and in vivo. The sGelMA-derived scaffolds reach high levels of stiffness, and the inclusion of porogens impacts positively the scaffold degradability and molecular diffusion, improved fitness of OACs, increased the expression of cartilage-related genes, increased glycosaminoglycan (GAG) synthesis, and improved remodeling toward cartilage-like tissues. Altogether, these data support the use of sGelMA solutions in combination with mammalian solid gelatin beads for highly injectable formulations for cartilage regeneration, strengthening the importance of the balance between mechanical properties and remodeling capabilities.
1. Introduction
Articular cartilage (AC) is made of specialized load-bearing tissue, is part of the diarthrodial joints, and is classified as hyaline cartilage. AC is a complex mechanical transductor of compressive forces and depends on the integrity of a collagen II microfibrillar arrangement, which interacts with other less represented types of collagens (such as VI, X, IX, XI, XII, and XIV). Proteoglycans, primarily aggrecan (ACAN), also interact with the collagen II fibrils. Their significant chondroitin sulfate content, a type of glycosaminoglycan (GAG), assists in preserving tissue hydrostatic pressure and integrity following deformation.1 Chondrocytes are responsible for synthesizing and maintaining the extracellular matrix (ECM). These cells are adapted to the constant compressive pulses maintaining the tissue's homeostasis through a balance between catabolism and anabolism of the matrix.2Due to its avascular nature, AC has a limited regenerative capacity.3 Even though small lesions can heal, hyaline tissue is usually replaced by fibrocartilage with lower mechanical resilience. Injuries, metabolic or inflammatory diseases can tilt the balance into a catabolic and degrading environment, leading to a slow but constant degeneration,4 and could derive in chronic lesions or diseases, such as arthrosis or osteoarthritis (OA).5
Important factors are necessary to be considered when it comes to designing an ideal scaffold for hyaline cartilage regeneration. First, vertical supports that fill chondral lesions would guide the anatomically correct formation of new tissue, while a sufficiently tough scaffold material would avoid the concentration of deformation at the edge of lesions during normal use of articular joints. These deformations can cause damage to the resident chondrocytes, resulting in metabolic imbalance and further progression of the lesion.6,7 Surgical interventions for chondral lesion restoration via scaffold implantations usually involve open surgeries generating additional problems that prolong post-operational hospitalization.8 Arthroscopic surgeries are less invasive and preferred for cartilage treatments, but scaffolds need to be injectable or extrudable for this type of procedure. In regard to the scaffold, a porous design would be more suitable for recapitulating organogenic-like processes that could lead to the development of healthy new hyaline cartilage. A mechanism of this kind would require spaces or pores for the cells to adhere, proliferate and secrete new extracellular matrices while an active and balanced degradation of the original scaffold material is carried out by the same cells.9
Due to the high water content of cartilage, hydrogels have been extensively studied as fillers for the restoration and host cell-driven regeneration of AC lesions. Mammalian-derived gelatins, especially the covalently photocrosslinkable methacrylated porcine gelatin (pGelMA),10 have been widely studied in tissue engineering, including cartilage regeneration.11–14 However, none of them have reached a substantially higher mechanical resistance (<0.199 MPa in compressive modulus), and they do not incorporate porosity within the scaffold structure after crosslinking. Unfortunately, pGelMA polymer solutions cannot be prepared at higher than 20% (w/v) concentrations while keeping a reasonable viscosity and liquid state stability at room temperature to facilitate manipulation, extrudability, and controlled shape hydrogel fabrication via specialized biofabrication systems. Therefore, it is undoubtedly complex to obtain a tough scaffold structure made of highly concentrated pGelMA solutions without blocking the extrusion or biofabrication system or without using elaborated temperature-controlled devices.11
Psychrophilic gelatins derived from cold-adapted species possess additional properties that are advantageous for applications in tissue engineering.15,16 For instance, salmon skin-derived gelatin (sGel) possesses less proline and hydroxyproline in its aminoacidic sequence than mammalian gelatins, and consequently, cold-adapted gelatins in solution have a very low-temperature melting point (∼4 °C), unlike mammalian gelatin solutions that melt above 30 °C, allowing for low viscous, highly concentrated, and stable liquid phase solutions at room temperature, properties that cannot be achieved with mesophilic gelatins.15–17
Methacrylated sGel (sGelMA) has demonstrated interesting applications in high-resolution 3D bioprinting due to its injectability properties,18 allowing the preparation of solutions with high concentrations of the polymer while maintaining the liquid state, making the biofabrication of scaffolds with high resistance to compression plausible for cartilage regeneration applications. Additionally, polymerization could be induced in situ with UV light, allowing the complete filling of a lesion, even for lesions with intricate edges.
AC is commonly subjected to constant biomechanical loads, with a resistance to compression in the compressive modulus range of 0.2–0.7 MPa.19 High-stiffness hydrogels could reach such values only after using a substantial concentration of the polymer component and a high degree of functionalization. After crosslinking highly concentrated biopolymer solutions, a tough and highly intercalated network will form, and it will require a certain degree of porosity to facilitate cell infiltration, growth, exchange of nutrients/waste, and remodeling toward new healthy tissue. A strategy is to add a sacrificial porogenic component to the formulation, which can be removed after the bulk hydrogel solution is polymerized, obtaining porous and highly stiff hydrogels. The advantage of using a porogenic component has been previously described in different models, including in cartilage regeneration, but the parameter of high mechanical resistance in the same formulation has not been achieved.9,20 One of these approaches uses surface-crosslinked gelatin beads to maintain the porogen stability when mixed with the liquid bulk hydrogel solution; otherwise, they could both melt and mix, and not form a porous structure after thermal removal of the porogen.9 This approach claims to be injectable, however it uses either warm liquid agarose or alginate solutions as the bulk hydrogel material, which requires a cooling step or a bath in calcium chloride to induce the sol–gel transition. This limits the application in cartilage lesion reconstruction and crosslinking through injection or minimally invasive arthroscopic approaches. A second approach also uses thermo-reversible solid mammalian gelatin beads but in combination with photocrosslinkable GelMA solution as the bulk hydrogel material.20 In this case, the temperature ranges in which the sol–gel and gel–sol transition occurs for both gelatins almost overlap; therefore, porous scaffold formation is only possible through careful and quick handling, and temperature control after mixing, which makes this approach impracticable for in situ repair of cartilage lesions.
To obtain an extrudable biopolymer mixture that after filling the lesion and crosslinking is capable of forming an implanted porous scaffold, the solid porogenic gelatin element and the dispersing liquid phase need to be stable and separated in different aggregation phases all through the process of mixing, extrusion system loading, and during the deposition and crosslinking of the solution into the lesion. Therefore, it is ideal that both components present very distinguishable transition states at very different temperature ranges; otherwise, they could both transit toward a liquid aggregation state and mix without forming pores after implantation.
Although there are many potential strategies to treat cartilage lesions using injectable scaffolds, none fulfill the requirements of a chondroinductive, remodeling-susceptible, porous and stiff cartilage-like scaffold. The proposal is to develop a biphasic combination to obtain an injectable porous biopolymeric formulation, stable within a wide range of temperatures before crosslinking. The potential application of this composed formulation is to extrude and fill a cartilage lesion with a tough and porous scaffold, giving mechanical support, whereas a remodeling process occurs, eventually replacing the material with healthy tissue.
2. Materials and methods
Ethics statement
Cartilage specimens were collected from individuals with OA who had undergone hip replacement surgery, after obtaining informed consent. The experiments were conducted in compliance with the guidelines of Good Clinical Practice and the Declaration of Helsinki. The Institutional Ethics Committee (Ethical and Scientific Committee, University of the Andes) and the institutional review board at the University of the Andes Clinic granted approval for the experiments.Immunocompromised mice (NOD·Cg-Prkdcscid Il2rgtm1Wjl/SzJ or NSG) were provided by the Cells for Cells Animal Facility, and the studies were designed following the Institutional Animal Care and Use Committee of University of the Andes, also in accordance with the recommendations of the Agencia Nacional de Investigación y Desarrollo (ANID). Protocols and experiments using mice were also revised and approved by the institutional Ethical Committee (Ethical and Scientific Committee, University of the Andes). The animals were maintained in an SPF (specific pathogen-free) controlled environment under a temperature of 24 ± 2 °C, humidity of 50 ± 10%, and 12 h![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 h light/dark cycle and housed in individually ventilated caging (IVC) systems.
:12 h light/dark cycle and housed in individually ventilated caging (IVC) systems.
Fabrication of methacrylamide salmon gelatin (sGelMA)
Salmon gelatin (sGel) was extracted from the skin of Atlantic salmon specimens (Salmo salar) as previously described.21 Salmon methacrylamide gelatin (sGelMA) was synthesized based on a previously described protocol.22 Briefly, 10% (w/v) salmon gelatin solution was prepared in phosphate-buffered saline (PBS) 1×, dissolving the gelatin under agitation at 60 °C. Methacrylic anhydride (Sigma-Aldrich, USA) was added dropwise until reaching a final concentration of 8% (v/v) in the volume of the gelatin solution, maintaining the functionalization reaction for 3 h under constant stirring at 60 °C. Afterwards, 3 volumes of PBS 1× were added to stop the methacrylation reaction. The solution was dialyzed for 7 days at 40 °C to remove any unreacted methacrylic anhydride and then freeze-dried, protected from light, and stored at −20 °C for later use. The obtained degree of functionalization (DoF) at the free amine of the lysine side chain was estimated to be 90% by 1H NMR spectroscopy and the OPA assay.16Preparation of porcine gelatin beads
Porcine gelatin beads were fabricated using a previously described water-in-oil emulsification protocol.23 Briefly, 10% w/v solution of non-functionalized type-A porcine gelatin (Sigma-Aldrich) was prepared by dissolving 0.5 g in 5 mL PBS 1× at 37 °C with gentle agitation. The gelatin solution was adjusted to pH 7.4 using NaOH 1 M. While still warm, the solution was filtered using a 5 μm pore nitrocellulose filter and subjected to a heat–cold sterilization protocol (3 series of incubation at 80 °C × 20 min and followed by 5 min at 4 °C). The porcine gelatin and the mineral oil (Sigma-Aldrich) were pre-warmed at 37 °C before conducting the emulsification protocol. In a biosafety cabinet, 5 mL of the gelatin solution was slowly added (1 mL min−1) into a mixture of 25 mL of mineral oil and 125 μL of Tween 20 (Sigma-Aldrich), the last kept under continuous stirring at 600 rpm for 10 minutes. Both the mineral oil and the Tween 20 were cell culture-quality to secure the sterility of the beads. Afterwards, the emulsion was cooled in an ice bath for 10 min at 700 rpm and was collected in a 50 mL conic tube containing cold PBS 1× and centrifuged for 3 min at 300g. The supernatant was eliminated, and the beads were washed 4 times to eliminate traces of mineral oil. The beads were maintained at 4 °C in PBS 1× supplemented with 100 U mL−1 of penicillin-100 μg mL−1 streptomycin until further use. To prepare the porous hydrogels, the PBS was first drained using a 100 μm cell strainer, and then the beads were pipetted into the respective formulations (v/v%). The beads remain slightly wet, yet the content of PBS was negligible and did not alter the final dilution of the formulations.Preparation of sGelMA solution
A highly concentrated solution of sGelMA was prepared by dissolving 12.7 g of the lyophilized material in 14 mL of PBS 1× supplemented with 3 mL of 1 M NaOH (to adjust pH to 7.4) under gentle agitation at 45 °C. Due to the high viscosity, the exact final concentration was estimated using the dry weight, and the obtained values were 40–50% w/v. The sGelMA solution was filtered while still warm with a 5 μm pore nitrocellulose filter and subjected to an additional heat–cold sterilization protocol for the construction of cellularized hydrogels.Preparation of sGelMA porous hydrogels
Different formulations of sGelMA solutions were prepared by mixing sGelMA, PBS 1×, and 0.2% (w/v) of lithium phenyl-2,4,6-trimethyl-benzoyl phosphinate or LAP (Sigma-Aldrich), as the photoinitiator. For porous formulations, 20, 40, or 60% v/v of beads was added to the preparation and the remaining volume (80, 60, and 40% respectively) was occupied by the sGelMA solution at final concentrations of 20, 25 and 35% w/v. The formulations were homogenized and poured into PDMS cylindrical molds and covered with a PDMS cap. Subsequently, polymerization was induced by UV irradiation for 1 minute using a UV light guide at a distance of 8 cm between the source and the molds (UV lamp, OmniCure®S2000, Excelitas Technologies, USA). The UV light had a filtered wavelength of 365 nm and an intensity of 261 mW cm−2.Extrusion force assay
The force necessary to extrude pre-crosslinked suspensions (non-porous and porous formulations) was calculated using a TA·XTplus Texture Analyser (Stable Micro Systems, UK). 400 μl of each formulation was added into a 1 mL syringe, with an 18 G × 1′′ needle, and cannulated to tubing with an inner diameter of 1 mm and length of 30 cm. The force required to push the plunger was assessed using a velocity of 0.2 mm s−1 and was registered in N.DSC (differential scanning calorimetry)
The melting points of gelled gelatin solutions were determined using DSC-1 (Mettler Toledo, Switzerland). Samples weighing ∼70 mg having a total volume of 60 μl were hermetically sealed in aluminum pans. The protocol used for analysis was as follows: cooling from 25 °C to −10 °C at 10 °C min−1, holding at −10 °C for 15 min, and heating from −10 °C to 60 °C at 5 °C min−1. In the case of porcine gelatin and GelMA porcine gelatin, the melting temperature (Tm) was determined as the onset of the endothermic peak observed upon heating. In the case of salmon gelatin, only the temperature determined from the endset of the endothermic peak was reported (Te), which corresponds to completely melted gelatin. The DSC was previously calibrated using indium as a standard and an empty pan was used as a reference. All analyses were carried out at least in duplicate.Mechanical testing
The mechanical properties of the sGelMA hydrogels were tested in an unconfined compression setup. Disks of the materials were prepared by pipetting the prepolymer solution into PDMS molds (8 mm φ, 4 mm h) and were subsequently polymerized as mentioned. The compressive modulus was tested using a TA·XTplus Texture Analyser (Stable Micro Systems, UK) equipped with a 5000 g load cell (Stable Micro Systems, UK). A 25 mm diameter probe was used for axial compression of the hydrogels at a speed test of 2 mm s−1. Stress–strain curves were recorded, and the compressive modulus (Young's modulus) was determined from the slope at the linear phase in the range of 5 to 15% strain.Diffusion assay
The diffusion properties of the sGelMA hydrogels were evaluated as described.24 To fabricate the hydrogels, 100 μL of the different formulations (porous and non-porous) were polymerized into PDMS molds (8 mm φ, 4 mm h). A 15 μL BSA-FITC (1 mg mL−1) drop was placed in the upper surface of the hydrogel, incubated at room temperature for 10 minutes, avoiding direct light, and then washed thoroughly 3 times with PBS 1× to remove the non-diffused fluorescent protein. The core segment of each hydrogel was cut transversally using a scalpel and visualized using a confocal microscope (Sp8, Leica), and 3 photos per sample were obtained. The average BSA-FITC diffusion was quantified using the Leica Application Suite X (LAS X) software (V3.3.0.16799) measuring 5 points per photo.Swelling
Hydrogels were prepared as described, weighed (Wdry), and soaked with PBS 1× at 37 °C for 10 days. The weight of the swollen samples (Wwet) was measured at pre-determined time points after removing excess water. The swelling ratios (SR) of hydrogels were calculated using the following formula: SR = (Wwet − Wdry)/Wdry × 100%.Degradability
The degradability was evaluated using an in vitro digestion assay.24 Hydrogels were fabricated (8 mm φ, 4 mm h) and incubated with PBS 1× at 37 °C for 24 hours to reach total swelling. Then, each hydrogel was weighed and registered as time = 0 (W0). These samples were supplemented with 10 and 50 U of collagenase II (Worthington) prepared in PBS 1× (in 2 mL of solution) and incubated with agitation (80 rpm) at 37 °C for 24 hours. The weight of each sample was measured every hour (Wt), and the rate of degradation was calculated as a percentage of the initial weight using the following expression: (Wt/W0 × 100%), where W0 is the initial wet weight of the sample.Scanning electron microscopy (SEM)
Empty or cellularized hydrogels were fixed with 10% formalin for 48 hours under agitation. After several washes with PBS 1×, samples were washed with 0.1 M sodium cacodylate buffer, pH 7.2, and post-fixed with osmium tetroxide. Following several washes with distilled water, samples were dehydrated in ascendant alcohols: 50–70–95–100–100%, 5 min each. CO2 critical point drying was performed in an Autosamdri-815, Series A (Tousimis, USA). Finally, samples were gold sputter-coated (200 Å) with a Desk V coating system (Denton Vacuum, USA). Samples were examined using a JEOL (Tokyo, Japan) IT300LV microscope, and representative images were obtained.OAC isolation and cell culture
Osteoarthritis-derived chondrocytes (OACs) were isolated from the femoral head of patients who had undergone hip replacement. The samples were transported into the cell culture facility and processed under sterile conditions. After a cleaning wash with 70% ethanol and PBS 1×, the cartilage was removed with a scalpel, minced into 1–2 mm width fragments, and deposited in a conical tube containing a washing solution (PBS 1×, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin). The conical tube was centrifuged 200g × 3 min to eliminate the washing solution without losing tissue. Then, 10 mL of 2.5 mg mL−1 Pronase (Sigma-Aldrich) was added, and the suspension was incubated at 37 °C for 1 hour with gentle agitation. The suspension was centrifuged, the supernatant eliminated, and 10 mL of 250 U mL−1 collagenase II solution (Sigma-Aldrich) was added. Additionally, this digestion was supplemented with 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, and 2.5 μg mL−1 amphotericin B, and maintained overnight (16–20 hours) at 37 °C with gentle agitation. The next day, to stop the digestion, serum-enriched medium was added: Dulbecco's Modified Eagle's medium (DMEM, Biological Industries, Israel) supplemented with 10% fetal bovine serum (FBS), and the cellular suspension was centrifuged at 300g × 5 min. The cellular pellet was resuspended in the same medium and filtered through a 100 μm cell strainer to remove any remaining undigested tissue. After that, the cellular suspension was seeded at 30000 cells per cm2 in growth medium: DMEM supplemented with 10% FBS, 4 mM L-glutamine, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, 0.1 mM nonessential amino acids (NEAA) (all from Thermo Fisher), 1 mM sodium pyruvate (Biological Industries), 0.4 mM L-proline, and 50 μg mL−1 ascorbic acid (both from Sigma-Aldrich), and the medium was replaced 2–3 times per week. When OACs reached 80–90% confluence, they were detached using 0.5% trypsin/EDTA (Thermo Fisher), and the experiments were performed using cells subcultured 2–3 times (passage 2–3). All cell cultures were maintained in a humidified incubator at 37 °C and 5% CO2.
Human knee articular chondrocytes (NHAC-k, CC-2550, Lonza) were maintained in CGM™ chondrocyte growth medium (BulletKit™, Lonza) during the first few subcultures. Afterwards, we used the same growth medium mentioned before.
Cell-laden sGelMA hydrogel construction and chondroinduction
OACs were detached using 0.5% trypsin (Thermo Fisher) and, after centrifugation, were resuspended in the respective pre-crosslinked sGelMA-based formulations, with or without beads. A volume of 100 μL of the mixture, containing 7.5 × 106 cell per ml (equivalent to 0.75 × 106 cell in 100 μL), was poured into cylindric PDMS molds (6 mm φ, 5 mm h) and then exposed to 365 nm UV light with an intensity of 261 mW cm−2 (OmniCure®S2000, Excelitas Technologies, USA) at a distance of 20 cm for 20 s per side. Cellularized hydrogels were deposited in 12-well cell culture plates (1 hydrogel per well) containing 2 mL of chondroinductive medium (DMEM supplemented with 0.2 mM ascorbic acid 2-phosphate, 0.5% bovine serum albumin (Thermo Fisher), 1% v/v insulin–transferrin–selenium mixture (ITS-X, Corning), 4 mM L-glutamine, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, 1 mM sodium pyruvate and 5 ng mL−1 TGF-β2 (Prepotech, USA).11 The medium was changed twice a week.PrestoBlue™ and LIVE/DEAD viability assays
To evaluate the viability of encapsulated OACs by measuring the mitochondrial activity, first, 10% PrestoBlue™ regent (PrestoBlue™ cell viability reagent, Thermo Fisher) was diluted in chondroinductive medium and added into the hydrogel in each well. After 2 hours of incubation at 37 °C, 100 μL of the supernatant was transferred into a 96-well flat-bottomed plate to measure the absorbance at 570 nm (600 nm reference) in a spectrophotometer (Tecan Reader). The diluted PrestoBlue™ in empty wells was used as the blank. All absorbance measures were conducted in duplicate.For live/dead staining (LIVE/DEAD™ viability/cytotoxicity kit L3224, Thermo Fisher), hydrogels were washed with PBS 1× at 37 °C to eliminate phenol red traces. The staining mixture (4 μM EthD-1 and 12 μM calcein-AM) was prepared in PBS 1×, and 100 μL of it was added to each hydrogel and incubated for 20 min at RT protected from light. Afterwards, the hydrogels were washed with PBS 1× and analyzed under an Sp8 confocal microscope (Leica) using LAS X software. Viability (live/total cells × 100%) was estimated by counting green (live) and red (death) cells with the Cell Counting tool of FIJI software.25 Circularity was calculated using the Shape Descriptors tool from the same software.
RNA isolation and real-time PCR analysis
Samples were stored at −80 °C before RNA extraction. The following protocol has been previously described,26 and some modifications are included according to the extraction kit protocol (E.Z.N.A Total RNA Kit Omega Biotek, #OB.R6834-02CH). Briefly, samples were thawed and homogenized in the presence of 350 μL of TRK lysis buffer using a plastic pestle and by extruding the lysates 15 times through a 19 gauge needle using a 3 ml syringe. The lysate was transferred into a new tube, and the remaining pieces of the hydrogels were washed with an additional 350 μL of lysis buffer to complete 700 μL of lysate for each sample. Next, we followed the manufacturer's guidelines.RNA concentration was estimated using a Nanodrop, and 1 μg of RNA was used for reverse transcription with RevertAid reverse transcriptase (Thermo Fisher) according to the manufacturer's instructions. The cDNA samples were analyzed by qPCR to determine the expression of different genes related to the cartilage lineage and OA (Table 1), and the 18S gene was used as a housekeeping gene. Brilliant II SYBR Green qPCR Master Mix (Stratagene) was used for the PCR reaction, and the amplification was conducted using the qPCR System 3000× (Stratagene). The expression rate was calculated using OACs at day 0 as the control (before encapsulation), and in the same passage as encapsulated cells, using the cycle threshold (Ct) values for each gene obtained with the MxPro software. The fold-change expression was calculated using the formula 2(−ΔΔCt).27
| Gen | Forward (5′–3′) | Reverse (5′–3′) | bp |
|---|---|---|---|
| COL1A1 | GTCACCCACCGACCAAGAAAC | AAGTCCAGGCTGTCCAGGGAT | 121 |
| COL2A1 | GGCAATAGCAGGTTCACGTACA | CGATAACAGTCTTGCCCCACTT | 79 |
| ACAN | TACACAGGTGAAGACTTTGTG | GACTCGAAGATGGGCTTTAC | 176 |
| SOX9 | GGAATGTTTCAGCAGCCAAT | TGGTGTTCTGAGAGGCACAG | 115 |
| PRG4 | GAGTACCCAATCAAGGCATTATCA | TCCATCTACTGGCTTACCATTGC | 80 |
| COMP | TCAGGACTCTCGGGACAACT | CTGTCAGGGACTCCGTCATT | 117 |
| COL6A1 | ATCGGACCTAAAGGCTACC | TTCTCCCCTTTCACCCATC | 117 |
| COL3A1 | GGGAACAACTTGATGGTGCTACT | TCAGACATGAGACTCTTTGTGCAA | 72 |
| COL10A1 | GCTAAGGGTGAAAGGGGTTC | CTCCAGGATCACCTTTTGGA | 118 |
| TGFβ1 | ACAATTCCTGGCGATACCTCAGCA | TGCAGTGTGTTATCCCTGCTGTCA | 172 |
| TGFβ3 | TGGACTTCGGCCACATCAAGAAGA | TGTTGTAAAGGGCCAGGACCTGAT | 135 |
| ADAMTS4 | GGCTAAAGCGCTACCTGCTA | GAGTCACCACCAAGCTGACA | 93 |
| TIMP1 | GATACTTCCACAGGTCCCACAAC | GCAAGAGTCCATCCTGCAGTT | 72 |
| TIMP2 | CGACATTTATGGCAACCCTATCA | CAGGCCCTTTGAACATCTTTATCT | 71 |
| TIMP3 | AACTTGGGTGAAGGCTGAGTGT | CCTCACCAAGGCCTAACAGATG | 96 |
| MMP1 | AGGTCTCTGAGGGTCAAGCA | CTGGTTGAAAAGCATGAGCA | 111 |
| MMP3 | TGCTTTGTCCTTTGATGCTG | GGAAGAGATGGCCAAAATGA | 135 |
| MMP13 | TTGAGCTGGACTCATTGTCG | GGAGCCTCTCAGTCATGGAG | 172 |
| RUNX2 | TGGCAGCACGCTATTAAATC | TCTGCCACTAGAATTCAAAA | 103 |
| 18S | TAGAGGGACAAGTGGCGTTC | CGCTGAGCCAGTCAGTGT | 104 |
Quantification of sulfated glycosaminoglycans (GAGs)
GAGs were determined using the DMMB (dimethyl methylene blue) assay.28 Briefly, hydrogels were stored at −20 °C until further processing. For the assay, hydrogels were thawed, and 300 μL of 250 μg mL−1 papain (Sigma-Aldrich) solution (5 mM L-cysteine, 5 mM EDTA, in PBS 1×, pH 6) was added to each sample. Samples were incubated at 65 °C for 16 hours and diluted 4 times in PBS 1×. Afterwards, 20 μL of each sample was mixed with 200 μL of the DMMB regent (pH 1.5) in a 96-well plate, and absorbance at 525/595 nm was measured in a NanoQuant Infinite Tecan M200 PRO (Switzerland). A calibration curve of bovine chondroitin sulfate A (CS-A, Sigma-Aldrich, #C9819) was elaborated in parallel by using a concentration range of CS-A between 1 and 50 μg mL−1 and the concentration of GAGs in the hydrogel's lysates was obtained by extrapolating the data from the curve. Additionally, the GAG concentration was normalized against the total DNA content. For this, diluted samples were mixed with 100 μL of SYBR solution (SYBR Safe DNA gel stain #S33102) previously diluted (1/10000) in TE buffer (1 mM EDTA, 10 mM Tris-HCl, pH 8). Likewise, concentrations of DNA in the lysates were determined using a calibration curve of calf thymus DNA (Sigma-Aldrich, #D4522) between 5 and 1000 ng mL−1 in a fluorescence microplate reader (Tecan infinite M1000Pro, excitation: 502 nm; emission: 530 nm).
Molecular composition by Raman spectroscopy
To determine the synthesis of GAGs from encapsulated OACs after the differentiation protocol, empty and cellularized hydrogels from days 0 and 35 post differentiation were analyzed. After several washes, the hydrogels were lyophilized for 2–3 days (Freezone, LabLonco) and analyzed using a Raman spectrometer (Xplora PLUS, Horiba Scientific, Lille, France) equipped with a near-infrared laser (λ 785 nm), fixed at 70 mV and the spectra were acquired using a 600 g mm−1 diffraction grating. An optical microscope (Olympus BX41) was used to focus the laser over the samples with a 10× objective (NA = 0.25). Each spectrum was acquired in the 200–3200 cm−1 range, with 30 s exposition and 3 accumulations. All spectra were corrected using the instrument's software (LabSpec 6 V6.4, Horiba Scientific) and normalized using the Origin software (OriginPro 2019b). The average spectra of 3 different OAC donors with 6 measures per sample were used for analysis, using additionally 3 different sGelMA concentrations in the hydrogels. Bovine chondroitin sulfate A (Sigma-Aldrich), sGel, and sGelMA were used as reference samples.Histological and immunohistochemical staining
After the chondroinduction protocol, hydrogels were washed with PBS 1× and fixed with 10% neutral buffer formalin for 48 hours at room temperature, with gentle agitation. After several washes, samples were dehydrated with a battery of ascendant concentrations of alcohols and xylene. Afterwards, hydrogels were embedded in paraffin to obtain 5 μm cross-sections. Cross-sections were deparaffinized, rehydrated, and finally stained using different histological methods: hematoxylin/eosin, safranin O 0.1% (AppliChem), Fast Green 0.08% (Sigma-Aldrich), Picro Sirius Red (ab150681, Abcam) and alizarin red S 2% (MP Biomedicals).For immunostaining, the rehydrated slides were boiled in citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6) for antigen retrieval. After several washes with PBS 1×, the slides were blocked with 1.5% w/v BSA for 2 hours. The antibodies were prepared in blocking solution and incubated ON at 4 °C: rabbit anti-collagen II (ab34712); mouse anti-aggrecan (ab3778); and rabbit anti-collagen I (ab34710) (all from Abcam, UK). The respective fluorescent secondary antibodies used were anti-mouse Alexa 488 (1397999) and anti-rabbit DyLight 594 (35561), both from Thermo Fisher. 4′,6-Diamidino-2-phenylindole (DAPI, Sigma) was used for nuclear counterstaining, and the slides were mounted in FluorSave™ (Merck Millipore, USA) before confocal microscopy analysis (Sp8 confocal microscope, Leica).
In vivo implantation
Cylindrical (6 mm φ, 2 mm h) cellularized hydrogels (10 × 106 cell per ml) were prepared as mentioned previously and maintained for 10 days in a chondroinductive medium before the implantation. The hydrogels were washed with PBS 1× on the day of the surgery to eliminate traces of medium before subcutaneous implantation in 8-week-old immunocompromised mice (NOD·Cg-Prkdcscid Il2rgtm1Wjl/SzJ or NSG). Animals were anesthetized by 3–5% sevoflurane inhalation, and the hair was removed using depilatory cream and an electric shaver. The hydrogels were implanted through a 0.5 cm incision and opening of a subcutaneous pocket (2 per animal). The wound was closed with a 5/0 polyamide suture. The skin was covered with Tegaderm™, and the animals were maintained in cages (<4 animals) with water and food ad libitum. After 5 weeks, the animals were euthanized by CO2 inhalation, and the hydrogels were recovered for GAGs and histochemical analysis. In a similar assay, hydrogels were recovered after 12 weeks, for degradation evaluation. The degree of degradation was calculated as the reduction of the volume of hydrogels at the end of the assay.Statistical analysis
The assays were performed at least in triplicate (technical and biological) using different OAC donors. The data are presented as mean ± SEM and analyzed by Kruskal Wallis (non-parametric test) or one-way variance analysis (ANOVA) and Tukey's post hoc tests for parametric data distribution. A p-value of 0.05 was considered statistically significant. The software GraphPad Prism 8.0.2 was used for this analysis.3. Results
Structural and thermal characterization of sGelMA
The methacrylation protocol generates a functionalized photo-crosslinkable material that can undergo free radical chain polymerization in the presence of light and a photoinitiator. This process induces covalent crosslinking between adjacent functionalized lysines, generating a thermostable hydrogel (Fig. 1A).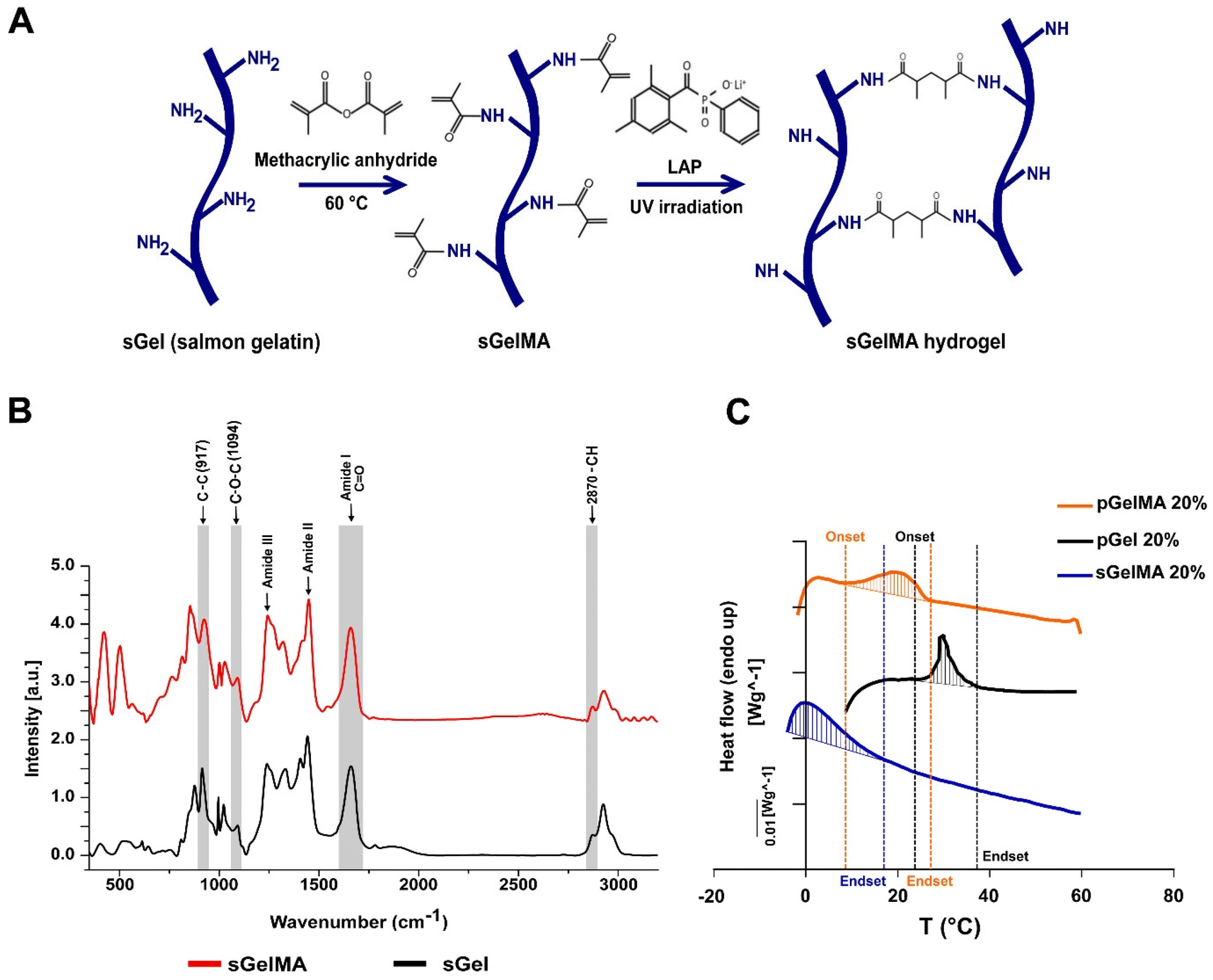 | ||
| Fig. 1 Molecular and thermal characterization of sGelMA. (A) Schematic representation of salmon gelatin (sGel) functionalization protocol. The lysine's ε-amine groups in the gelatin react with the methacrylic anhydride. The crosslinking of metacrylamide groups is mediated by free radical chain polymerization, using a photoinitiator (LAP) and UV irradiation. (B) Raman spectroscopy spectra from sGel and sGelMA in the region of 450–3200 cm−1. The prominent bands are associated with the amide groups (I, II, and III), characteristic of proteins. The intensity data were normalized and plotted in arbitrary units (a.u). (C) Typical DSC thermograms observed in sGelMA, pGel and pGelMA. Thermograms correspond to the heating run and the shaded area was defined by the DSC software. | ||
Changes in the Raman profiles of sGel and sGelMA
Previously, we showed an extensive characterization of sGelMA properties.16 The present study includes the additional characterization of molecular changes between sGel and sGelMA using Raman spectroscopy. The spectra of both sGel and sGelMA were evaluated in the spectral segment of 450–3200 cm−1 (Fig. 1B, and Table 2). The observed peaks were associated with the amide groups and showed similarity between them (amides I, II, and III), but some differences were detected at 917, 1094, 1444 cm−1 (amide II), and 1662 cm−1 (amide I). The 917 cm−1 signal corresponds to the stretching vibrations of the C–C bonds, associated with the helical conformation of the gelatin. However, the same peak in sGelMA was broader and more extended, with a displacement of the intensity toward 927 cm−1. Analogously, we observed an increment in the intensity of the amide III peak and a displacement in the absorption from 1242 to 1245 cm−1. This peak is assigned to the stretching vibrations of the C–N bond. These changes can be associated with the loss of the helical configuration in the sGelMA due to the bulky methacrylate groups. The absorption at 1094 cm−1 corresponds to the stretching vibrations of the C–O–C bonds, and it was augmented by 43% in sGelMA, related to the presence of the methacrylamide groups. The amide II signal had a slight increase (1.5%) in sGelMA, and it was displaced to 1451 cm−1 for the stretching and flexion (scissors) vibrations from the C–H bonds (CH3 and CH2 groups). The amide I signal had an increment in normalized intensity (1.54–1.60), possibly due to the overlap of the signals of the stretching vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond from the methacrylamide groups. In the 2820–3000 cm−1 region, the signal was extended at 2932 cm−1 in sGelMA, associated with the –CH bonds generated during the methacrylation. However, no increment in the intensity of the signal was observed. These data confirmed that the functionalization process alters the molecular structure of sGel. Nevertheless, methacrylation produces small changes and maintains the main biological and rheological properties.16
C bond from the methacrylamide groups. In the 2820–3000 cm−1 region, the signal was extended at 2932 cm−1 in sGelMA, associated with the –CH bonds generated during the methacrylation. However, no increment in the intensity of the signal was observed. These data confirmed that the functionalization process alters the molecular structure of sGel. Nevertheless, methacrylation produces small changes and maintains the main biological and rheological properties.16
| Assignment | sGel | sGelMA | ||
|---|---|---|---|---|
| (cm−1) | Normalized intensity (dimensionless) | (cm−1) | Normalized intensity (dimensionless) | |
| C–C | 917 | 1.50 | 927 | 1.75 |
| C–O–C | 1094 | 0.53 | 1094 | 0.73 |
| Amide III | 1242 | 1.58 | 1245 | 1.81 |
| Amide II | 1444 | 2.06 | 1451 | 2.09 |
| Amide I | 1662 | 1.54 | 1662 | 1.60 |
Variations in the melting profiles of mesophilic and psychrophilic gelatins
To evaluate which biomaterials can be mixed to obtain a stable formulation at room temperature and maintain the integrity of porogens, we analyzed the thermal profile of porcine gelatin (with and without functionalization with pGelMA and pGel respectively) and sGelMA using differential scanning calorimetry (DSC). pGel showed the initiation toward a liquid transition (onset) at around 27 °C and pGelMA finished this transition at 22–24 °C, whereas sGelMA is completely melted at temperatures around 14 °C, independent of the gelatin concentration (Table 3). These data suggest that a pGelMA/pGel mixture is not an optimal formulation to fabricate porous scaffolds in situ because the range of temperature in which the porogens remain gelled while the pGelMA is liquid is very narrow; therefore a special device would be required to control the temperature during mixture loading and in situ fabrication. In contrast, the “temperature window” between pGel and sGelMA is around 10 °C, meaning that sGelMA is capable of maintaining its liquid state whilst pGel remains gelled along a broad range of temperature (15–25 °C) (Fig. 1C, and Table 3).| Concentration (%) | Onset T° (°C) | Endset T° (°C) | |
|---|---|---|---|
| sGelMA | 10 | — | 14.8 (1.0) |
| 20 | — | 14.4 (0.7) | |
| 30 | — | 14.8 (0.3) | |
| 35 | — | 14.5 (0.4) | |
| Porcine Gel | 10 | 26.5 (0.2) | 34.9 (0.4) |
| 20 | 27.4 (0.2) | 32.4 (0.8) | |
| pGelMA | 10 | 9.5 (0.9) | 24.0 (0.6) |
| 20 | 8.6 (0.8) | 22.4 (3.6) |
Fabrication of porous hydrogels
To overcome the potentially detrimental effects of highly crosslinked hydrogels over resident cells, we add a sacrificial porogenic component (or beads) made of pGel to generate voids after the melting and diffusion of the gelatin when implanted or incubated at 37 °C. This process would not alter the covalent structure formed by the crosslinked sGelMA.pGel beads were between 50 and 600 μm in diameter (data not shown) and had a spherical appearance and smooth surface. Fig. 2A shows a schematic representation of the fabrication of a porous hydrogel using pGel beads.
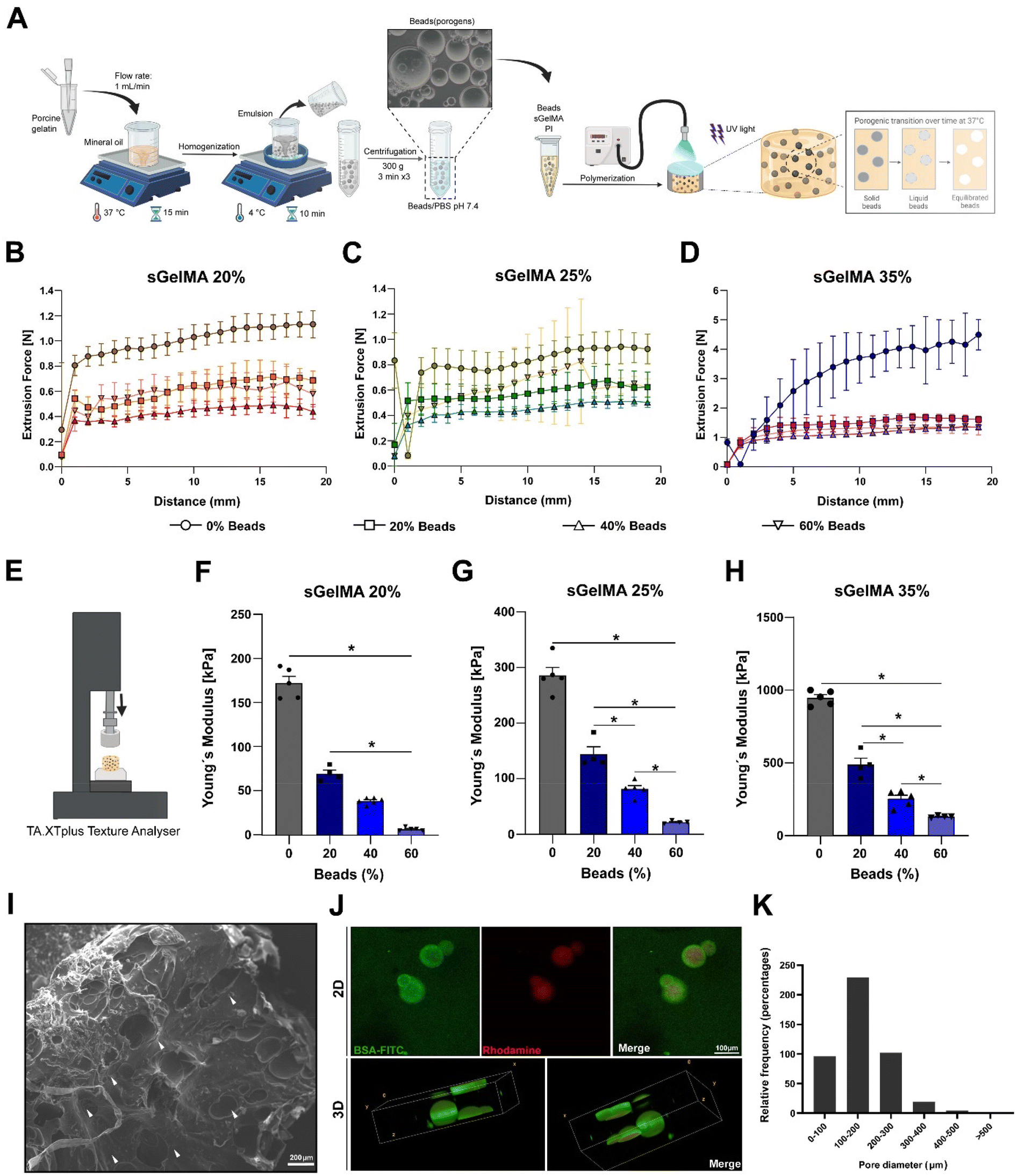 | ||
| Fig. 2 Fabrication and structural characterization of porous hydrogels. (A) Schematic representation of the fabrication protocol of porcine gelatin beads and porous sGelMA hydrogels. The beads are formed by a water-in-oil emulsion and the hydrogels are polymerized by UV irradiation. At 37 °C, the gelatin beads start to melt, reaching a completely liquid state, and a subsequent diffusion process and equilibrium with the surrounding hydrogel solvent. Extrudability of the different formulations with 20, 40, and 60% beads: (B) sGelMA 20%, (C) sGelMA 25%, and (D) sGelMA 35%, n = 4–6 hydrogels per formulation, data are shown as mean ± SD. (E) Representation of the experimental setup for calculating Young's modulus (YM) of sGelMA hydrogels. (F–H) Mechanical properties of the analyzed formulations: sGelMA 20, 25, and 35% w/v, supplemented with 20, 40, or 60% v/v of beads, n = 4–6 hydrogels per formulation, the data are shown as mean ± SEM and were analyzed by ANOVA test and Tukey's post hoc test, * p < 0.05. (I) SEM image shows the surface of a hydrogel fabricated with 40% v/v of gelatin beads. White arrowheads indicate the presence of voids generated by the porogenic beads. Scale bar = 200 μm. (J) Porous hydrogel composed of rhodamine-containing beads and BSA-FITC-containing sGelMA hydrogel; the upper panel shows the appearance of the sample in 2D, and in the lower panel, a 3D reconstruction of a 150 μm z-axis section of the hydrogel is represented. Scale bar = 100 μm. (K) Relative frequency of the diameters of the pores in the sGelMA hydrogels. | ||
Taking the advantages of the rheology and cold-adaptation of sGelMA,16 we were able to prepare biphasic polymer solutions with high concentrations of sGelMA (40–50% w/v) and a solid phase composed of pGel beads, keeping their liquid aggregation state throughout a broad window of temperatures (>7 °C). Then, 3 concentrations of sGelMA solutions were established for hydrogel composition in our study: 20, 25, and 35% w/v, supplemented with proportions of 0, 20, 40, and 60% v/v of the beads.
Extrudability of the biphasic porous formulations and Young's modulus of the crosslinked hydrogels
Because the ideal application of a cartilage-restoring scaffold should involve a minimally invasive approach for injecting directly into the lesion, measurements of the required forces to extrude the different formulations through a 1 mm diameter and 30 cm length tubing were conducted (Fig. 2B–D). Additionally, their mechanical properties post-crosslinking were also studied by measuring their Young's modulus (YM) in an unconfined setup (Fig. 2E).We observed that the higher the sGelMA concentration, the larger the force required to extrude the pre-polymerized solutions. On the other hand, supplementing formulations with beads reduces significantly the required force for extrusion (Fig. 2B–D). Likewise, the higher the sGelMA concentration, the stiffer the hydrogels (Fig. 2F–H, and Table 4), reaching a maximum YM value close to 1 MPa. The second observation was that the porogenic component negatively affected the YM in all conditions. There was a dose-dependence effect, where the formulations with the highest percentage of porogens had the lowest YM compared with their non-porous counterparts. According to the data, the hydrogels with the closest cartilage-like mechanical properties were the sGelMA formulations at 35% w/v concentration and supplemented with different amounts of porogenic beads (Fig. 2F–H, and Table 4).
| sGelMA (w/v) | YM (kPa) | |||
|---|---|---|---|---|
| Beads proportion (v/v) | ||||
| 0% | 20% | 40% | 60% | |
| 20% | 172 ± 17.1 | 69.07 ± 8.0 | 38.24 ± 3.5 | 6.30 ± 1.9 |
| 25% | 285.7 ± 31.7 | 144.2 ± 26.2 | 81.36 ± 13.8 | 21.79 ± 2.4 |
| 35% | 946.5 ± 50.5 | 487.8 ± 87.8 | 252.9 ± 54.9 | 130.1 ± 6.9 |
The porous structure of hydrogels
SEM showed a dense and compact surface of the hydrogel, interrupted by the voids left by the beads after liquefaction and diffusion at 37 °C (Fig. 2I), suggesting that cells would otherwise face an impenetrable material when encapsulated. Using beads prepared in a rhodamine solution and hydrogel formulations supplemented with BSA-FITC, we obtained a fluorescent construct to be analyzed by confocal microscopy. We observed the presence of beads in the hydrogel in images of 2D sections, and after analyzing a 150 μm z-axis segment to elaborate a 3D representation of the beads inside the hydrogels, the spherical shape of the beads could be identified (Fig. 2J). These samples were analyzed immediately after the hydrogel fabrication to avoid melting and diffusion. Using 2D confocal images, we quantified pore diameters and determined that the greater proportion of beads were in the diameter range of 100–200 μm (50.8%), followed by 200–300 μm (22.6%), <100 μm (21.2%), 300–400 μm (4.2%), 400–500 μm (0.9%) and >500 μm (0.2%) (Fig. 2K). It is important to note that the number of beads incorporated in the formulation generates essentially a non-interconnected porous structure (see Fig. 2I–J), with a porosity that is defined by the volumetric percentage of incorporated beads.Physical properties of porous sGelMA hydrogels
Following the mechanical characterization of the whole spectrum of discrete porous formulations previously generated, and their preliminary biological responses (mainly production of GAGs during an in vitro experiment of cartilage formation, addressed later in this work) (Fig. S1, ESI†), selection and the physical evaluations were conducted for 20, 25 and 35% w/v sGelMA with an intermediate level of the porogenic component: 40% v/v. The formulations were denominated G20B, G25B, and G35B. Physical parameters are relevant to understand the performance of sGelMA-derived formulations as scaffolds to harbor cells and evaluating the effect of porosity on cell activity performance and chondroinductivity.Diffusion
One fundamental parameter for the well-being of encapsulated cells in scaffolds is the molecular diffusion capacity through the scaffold material. It allows the optimum exchange of gases, nutrients, and waste with the surrounding. Diffusion of BSA-FITC in the different formulations was measured to evaluate the exchange of a medium-size protein (66 kDa approximately) across these types of scaffolds (Fig. 3A–G). Within the non-porous formulations, the highest diffusion level was observed in scaffolds with the lowest concentration of sGelMA. The same applies to porous hydrogels, but porosity in the hydrogels remarkably favors diffusion compared with the non-porous formulations. For instance, pores close to the surface can be visualized as filled with BSA-FITC (Fig. 3D′–F′ and G). The estimated increment in the diffusion compared with that of non-porous hydrogels was 2.6, 2.8, and 6.5 times for G20B, G25B, and G35B, respectively. This can be explained by the presence of pores but could also be explained to a certain degree by the presence of liquefied porogens affecting the net number of positive charges in the hydrogel. This favors the diffusion of a negatively charged protein, such as the BSA at pH 7.4. At this point, it is important to recognize that although the porous structure is essentially not interconnected, it contributes to a higher molecular diffusion and space for cells to carry out their cell-signaling activities.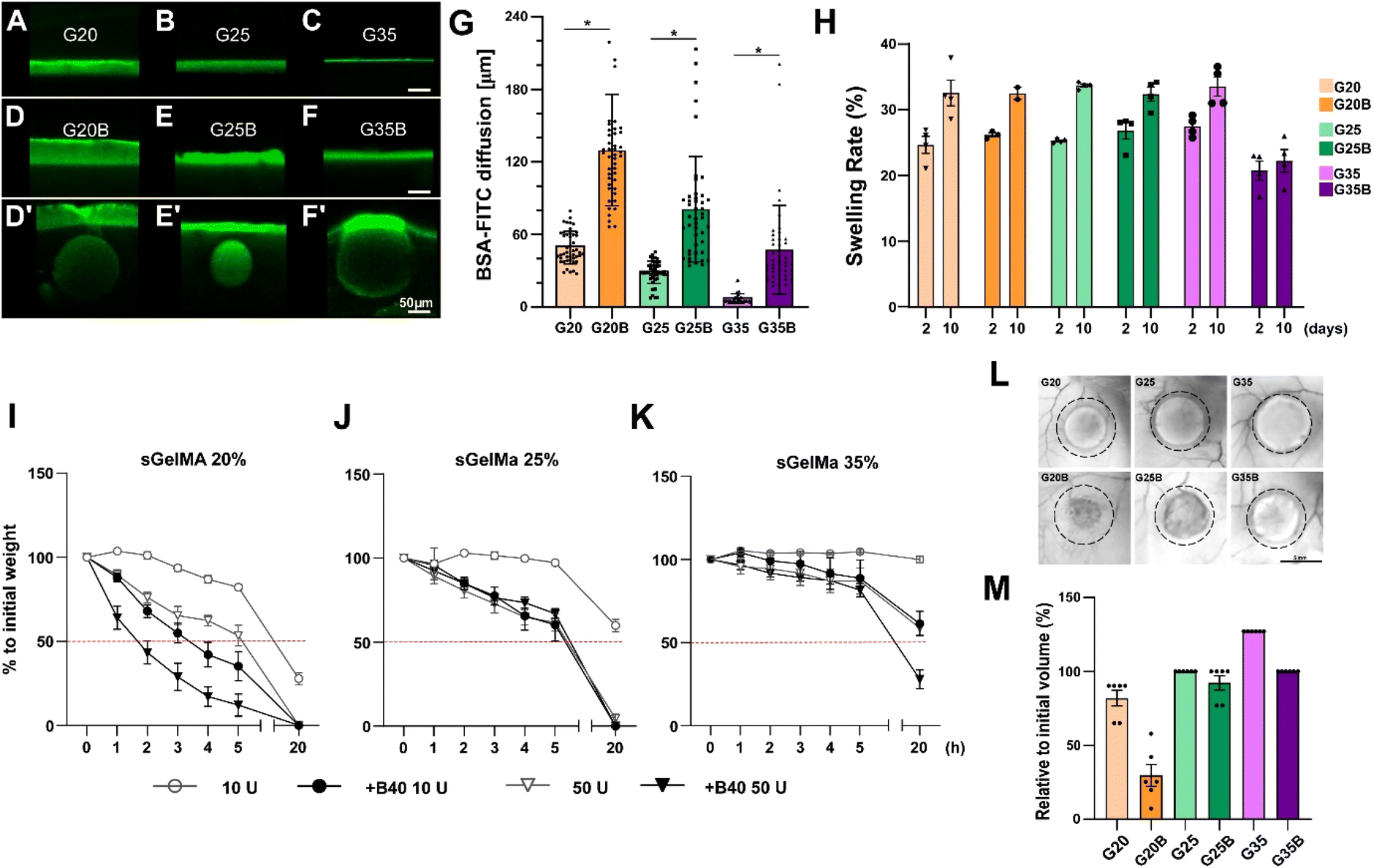 | ||
| Fig. 3 Characterization of the physical properties of sGelMA porous hydrogels. BSA-FITC diffusion assay in (A) G20, (B) G25, (C) G35, (D) G20B, (E) G25B, (F) G35B. (D′–F′) The images show specific segments of the hydrogels where the presence of beads enhanced the diffusion of BSA-FITC (outliers in the quantification in G). Scale bar = 50 μm. (G) Quantification of the diffused BSA-FITC, n = 3 hydrogels per formulation, the data are shown as mean ± SEM and were analyzed by the Kruskal–Wallis test and Dunn's multiple comparisons tests, *p < 0.05. (H) Swelling rate of formulations after 2 and 10 days of incubation at 37 °C. n = 4 hydrogels per formulation. The data are shown as mean ± SEM. An in vitro degradation test was made to evaluate the effect of pores in the formulations: (I) G20/G20B, (J) G25/G25B, and (K) G35/G35B using 10 and 50 U of collagenase II. n = 4 hydrogels per formulation. In vivo degradation assay for G20/G20B, G25/G25B, and G35/G35B: (L) Representative images of the recovered hydrogels after 12 weeks of subcutaneous implantation in NSG mice. Scale bar = 5 mm. (M) Degree of degradation, n = 6 hydrogels per formulation (2 per animal). The data are shown as mean ± SEM. | ||
Swelling
Hydrogels have high water content, and due to the charged polymers, they can reach a significant degree of swelling. Therefore, it is essential to determine how the volume can change when the hydrogels are exposed to wet environments and restricted spaces, such as a lesion on the cartilage surface.To study the effect of porosity on water absorption and swelling, different formulations were evaluated using an in vitro approach. After 2 days of incubation at 37 °C, all formulations showed a swelling rate of 20–30%, meaning there was an increment in the weight of the hydrogels by water absorption. Interestingly, the less-swelled hydrogel was G35B (Fig. 3H). After 10 days, there was an additional increment in the absorbed water (2–10%). Non-porous formulations showed a larger increment compared to the porous ones, and this increment was almost absent in G35B.
Degradability
Due to the high concentration of sGelMA and the high degree of functionalization, hydrogels can be refractory to be remodeled by cells. In this regard, the collagenase II-driven degradability of different formulations was evaluated.As expected, the formulations with the lowest sGelMA concentration (G20, G20B) had the fastest degradation rate. This degradation was more prominent in the presence of 40% v/v beads. After 20 hours of incubation, the groups using sGelMA 20% showed a decrease in weight greater than 50% (Fig. 3I). A similar result was obtained for G25/G25B, but the degradation rate was slower, with not all conditions reaching 50% degradation (Fig. 3J). The delay in the degradation was remarkable for the G35 formulations. After 20 hours, only G35B using 50 U of collagenase II was degraded below 50%, and there was almost no degradation in G35 (Fig. 3K). The same trend was observed when degradability was evaluated in vivo, by measuring the volume of hydrogels before and after 12 weeks of subcutaneous implantation in NSG mice. A considerable degradation occurred for G20, G20B, and G25B. G25 and G35B were more resistant to degradation, whereas G35 had an increase in volume, probably explained by a combination of resistance to degradation and swelling (Fig. 3L and M).
In summary, these data indicate that hydrogel degradation or remodeling capacity depends on the concentration of sGelMA. Additionally, the presence of pores cooperates by favoring the process.
Chondroinductive properties of porous sGelMA hydrogels
As mentioned previously, the clinical application of scaffolds for chondral lesion restoration requires remodeling and replacement by new cartilage tissue. To determine if the formulations can sustain cellular viability and metabolism, chondrocytes were encapsulated and subjected to an in vitro chondroinductive protocol.If we consider that the deformation of the AC and chondrocytes damage during pressure loading is highly accentuated at the defect rim, in a clinical setting, the empty scaffold would be invaded primarily by sick chondrocytes located at the edge of this lesion and with an osteoarthritic profile.7,29 Therefore, the actual study has aimed to use OACs. However, a high number of undifferentiated OACs at a low passage is difficult to obtain. To solve this problem, the initial experiments were conducted using commercially available healthy human chondrocytes (NHACk). Additionally, although the incorporation of cells in the surrounding of empty hydrogels resemble better the clinical conditions in which chondral lesions are expected to be filled with an empty scaffold, to accelerate the experimental read-out, viability and chondroinductive protocols in this whole investigation were conducted using an experimental setting that included the fabrication of chondrocyte-laden hydrogels instead.
The viability of embedded cells was followed for 5 weeks by measuring the mitochondrial activity (Fig. S1A–C, ESI†), and by live/dead staining (Fig. S1D, ESI†). It was first noted that the lowest viability over time was observed in the non-porous formulations and depended on the concentration of sGelMA. Second, pores had a positive effect on metabolic activity and cell viability, and the higher the porosity, the more improvements were observed.
The live/dead staining showed a high proportion of live cells in all formulations, but a higher number of dead cells are found in formulations with a higher concentration of sGelMA and lower porosity (Fig. S1A–D, ESI†). Microscopy images demonstrate that pores can allow cells to adhere individually, form cell conglomerates, or simply remain empty (Fig. S1E ESI†).
An additional fundamental parameter to evaluate the chondroinductivity of chondrocyte-laden hydrogels is the synthesis of glycosaminoglycans (GAGs), an important component of the cartilage ECM. Lower levels of GAGs were synthesized at higher stiffness, suggesting a negative correlation between stiffness or the higher stringent network of concentrated hydrogels and the synthesis of GAGs. The presence of pores in the hydrogels induced a significant increase in the levels of GAGs in a dose-dependent manner, showing the functional consequences of the porosity in the hydrogels (Fig. S1F, ESI†). The same results were observed after normalizing the levels of GAGs to the total DNA, which is an indirect measure of the total number of cells (Fig. S1G, ESI†).
The cellular data indicate that a higher proportion of pores showed better results in terms of GAG production, but also involved the reduction in mechanical properties (Fig. 2F–H, and Table 4). Therefore, we had to reflect that obtaining an ideal formulation with the highest chondroinductivity and tissue-like mechanical properties was not feasible with the adopted approach. So, we made some compromises and studied formulations at 3 different sGelMA concentrations but maintaining a 40% v/v proportion of beads and they were named G20B, G25B, and G35B.
Cell viability, distribution, and morphology of OACs encapsulated in porous sGelMA hydrogels
OACs were isolated from different donors and, after expansion for 1–2 subcultures were encapsulated in the previously defined and selected formulations (Fig. 4A). On the following day, live/dead staining showed similar morphology and viability among the 3 formulations, where the predominant morphology was rounded. After 35 days, there was a significant decline in viability in G35B relative to that of G20B and G25B (Fig. 4A–C).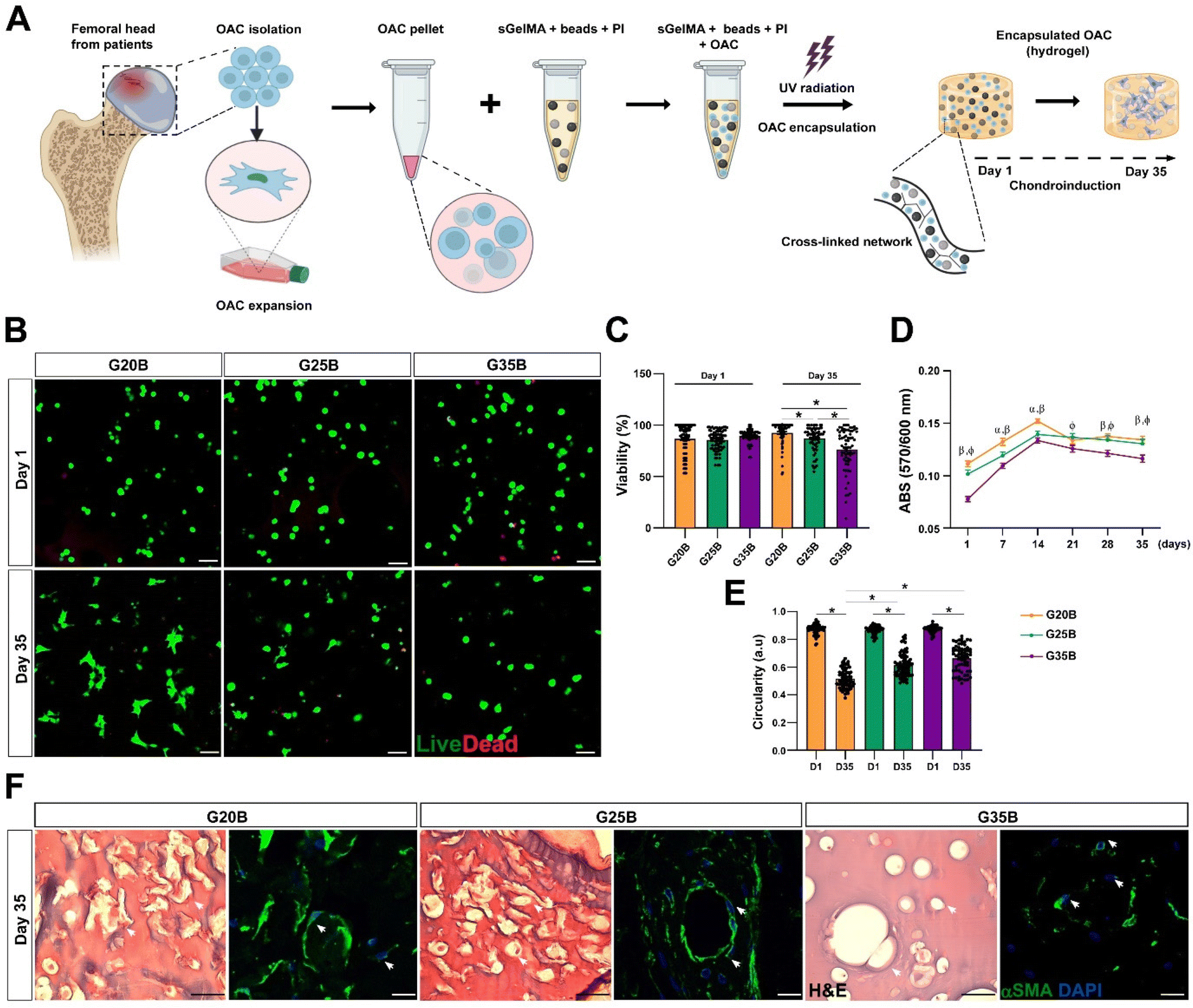 | ||
| Fig. 4 Viability and distribution of OACs encapsulated in G20B, G25B, and G35B. (A) Schematic representation of the protocol for OACs isolation, expansion, and encapsulation. (B) Live/dead staining on days 1 and 35 after the encapsulation. Scale bar = 100 μm. (C) Quantification of the cell viability (each point represents the mean of one image), n = 3 OACs donors. (D) The metabolic activity was evaluated on a week-to-week basis by the PrestoBlue assay. n = 3 hydrogels per formulation, 4 OACs donors. The data are shown as mean ± SEM and were analyzed by 2-way ANOVA and Tukey's post hoc test, * p < 0.05 (α: G20B vs. G25B; β: G20B vs. G35B; φ: G25B vs. G35B). (E) Circularity of OACs measured in the live/dead staining in G20B, G25B, and G35B after 1 and 35 days of encapsulation, n = 3 OAC donors. (F) H&E and αSMA staining of OACs after 35 days. Scale bar = 50 μm. | ||
Another related measurement was the determination of the metabolic activity of OACs during the assay. On day 1, G35B showed lower metabolic activity than G20B and G25B; however, there was a sustained increment until day 14 in all conditions, reaching a plateau that remained until day 35. Overall, G35B was significantly lower than the other conditions, probably due to initial loss in viable cells or a diminished cellular metabolism derived from the harsh confinement of cells within the hydrogels (Fig. 4D).
Cell circularity was determined on days 1 and 35 as a measure of cell spreading and adaptation to these formulations. In G20B, the cells spread and acquired a fibroblastoid phenotype, with a corresponding decrease in cell circularity, a similar result was observed for G25B, whereas for G35B, cell circularity did not decrease as much as in the case of the other formulations, suggesting the inability to adapt and remodel under this stiff environment (Fig. 4E).
In concordance with the live/dead images, H&E, and smooth muscle alpha-actin (αSMA) staining, a component of the chondrocyte cytoskeleton, important differences in cell morphology among the 3 formulations were observed. In G20B, OACs appeared mainly with fibroblastoid morphology and were surrounded by a fibrous halo, suggesting the deposition of ECM. In G25B, a similar distribution and morphology were observed. In G35B, there was a lack of fibrous deposits, and the cells were contained in lacunae-like structures reminiscent of the cartilage tissue (Fig. 4F).
Molecular characterization of the OACs’ responses to the different formulations
After 35 days of the chondroinductive protocol, mRNA was extracted, and the respective cDNA was analyzed for cartilage-related genes. To show the differences among donors and formulations, data were normalized relative to the G20B group; however, this masks the fold-change expression of the transcripts in the samples at day 35 compared to day 0, which is why we added a complementary table where these data are provided (Table S1, ESI†). The data are shown in terms of percentage relative to G20B.First, we evaluated the expression of COL1A1, COL2A1, and ACAN genes characteristic of the hyaline cartilage. COL1A1 is expressed in hyaline cartilage, and its upregulation has been correlated with the formation of fibrocartilage (in vivo). COL1A1 expression increased slightly in all 3 formulations with respect to day 0 (Table S1, ESI†), and there was a stiffness-related trend with a lower expression in G25B and G35B relative to G20B (Fig. 5A). Likewise, COL2A1, the most relevant collagen present in AC, increased in all 3 formulations, but with a pronounced difference among groups, showing a higher expression in G20B compared with G25B and G35B (Fig. 5B). Interestingly, the response intensity to our protocol was different among the 4 OAC donors but still conserved the stiffness-related effect when the three different formulations are compared (Fig. S2A, ESI†). Additionally, ACAN, the most prominent proteoglycan in the hyaline cartilage, was almost unchanged with respect to day 0 of the chondroinductive protocol but also showed a stiffness-related trend, similar to the expression pattern of COL2A1 (Fig. 5C).
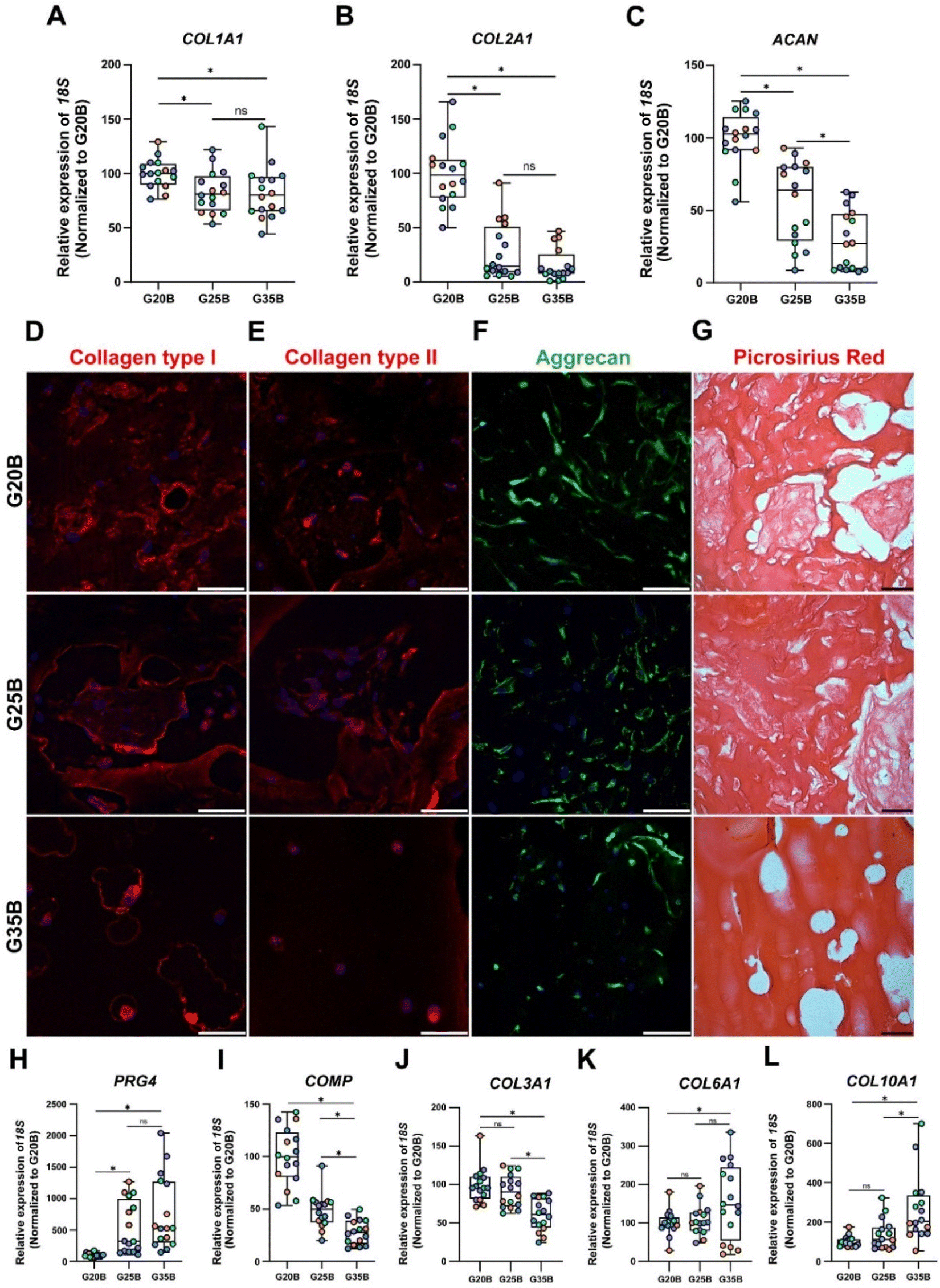 | ||
| Fig. 5 Expression of cartilage-related markers in OACs in G20B, G25B, and G35B. Expression of (A) COL1A1, (B) COL2A1, and (C) ACAN, genes related to hyaline cartilage, n = 4 hydrogels per formulation and 4 OACs donors (different colors), the data are shown as mean ± SEM and were analyzed by the Kruskal–Wallis test and Dunn's multiple comparisons test, *p < 0.05. The data were normalized relative to G20B. Expression of (D) collagen type I, (E) collagen type II, and (F) Aggrecan. (G) Picrosirius Red staining in samples after 35 days. Scale bar = 50 μm. The differentiation process was also evaluated through the expression of additional genes related to the chondrogenic lineage: (H) PRG4, (I) COMP, (J) COL3A1, (K) COL6A1, and (L) COL10A1. n = 4 hydrogels per formulation and 4 OACs donors (different colors), the data are shown as mean ± SEM and were analyzed using the Kruskal–Wallis test and Dunn's multiple comparisons test, *p < 0.05. The data was normalized relative to G20B. | ||
As mentioned, the responses in the 4 studied donors were similar in regard to the stiffness-related trend but not in the magnitude of the fold change relative to the mRNA content from the respective cells on the first day of the encapsulation. COL2A1/COL1A1 ratios, an indicator of cartilage type, must be >1 to consider the process acting more toward a hyaline development than a fibrocartilage commitment.30,31 For donors #1, #2, and #4, there was an upregulation of the COL2A1 transcript with COL2A1/COL1A1 ratios >1. The exception was donor #3; although the relative magnitudes of the responses were still stiffness-dependent, a higher level of COL2A1 transcript was present at the beginning of the differentiation protocol, and there was not a net increment at the end of the experiment (Fig. S2A, ESI†).
TGFβ1 and TGFβ3 signaling are associated with the initial stages of chondrocyte differentiation, and showed a stiffness-related response similar to that observed in COL2A1 and ACAN transcripts (Fig. S2E and F ESI†).32SOX9 levels were twofold relative to day 0 (Table S1, ESI†), but there were no differences among formulations or donors (Fig. S2B, ESI†). The expression of RUNX2, associated with osteogenic differentiation, was very heterogeneous among donors, but globally, there was not a stiffness-related response (Fig. S2D, ESI†).
All these transcriptional patterns correlate well with the expression and secretion of the ECM after 35 days of chondroinduction. For instance, collagen I expression was present in all 3 formulations but without distinguishable differences between G20B and G25B. However, there was a remarkable differential distribution in G35B, where OACs and their lacunae were surrounded by collagen I protein deposits (Fig. 5D). Collagen II distribution was similar to that of collagen I, including G35B; however, we did not observe fibers in the G35B group (Fig. 5E). Aggrecan was distributed in the cell's cytoplasm, showing the different cell morphologies among the different formulations (Fig. 5F). Picrosirius Red staining showed a strong presence of collagen fibers surrounding the cells and also filling some of the pores in G20B and G25B. However, there was almost an absence of collagen fibrillar structures in G35B, suggesting that the formulations with less stiffness are more suitable for ECM formation and remodeling (Fig. 5G).
Additionally, we analyzed other cartilage-related genes. PRG4 (Proteoglycan 4 or lubricin) is expressed abundantly by superficial chondrocytes of hyaline articular cartilage. This gene was poorly expressed in G20B, but there was a substantial increment in gene expression in G25B and G35B, suggesting an induced response mediated by stiffness (Fig. 5H). COMP (cartilage oligomeric matrix protein), with a key role in the assembly and stabilization of the ECM, had a significant increase in expression but was inversely proportional to the stiffness of formulations (Fig. 5I). A similar response was observed for COL3A1, also an important mediator of the assembly of the collagen fibers in the cartilage ECM (Fig. 5J). An inverse stiffness response was observed for COL6A1 and COL10A1; the first codifies a type of collagen specific for the pericellular matrix and with a role in mechanical signal transduction from the surrounding tissue, whereas the second corresponds to a collagen type expressed by hypertrophic chondrocytes (Fig. 5K–L) and has been reported to increase in tissue engineered cartilages.33
Synthesis of GAGs is affected by stiffness
GAGs are synthesized by chondrocytes, and we evaluated if encapsulated OACs also exhibited this property. After 35 days, we were able to detect the presence of sulfate groups from GAGs in the hydrogels after the differentiation protocol using Raman spectroscopy. There was a significant increment in the vibration of OSO3− (the axial orientation in the aromatic ring) at 1085 cm−1 in the following order: G20B > G25B > G35B, but there was not a significant difference in the vibration at 867 cm−1 (attributed to C–O–S asymmetric vibrations) and 757 cm−1 (to OSO3− asymmetric stretching). The signals for the amide bonds (I, II, III) changed according to the content of sGelMA in G20B, G25B, and G35B (data not shown) (Fig. 6A–D). We tried to detect the appearance of collagen after the chondroinductive protocol, but the signals from the amides of the sGelMA were too high and could have masked the signals from new collagen fibers.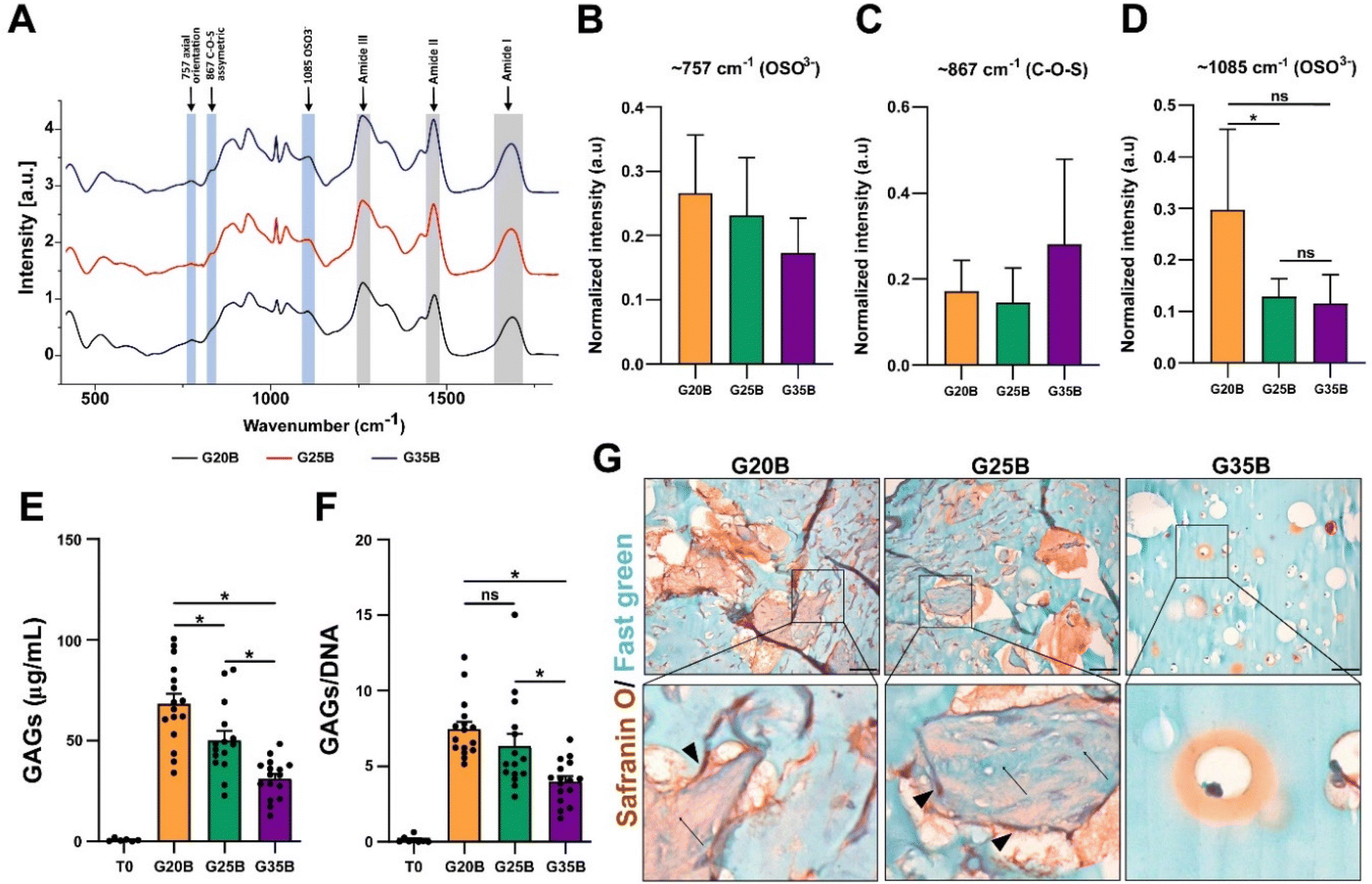 | ||
| Fig. 6 Encapsulated OACs synthesize GAGs in a stiffness-dependent way. (A) The spectra of samples were analyzed in the 350–1800 cm−1 range to detect molecular vibrations of the C4S sulfated groups present in GAGs. Bands from amide groups are depicted in gray bars and the vibration from C4S bonds is in light blue. These data are shown comparing the spectra from day 35 to day 0 and then were normalized internally to compare different formulations and plotted as arbitrary units (a.u). Quantification of main bands for GAGs in G20B, G25B, and G35B at (B) 757, (C) 867, and (D) 1085 cm−1, n = 3 hydrogels per formulation and 3 OACs donors, (Stratigraphic software was used for statistical analyses, * p < 0.05). (E) Biochemical detection of GAGs in hydrogels from day 0 and day 35. (F) GAG data normalized by DNA content. n = 4 hydrogels per formulation and 4 OACs donors, ANOVA test and Dunn's post-test, * p < 0.05. (G) Safranin O/Fast green staining in hydrogels of G20B, G25B, and G35B after 35 days of differentiation. Scale bar = 50 μm. Arrows indicate protein fibers inside pores and arrow-heads cells in the surrounding. | ||
The molecular detection of GAGs was accompanied by biochemical quantification using the DMMB assay. The synthesis of GAGs was dependent on the stiffness, with the highest yield for G20B and the lowest for G35B. The same pattern was observed when data were normalized with the DNA content (Fig. 6E and F).
Histological staining showed an abundant secretion of GAGs (in red) surrounding cells and filling the void spaces generated by the porogens, showing a strong remodeling of the matrix mostly in G20B and G25B-based hydrogels. Remarkably, we observed depositions of ECM in the form of “wool balls”, filled with protein fibers positive for Fast green staining (most probably, collagens) mixed with GAG depositions and surrounded by OACs. In G35B, there were also red deposits surrounding cells, but it was limited to the individual cell area and there were no deposits in the rest of the hydrogel (Fig. 6G).
Matrix remodeling and ECM synthesis
The expression of ECM components such as collagens, proteoglycans, and GAGs implies that OACs could remodel the hydrogel and replace the hydrogel material with new ECM components. Then, we hypothesized that the mechanical properties of the hydrogels should change after the chondroinductive protocol. To address this issue, we measured the YM before and after the chondroinductive protocol (35 days) (Fig. 7A). Interestingly, the YM increased for G20B and G25B (5.7 and 1.7-fold change respectively) suggesting that OACs remodeled the hydrogel by replacing partially the hydrogel material with new ECM components. In G35B, there were no changes in the YM, meaning that the remodeling could have been poor or absent (Fig. 7B). These results were in line with the levels of GAGs, measured at the same time points when YMs were determined (Fig. 7C).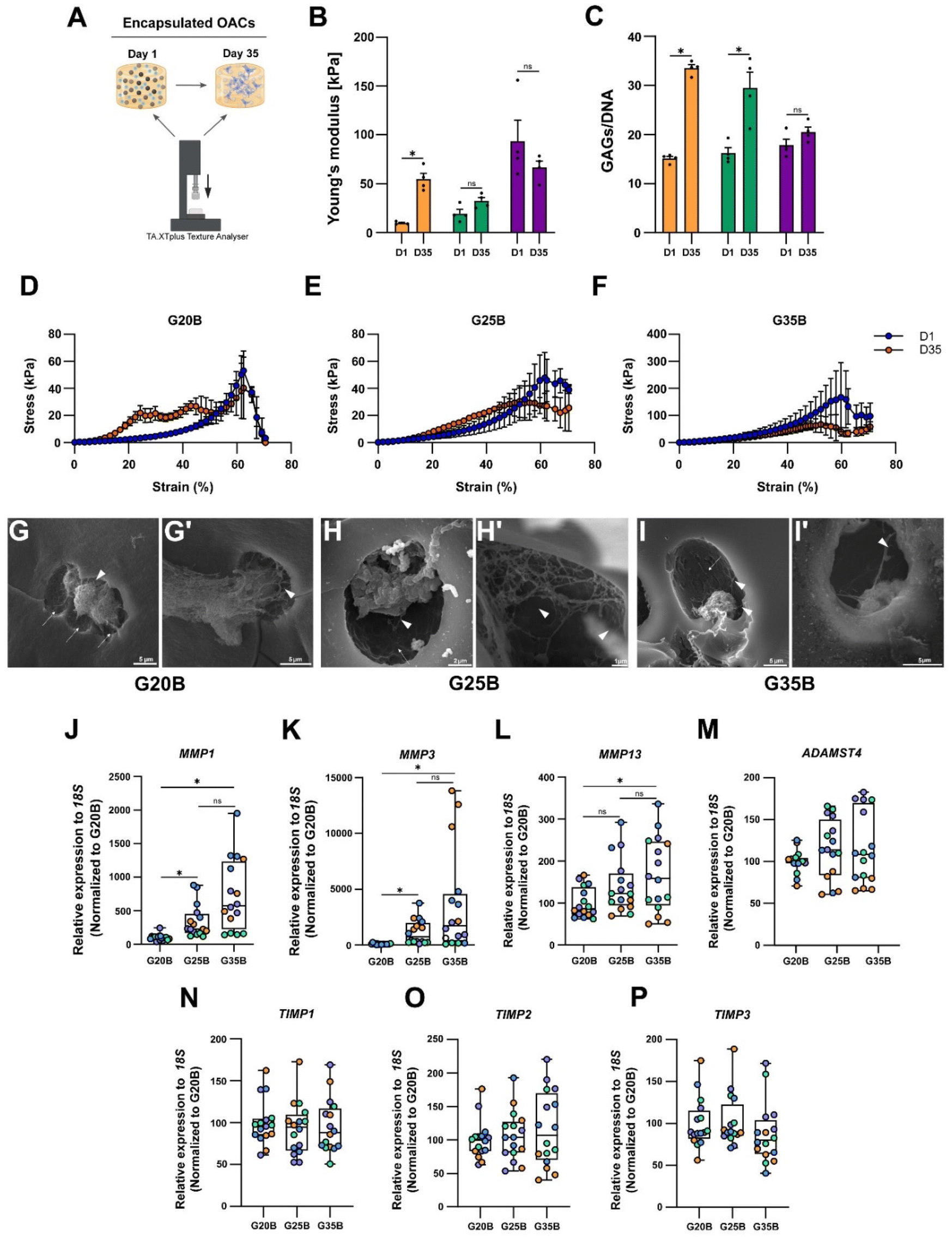 | ||
| Fig. 7 Functional consequences of the remodeling in the sGelMA hydrogels (A) experimental setup designed to evaluate the change in the YM in hydrogels before and after the chondroinductive protocol. (B) YM of hydrogels at days 1 and 35. (C) GAGs determination before and after the chondroinductive protocol, normalized by total DNA. n = 4 hydrogels per condition, 2-way ANOVA, and Sidak's multiple comparisons test, *p < 0.05. Stress vs. strain curves shows the break profile of the hydrogels before and after the differentiation protocol of (D) G20B, (E) G25B, and (F) G35B. SEM images show the ultrastructure of hydrogels after the chondroinductive protocol. (G–I) At high magnification, we can observe the presence of ECM fibers (arrowheads) and OACs occupying cavern-like structures (arrows). Scale bars are described for each image. The remodeling of the hydrogels was also characterized in terms of the expression of proteolytic enzymes (J) MMP1, (K) MMP3, (L) MMP13, and (M) ADAMTS4, and their inhibitors (N) TIMP1, (O) TIMP2, and (P) TIMP3; n = 4 hydrogels per formulation and 4 OACs donors (different colors), the data are shown as mean ± SEM and were analyzed using the Kruskal–Wallis test and Dunn's multiple comparisons test, *p < 0.05. The data were normalized relative to G20B. | ||
Another piece of evidence confirming that hydrogels had improved mechanical properties is the profile of breaking in the stress vs. strain plots. Even though all hydrogels broke at 60% strain, the remodeled conditions conferred more resistance (orange curve over the blue curve, day 1 and day 35 respectively), especially for G20B (Fig. 7D) where the curve representing the 35-day hydrogel is above the curve of the 1-day hydrogel. This means that it had more resistance to compression after the chondroinductive protocol. Something similar was observed for G25B, but the endured stress was less compared to that of G20B (Fig. 7E). For G35B, both curves were similar, in agreement with the calculated YM, suggesting no changes in the composition of the hydrogel occurred (Fig. 7F).
To characterize the remodeling at the ultrastructure level, samples were analyzed with SEM microscopy. After 35 days, the porous structure of hydrogels was observed and individual or cell conglomerates were also present. In the 3 conditions, we observed the presence of OACs immersed in a lacunae-like structure and with corresponding space between the cell and the lacunae walls. Because these cells were encapsulated in a formulation with a high concentration of polymer, the presence of these “caves” (Fig. 7G–I, arrows) suggests that OACs remodeled their environment by digesting the polymer network and replacing it with ECM-based fibers (Fig. 7G–I, arrowheads). Due to the stringent processing of the samples during the execution of the SEM technique, a lot of cells disappeared from the surfaces, and it was not possible to quantify the number of cells inside the lacunae.
Chondrocytes are capable of anabolizing ECM components, but also of catabolizing them to maintain homeostasis in the tissue. For this, they express several proteolytic enzymes such as metalloproteinases (MMPs) and disintegrin metalloproteinases with thrombospondin motifs (ADAMTSs). The expression of MMP1, MMP3, MMP13, and ADAMTS4 in the formulations was evaluated, and a substantial increment in the transcript of these 4 enzymes was detected, especially for the MMPs (up to 2000-fold change respect to day 0). There was also a strong correlation between the stiffness and the expression of MMP1, MMP3, and MMP13 (Fig. 7J–L). ADAMTS4 was upregulated but there was no clear correlation with the formulation's stiffness (Fig. 7M).
Tissue inhibitors of metalloproteinases (TIMPs) are the counterpart of the proteolytic enzymes to maintain the turnover and homeostasis of the tissues. We evaluated the most prominent in the cartilage tissue: TIMP1, TIMP2, and TIMP3. For TIMP types 1 and 2, there was almost no response, but there was a significant upregulation for TIMP3 after 35 days of encapsulation, without a specific stiffness-associated trend (Fig. 7N–P).
In vivo evaluation of the chondroinductive properties of G20B, G25B, and G35B
When conducting the evaluation of scaffold designs in vitro, hydrogels are not subjected to a degradative environment as would be the case for hydrogels within a cartilage lesion and surrounded by the synovial fluid. In this regard, it is relevant to understand if the scaffold design can also support remodeling and formation of new ECM while the environment exerts a degradative pressure derived from enzymes and the action of cells in the near space. We evaluated the in vivo degradation and in a separate experiment if the chondroinduction observed in vitro could be recapitulated when hydrogels were subcutaneously implanted in NSG mice (Fig. 8A). In the formulations with low and medium content of sGelMA (G20 and G25) and their porous counterparts (G20B, G25B), a degradative response was observed with a decrease in the volume (height and diameter). This degradation was remarkably larger for G20B: the image shows the fusion of 2 hydrogels and one of them is significantly smaller (Fig. 8B). After the macroscopic analysis of the recovered hydrogels, GAGs were quantified and detected under all conditions. Also, the porous formulations have a higher concentration of GAGs than their respective non-porous condition. Comparing these results with those of hydrogels analyzed on the same day of the surgery (D10), there was a slight decrease in GAGs in some of the formulations, excluding G20B. When the GAG data were normalized for total DNA, the conclusions changed, showing for all subcutaneously implanted formulations higher levels of GAG deposition than in the D10 controls, except for G35 (ESI Fig. 3†). Additionally, unlike the G20/G20B pair, the ratio of GAGs/DNA was higher in the porous formulations than in the non-porous one. In G20/G20B the trend was inverted because the G20B had an enormous amount of DNA due to extensive invasion and encapsulation by the host's cells, resulting in a highly remodeled and degraded hydrogel. The reduced amount of total GAGs and the less DNA observed in the rest of the formulations compared to that in the D10 control suggest that some level of cell death and halt in the differentiation/remodeling process occurred after subcutaneous implantation (Fig. S3A and B, ESI†).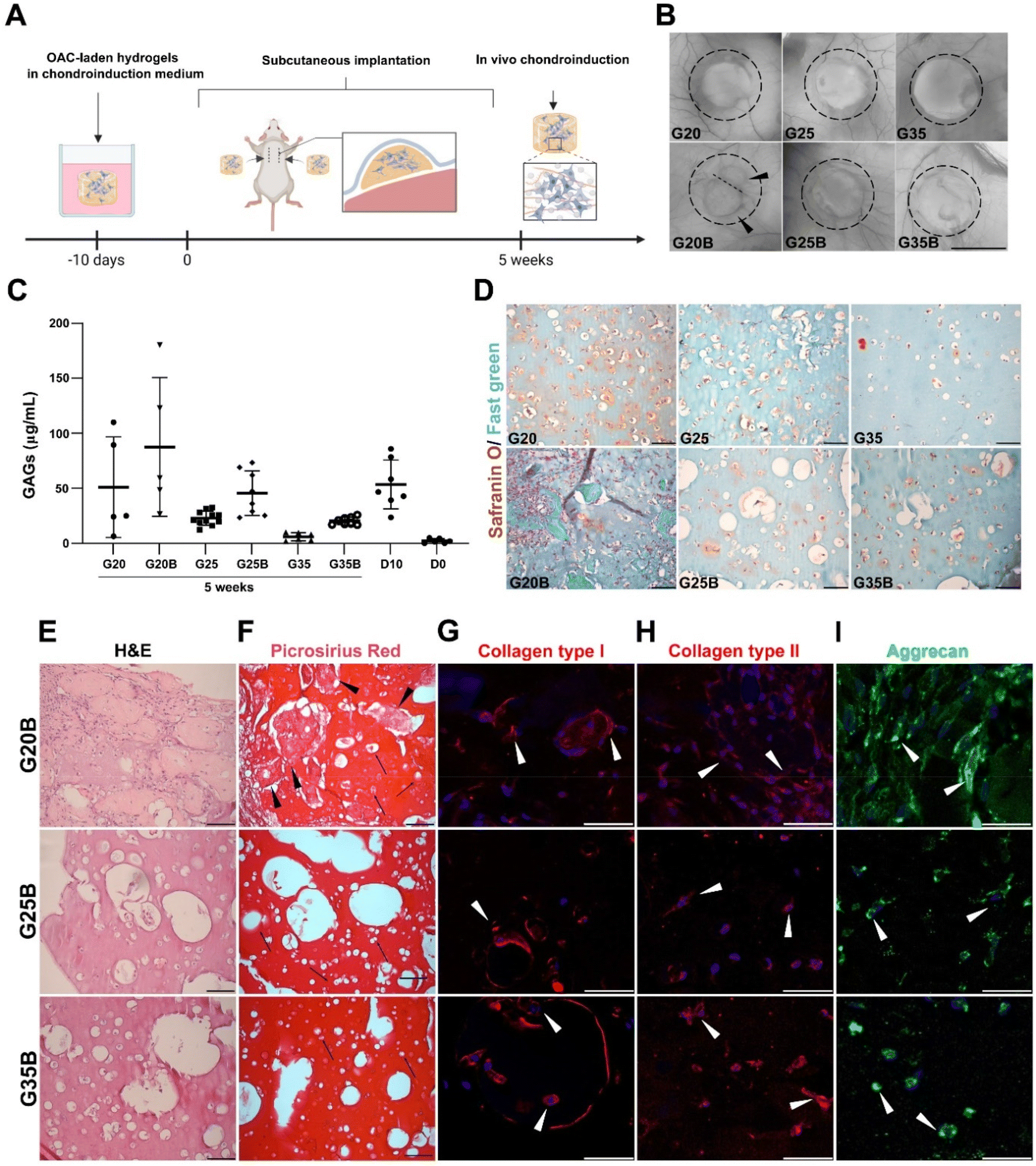 | ||
| Fig. 8 In vivo evaluation of ECM synthesis and remodeling. (A) Schematic representation of the experimental setup. Hydrogels were implanted subcutaneously in the back of NSG mice. After 5 weeks, the hydrogels were recovered and analyzed. (B) Macroscopic appearance of the explanted hydrogels, black arrowheads indicated the presence of 2 fused hydrogels in G20B. Scale bar = 1 mm. (C) Determination of GAGs in the recovered samples, each point represents a unique hydrogel. D0 corresponds to hydrogels saved the same day of encapsulation and D10 to hydrogels recovered the same day of the surgery, after 10 days maintained in a chondroinductive medium. (D) Safranin O/Fast green staining shows the deposition of GAGs and proteins in the porous and non-porous formulations, scale bar = 50 μm. (E) H&E staining in the porous formulations shows the grade of remodeling and cellularity. (F) Collagen deposition depicted by Picrosirius Red staining, black arrowheads show protein deposits, arrows indicate individual OACs inside lacunae-like spaces. Expression of proteins related to the hyalin cartilage's ECM: (G) Collagen type I, (H) Collagen type II, and (I) Aggrecan, arrowheads show OACs positive staining for the respective proteins. DAPI was used for nuclear staining, scale bar = 50 μm. | ||
The safranin O/Fast green staining showed the presence of GAGs surrounding the OACs in G20, G25, and G35, and in that order of intensity. A similar staining pattern was observed for the porous formulations, with the exception of G20B, which looked more cellularized and degraded. In the latter, the porous structure was lost, and large deposits of proteins were present surrounding the hydrogel. In G25B, GAGs were detected in the proximity of OACs and inside the pores (Fig. 8D).
H&E staining showed the ECM remodeling and increment in cellularity, more pronounced in G20B due to the extensive tissue encapsulation observed in this formulation (Fig. 8E). Accordingly, collagen fibers were detected by Picrosirius Red inside pores and surrounding OACs (Fig. 8F). In both staining, the OACs surrounded by a lacunae-like space were observed. Additionally, we evaluated the expression of collagens type I, type II, and aggrecan, characteristics of the hyaline phenotype. Collagen type I was distributed inside OACs (cytoplasm) in all formulations and was detected in fibrous deposits similar to those described for the Picrosirius Red staining. Interestingly, in G25B and G35B, the lacunae-like spaces were surrounded by an intense deposit of collagen type I (Fig. 8G). Collagen type II was detected mainly in the cytoplasm, and we did not observe fibrous deposits (Fig. 8H). Their distribution was similar to the aggrecan (Fig. 8I). The pattern observed in the non-porous formulations showed a less remodeled hydrogel and almost no deposition of collagens (Fig. S3C and D, ESI†). Collagen types I and II, along with aggrecan, were expressed following a similar cytoplasmatic pattern (Fig. S3E–G, ESI†).
4. Discussion
The restoration of the structure and functionality of AC has been an elusive goal for physicians and tissue engineers. The most common treatment for AC lesions, microfracture, induces the formation of fibrocartilage; however, its suboptimal properties are more consistent with reparative rather than regenerated tissue. ACI/MACI (autologous chondrocyte implantation and matrix-seeded ACI, respectively) treatment represents an improvement regarding the overall outcome for medium- and large-sized lesions, delaying the need for a prosthesis. However, they possess some downsides, such as the requirement of 2 surgical procedures, open surgery, and the in vitro expansion of the patient's isolated chondrocytes, which entails the loss of the hyaline transcriptomic profile of cells, compromising the outcome of ACI/MACI.34Due to the lack of effective treatments, significant research efforts have been allocated to developing new scaffolds and promoting the healthy regeneration of the cartilage. An ideal and inexpensive approach involves the use of an acellular scaffold, promoting migration and new tissue formation by surrounding cells in the cartilage lesion. Concerning the scaffolds’ material, several alternatives have been proposed for this application.35–37
sGelMA, derived from a cold-adapted fish, has unique rheological features.21,38 The combination of sGelMA and porogens from non-functionalized porcine gelatin, allows the generation of a highly concentrated sGelMA liquid phase with solid gelatin beads that remain gelled at room temperature, which is possible due to the different melting points. This biphasic composition can form porous hydrogels and fill articular lesions through minimally invasive procedures to be covalently polymerized in situ after the UV light exposition. The porogens melt at 37 °C, leaving voids that will positively affect the regeneration process.39
High mechanical resistance is a desirable scaffold characteristic for cartilage regeneration,24 and has been successfully achieved by Zhao et al.40 using crystallized polyvinyl alcohol reinforced with bacterial cellulose, reaching a compressive strength of 98 MPa, even higher than cartilage. In this case, the design is closer to an implant instead of a scaffold and lacks remodeling capabilities. In this proposal, both, mechanical resistance and remodeling are considered. In this regard, scaffolds porosity is fundamental for cell maintenance and the efficient exchange of nutrients and gases between resident cells and their surroundings.41 We aim to develop a stiff material, but with good diffusion properties, which is why we have chosen to use porous and concentrated hydrogel with proven better diffusion and mechanical capacity instead of less concentrated non-porous hydrogels with weak mechanical properties. We analyzed our data, comparing YM and GAGs levels in all formulations, and we have found that an appropriate cell response is not only a matter of stiffness but also porosity. For formulations with the same stiffness but with different levels of porosity, the one with higher porosity will show better cell response.
Pores positively affected the fitness of OACs, however, there was an inverse relationship between the anabolic activity of OACs and the stiffness of the formulations. For example, in formulation G35B, we observed a rounded morphology and a lacunae-like space surrounding the chondrocytes, which reminds the aspect of healthy hyaline cartilage. However, the molecular data indicated that it is challenging for cells to remodel this material, as suggested by the mRNA levels of metalloproteinases and the scarcity of ECM deposition. In contraposition with G35B, the less stiff formulation, G20B, showed higher levels of anabolic activity (COL2A1, ACAN, COL6A1, COMP) with a significant increase in the YM and mechanical strength after differentiation. This article shows that the latter is consistent with the recapitulation of a healthy hyaline profile and cartilage-like tissue formation.
On the other hand, pores have an unfavorable effect on the mechanical properties of the formulations, reaching a reduction of more than 50% in the YM when 40% v/v of beads was used. However, the material itself gains in degradability, diffusion capacity, and remodeling, either in vitro or in vivo. In vivo data showed extensive degradation, possibly detrimental for cartilage regeneration if the rate of ECM synthesis is slower than the hydrogel degradation rate. Although there is not much information about the proteolytic activity in the subcutaneous fluid, and how this can vary in the presence of an incision or implanted hydrogels in our in vivo model,42 it is well-known that a relatively high concentration of proteases can be found in the synovial fluid of osteoarthritic knees.43 In this regard and taking into consideration that cartilage regeneration requires months for remodeling and new tissue formation,44 scaffolds for lesion restoration should have a prolonged residence time on the site of injury. Our data showed that at least those formulations with high concentrations of the biopolymer are substantially more resistant to degradation (see Fig. 3) than the lower concentrated hydrogels under high enzymatic degradative conditions. On the other hand, the subcutaneous in vivo results showed that most of the formulations, except for the G20B, stay volumetrically intact after 12 weeks, and that the feasibility of conducting cartilage induction is still present after implantation. However, unlike the observed in vitro results in G20B, the excessive volumetric loss and cell invasion in the G20B group in the subcutaneous cartilage formation experiment suggest that G25B design would be a more appropriate scaffold for cartilage restoration in vivo. Because the subcutaneous approach is certainly not an indicator of a successful cartilage repair in a clinical or in vivo setting, still more research is needed to specifically define the optimal formulation suitable for intraarticular application. This is why our next step will be the evaluation of this issue in an in vivo model of cartilage lesions in sheep.
The excellent work from Hua et al.45 shares similar considerations to our proposal, and demonstrated the strong adhesion of their material to the cartilage, fundamental to an arthroscopic application, such as ACI, but lacks in the description of the behavior of patients’ cells inside this material. To our knowledge, this is the first time that a formulation considers the mechanical resistance to compression, chondroinductivity of patient's cells, porosity, and remodeling capabilities in a biomaterial-based formulation. Additionally, the dedifferentiation observed in chondrocytes during cell expansion in 2D cell cultures could be reverted if chondrocytes are encapsulated in the formulations presented here, as we showed with the recovery of synthesis of GAGs, and the expression of several genes, such as COL2A1. These results support a possible application of sGelMA-derived formulations in ACI.
A limitation in the application of these sGelMA-based formulations is the requirement of the invasion of neighboring cells to replace the scaffold. But it is unknown whether these cells maintain their mobility and regenerative properties if the tissue has been damaged. If that were the case, the potential application of these formulations could be restricted to tissue of patients who are not terminally ill such as in severe osteoarthritic patients.
In general terms, there is no optimal formulation in terms of mechanical and biological support. A stiff scaffold can mimic the mechanical properties of cartilage, but it is less prone to remodeling and replacement by new tissue. Concurrently, a softer scaffold might be replaced with new tissue, but lacking the needed mechanical resistance to avoid exacerbated deformation of the cartilage at the lesion rim, which could cause additional chondrocytes damage and progression of the defect. Besides, it also will depend on the specific characteristics of each patient: size and grade of the lesion, chondrocyte metabolism, and migratory capacity of cells, among others. It opens the door to a technology where individualized formulations can be designed for each patient, based on these customizable formulations and according to the specifications of the lesions.
5. Conclusion
In this study, sGelMA has been postulated as a versatile biomaterial with improved properties due to its psychrophilic nature, making it suitable for the generation of scaffolds and regenerative applications. The capacity to maintain the stability of the liquid aggregation state, extrudability, and separated phases when combined with solid mammalian gelatin beads within an extended range of temperatures makes these formulations truly injectable for filling and forming porous scaffolds in a cartilage lesion using minimally invasive procedures. The stable liquid state of sGelMA solutions allows the appropriate filling of the lesions and resembles the continuous, smooth, and unscathed profile of the cartilage surface, without the need for the precise manual carving of a solid scaffold. Additionally, the capacity of concentrating the sGelMA solution makes it possible to reach mechanical properties (0.25 MPa, G35B formulation) that could, to a certain degree, protect from intensive accumulation of deformation. The chondroinductivity of the presented hydrogel formulations, and their potential of being replaced with new hyaline cartilage by the action of infiltrated host cells, must be evaluated in a pre-clinical model of joint cartilage damage; however, we hypothesize that the resistance to compression and remodeling capabilities will have a cooperative regeneration effect in vivo.Conflicts of interest
MK and JPA receive stipends from Cells for Cells, a Chilean Biotechnology Company, for cell therapy development. GZ, CH, and JPA are coinventors of PCT (PCT/CL2022/050086) for the generation of porous and tough scaffolds.SV, JN, IC, PD, and WG declare that they have no competing interests.
Acknowledgements
This work was supported by ANID—Basal funding for the Scientific and Technological Center of Excellence, IMPACT, #FB210024. We also acknowledge the financial support from CORFO through grant 20CVID-128078.References
- J. Eschweiler, N. Horn, B. Rath, M. Betsch, A. Baroncini, M. Tingart and F. Migliorini, Life, 2021, 11(4), 302–315 CrossRef CAS PubMed.
- S. J. Gilbert and E. J. Blain, Cartilage mechanobiology: How chondrocytes respond to mechanical load, editor:Mechanobiology in Health and Disease, Elsevier, 2018, pp. 99–126 Search PubMed.
- A. J. S. Fox, A. Bedi and S. A. Rodeo, Sports Health, 2009, 1(6), 461–468 CrossRef PubMed.
- Z. Zhao, Y. Li, M. Wang, S. Zhao, Z. Zhao and J. Fang, J. Cell. Mol. Med., 2020, 24(10), 5408–5419 CrossRef CAS PubMed.
- D. Martínez-Moreno, G. Jiménez, P. Gálvez-Martín, G. Rus and J. A. Marchal, Biochim. Biophys. Acta, Mol. Basis Dis., 2019, 1865(6), 1067–1075 CrossRef PubMed.
- M. S. Venäläinen, M. E. Mononen, J. Salo, L. P. Räsänen, J. S. Jurvelin, J. Töyräs, T. Virén and R. K. Korhonen, Sci. Rep., 2016, 6, 37538 CrossRef PubMed.
- L. Zevenbergen, W. Gsell, D. D. Chan, J. Vander Sloten, U. Himmelreich, C. P. Neu and I. Jonkers, Osteoarthritis Cartilage, 2018, 26(12), 1710–1721 CrossRef CAS PubMed.
- P. K. Edwards, J. R. Ebert, G. C. Janes, D. Wood, M. Fallon and T. Ackland, J. Sport Rehabil., 2014, 23(3), 203–215 Search PubMed.
- Y. Gong, K. Su, T. T. Lau, R. Zhou and D.-A. Wang, Tissue Eng., Part A, 2010, 16(12), 3611–3622 CrossRef CAS PubMed.
- J. W. Nichol, S. T. Koshy, H. Bae, C. M. Hwang, S. Yamanlar and A. Khademhosseini, Biomaterials, 2010, 31(21), 5536–5544 CrossRef CAS PubMed.
- W. Schuurman, P. A. Levett, M. W. Pot, P. R. van Weeren, W. J. A. Dhert, D. W. Hutmacher, F. P. W. Melchels, T. J. Klein and J. Malda, Macromol. Biosci., 2013, 13(5), 551–561 CrossRef CAS PubMed.
- X. Li, J. Zhang, N. Kawazoe and G. Chen, Polymers, 2017, 9(8), 309–322 CrossRef.
- S. Pahoff, C. Meinert, O. Bas, L. Nguyen, T. J. Klein and D. W. Hutmacher, J. Mater. Chem. B, 2019, 7, 1761–1772 RSC.
- R. Levato, W. R. Webb, I. A. Otto, A. Mensinga, Y. Zhang, M. van Rijen, R. van Weeren, I. M. Khan and J. Malda, Acta Biomater., 2017, 61, 41–53 CrossRef CAS PubMed.
- H. J. Yoon, S. R. Shin, J. M. Cha, S. H. Lee, J. H. Kim, J. T. Do, H. Song and H. Bae, PLoS One, 2016, 11(10), e0163902 CrossRef PubMed.
- A. Zaupa, N. Byres, C. Dal Zovo, C. A. Acevedo, I. Angelopoulos, C. Terraza, N. Nestle, P. N. Abarzúa-Illanes, F. Quero, P. Díaz-Calderón, Y. Olguín, T. L. Akentjew, C. A. Wilkens, C. Padilla, F. C. Zacconi, K. Pino-Lagos, J. J. Blaker, M. Khoury, J. Enrione and J. P. Acevedo, Mater. Sci. Eng., C, 2019, 102, 373–390 CrossRef CAS PubMed.
- M. C. Echave, L. Saenz del Burgo, J. L. Pedraz and G. Orive, Curr. Pharm. Des., 2017, 23(24), 3567–3584 CrossRef CAS PubMed.
- A. Zaupa, C. Terraza, P. N. Abarzúa-Illanes, N. Byres, G. Zavala, J. Cuenca, C. Hidalgo, S. M. Viafara-García, B. Wolf, K. Pino-Lagos, J. J. Blaker, M. Rumbak, M. Khoury, J. Enrione and J. P. Acevedo, Biomacromolecules, 2023, 24(1), 150–165 CrossRef CAS PubMed.
- F. Boschetti, G. Pennati, F. Gervaso, G. M. Peretti and G. Dubini, Biorheology, 2004, 41(3–4), 159–166 Search PubMed.
- X. Li, Y. Chen, N. Kawazoe and G. Chen, J. Mater. Chem. B, 2017, 5, 5753–5762 RSC.
- P. Díaz, D. López, S. Matiacevich, F. Osorio and J. Enrione, J. Sci. Food Agric., 2011, 91(14), 2558–2565 CrossRef PubMed.
- T. L. Akentjew, C. Terraza, C. Suazo, J. Maksimuck, C. A. Wilkens, F. Vargas, G. Zavala, M. Ocaña, J. Enrione, C. M. García-Herrera, L. M. Valenzuela, J. J. Blaker, M. Khoury and J. P. Acevedo, Nat. Commun., 2019, 10(1), 3098 CrossRef.
- C. M. Hwang, S. Sant, M. Masaeli, N. N. Kachouie, B. Zamanian, S.-H. Lee and A. Khademhosseini, Biofabrication, 2010, 2(3), 035003 CrossRef PubMed.
- J. Yang, Y. Xiao, Z. Tang, Z. Luo, D. Li, Q. Wang and X. Zhang, J. Mater. Chem. B, 2020, 8, 4680–4693 RSC.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9(7), 676–682 CrossRef CAS PubMed.
- D. Loessner, C. Meinert, E. Kaemmerer, L. C. Martine, K. Yue, P. A. Levett, T. J. Klein, F. P. W. Melchels, A. Khademhosseini and D. W. Hutmacher, Nat. Protoc., 2016, 11, 727–746 CrossRef CAS PubMed.
- M. W. Pfaffl, Nucleic Acids Res., 2001, 29(9), e45 CrossRef CAS PubMed.
- C. H. Zheng and M. E. Levenston, Eur. Cells Mater., 2015, 29, 224–236 CrossRef CAS PubMed.
- J. Southan, E. McHugh, H. Walker and H. M. Ismail, Front. Mol. Biosci., 2020, 7, 592905 CrossRef CAS PubMed.
- S. Marlovits, M. Hombauer, M. Truppe, V. Vècsei and W. Schlegel, Joint Surg. Br., 2004, 86-B(2), 286–295 CrossRef PubMed.
- A. Barlič, M. Drobnič, E. Maličev and N. Kregar-Velikonja, J. Orthop. Res., 2008, 26(6), 847–853 CrossRef PubMed.
- W. Wang, D. Rigueur and K. M. Lyons, Birth Defects Res., Part C, 2014, 102(1), 37–51 CrossRef CAS PubMed.
- P. Yeung, K. H. Cheng, C. H. Yan and B. P. Chan, Sci. Rep., 2019, 9(1), 12453 CrossRef CAS PubMed.
- A. Colombini, F. Libonati, S. Lopa, G. M. Peretti, M. Moretti and L. de Girolamo, Knee Surg., Sports Traumatol., Arthrosc., 2023, 31(6), 2338–2348 CrossRef PubMed.
- M. Wasyłeczko, W. Sikorska and A. Chwojnowski, Membranes, 2020, 10(11), 348 CrossRef PubMed.
- H. Kwan, E. Chisari and W. S. Khan, Materials, 2020, 13(2), 306–316 CrossRef CAS PubMed.
- E. Kon, A. Roffi, G. Filardo, G. Tesei and M. Marcacci, Arthroscopy, 2015, 31(4), 767–775 CrossRef PubMed.
- H. Yang, H. Wang, M. Huang, G. Cao, F. Tao, Q. Shen, G. Zhou and H. Yang, Compr. Rev. Food Sci. Food Saf., 2022, 21(2), 942–963 CrossRef CAS PubMed.
- M. M. Nava, L. Draghi, C. Giordano and R. Pietrabissa, J. Appl. Biomater. Funct. Mater., 2016, 14(3), e223–e229 CAS.
- J. Zhao, H. Tong, A. Kirillova, W. J. Koshut, A. Malek, N. C. Brigham, M. L. Becker, K. Gall, B. J. Wiley, J. Zhao, H. Tong, A. Malek and B. J. Wiley, Adv. Funct. Mater., 2022, 32(41), 2205662 CrossRef CAS.
- Q. L. Loh and C. Choong, Tissue Eng., Part B, 2013, 19(6), 485–502 CrossRef CAS PubMed.
- T. Nakade, H. Yokota, H. Taniyama, Y. Hori, N. Agata, T. Ikeda, H. Furusaki, Y. Yamada, Y. Uchida, A. Yuasa, M. Yamaguchi and K. Otomo, Carbohydr. Polym., 2000, 41(4), 327–329 CrossRef CAS.
- M. Palmer, E. Stanford and M. M. Murray, Materials, 2011, 4(8), 1469–1482 CrossRef CAS PubMed.
- L. Peng, B. Zhang, X. Luo, B. Huang, J. Zhou, S. Jiang, W. Guo, G. Tian, Z. Tian, S. Shen, Y. Li, X. Sui, S. Liu, Q. Guo and H. Li, Stem Cells Int., 2021, 2021, 5590479 Search PubMed.
- Y. Hua, H. Xia, L. Jia, J. Zhao, D. Zhao, X. Yan, Y. Zhang, S. Tang, G. Zhou, L. Zhu and Q. Lin, Sci. Adv., 2021, 7(35), eabg0628 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm00703k |
| This journal is © The Royal Society of Chemistry 2023 |