
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication of hydrogel microspheres via microfluidics using inverse electron demand Diels–Alder click chemistry-based tetrazine-norbornene for drug delivery and cell encapsulation applications†
Rubén
Pareja Tello
 a,
Shiqi
Wang
a,
Flavia
Fontana
a,
Alexandra
Correia
a,
Giuseppina
Molinaro
a,
Sandra
López Cerdà
a,
Sami
Hietala
b,
Jouni
Hirvonen
a,
Goncalo
Barreto
cde and
Hélder A.
Santos
*afg
a,
Shiqi
Wang
a,
Flavia
Fontana
a,
Alexandra
Correia
a,
Giuseppina
Molinaro
a,
Sandra
López Cerdà
a,
Sami
Hietala
b,
Jouni
Hirvonen
a,
Goncalo
Barreto
cde and
Hélder A.
Santos
*afg
aDrug Research Program, Division of Pharmaceutical Chemistry and Technology, University of Helsinki, Helsinki FI-00014, Finland. E-mail: h.a.santos@umcg.nl
bDepartment of Chemistry, University of Helsinki, Helsinki FI-00014, Finland
cClinicum, Faculty of Medicine, University of Helsinki and Helsinki University Hospital, Haartmaninkatu 8, 00014, Helsinki, Finland
dOrton Orthopedic Hospital, Tenholantie 10, 00280, Helsinki, Finland
eMedical Ultrasonics Laboratory (MEDUSA), Department of Neuroscience and Biomedical Engineering, Aalto University, 02150, Espoo, Finland
fDepartment of Biomedical Engineering, University Medical Center Groningen, University of Groningen, Ant. Deusinglaan 1, 9713 AV Groningen, The Netherlands
gW.J. Kolff Institute for Biomedical Engineering and Materials Science, University Medical Center Groningen, University of Groningen, Ant. Deusinglaan 1, 9713 AV Groningen, The Netherlands
First published on 13th June 2023
Abstract
Microfluidic on-chip production of polymeric hydrogel microspheres (MPs) can be designed for the loading of different biologically active cargos and living cells. Among different gelation strategies, ionically crosslinked microspheres generally show limited mechanical properties, meanwhile covalently crosslinked microspheres often require the use of crosslinking agents or initiators with limited biocompatibility. Inverse electron demand Diels Alder (iEDDA) click chemistry is a promising covalent crosslinking method with fast kinetics, high chemoselectivity, high efficiency and no cross-reactivity. Herein, in situ gellable iEDDA-crosslinked polymeric hydrogel microspheres are developed via water-in-oil emulsification (W/O) glass microfluidics. The microspheres are composed of two polyethylene glycol precursors modified with either tetrazine or norbornene as functional moieties. Using a single co-flow glass microfluidic platform, homogenous MPs of sizes 200–600 μm are developed and crosslinked within 2 minutes. The rheological properties of iEDDA crosslinked bulk hydrogels are maintained with a low swelling degree and a slow degradation behaviour under physiological conditions. Moreover, a high-protein loading capacity can be achieved, and the encapsulation of mammalian cells is possible. Overall, this work provides the possibility of developing microfluidics-produced iEDDA-crosslinked MPs as a potential drug vehicle and cell encapsulation system in the biomedical field.
Introduction
Hydrogel microspheres constitute micron-size hydrogel particles developed as microcarriers, drug delivery and tissue regeneration systems.1 As hydrogels, they constitute three-dimensional polymer-based structures with the ability to show a high swelling nature in aqueous media.2,3 Their advantages over other nano- and micro-sized counterparts include their considerable surface area suitable for bioconjugation, inner functionalized porous network for encapsulation, high colloidal stability, and mechanical flexibility.4 Moreover, they have been developed as promising platforms for different biomedical purposes, such as DNA or siRNA delivery,5 cell encapsulation,6 drug delivery7 and tissue regeneration.8Several production methods of hydrogel microspheres either based on top-down or bottom-up approaches have been described in the literature, including mechanical agitation,9 extrusion,10 atomization,11 and high-pressure homogenization,12 among others. However, microfluidics remains a highly popular technique for their fabrication due to: (i) high control of the size and shape of the microspheres (MPs) during their production;13 (ii) high encapsulation efficiency depending on the droplet generation-caused shear forces;14 (iii) continuous production, high batch-to-batch homogeneity,15 easily maintenance of sterile conditions; and (iv) in the case of cell encapsulation, even accurate control of the number of encapsulated cells per microsphere.16,17 Microfluidic devices can be produced by different materials, such as glass, polydimethylsiloxane (PDMS) or silica. However, glass remains highly convenient due to its high compatibility with organic solvents, easy manufacturing of devices compared to the other materials and low cost.18–20
Different gelation and crosslinking strategies have been studied for the fabrication of MPs depending on the used precursor materials,21 the production method,22 and the applicability of the hydrogel.23 For example, common physical crosslinking methods include ionic crosslinking and thermal crosslinking.24 Ionically crosslinked MPs, formed by a polyelectrolyte polymer solution crosslinked after exposition to counter ions, have been extensively studied25–27 and are usually characterized by their limited gelation28 and high burst release.29 Thermally crosslinked MPs are formed without any additional agent due to temperature-based induction of hydrogen bonds, van der Waals forces and hydrophilic/hydrophobic interactions.4 On one hand, the poor mechanical properties of the systems and the difficulties to apply temperature control systems in commonly used MPs production methods, such as microfluidics, remain their main disadvantages.30 On the other hand, chemical crosslinking using crosslinking agents and initiators remains a common alternative to produce MPs with better mechanical properties.31,32 Photo-induced crosslinking has recently gained attention due to the addition of photo-initiators,33 its fast kinetics and the easiness to combine photo-inducing systems with MPs production methods. However, the exposure to UV-light can be incompatible with cells or biological agents.34 Click chemistry remains a common alternative for the synthesis of covalently crosslinked hydrogels due to its high chemoselectivity, fast kinetics and high efficiency in intricate aqueous media under physiological pH and temperature conditions.35 Among different biorthogonal click copper-free reactions alternatives that have been described, inverse electron demand Diels–Alder (iEDDA) reaction between tetrazine and a suitable dienophile, such as norbornene becomes a highly promising option.36 iEDDA has considerably fast kinetics without the need of additional initiators or catalysts, considerably simple synthetic routes, no cross-reactivity with possible payloads such as proteins and high compatibility with cell encapsulation.37
To the best of our knowledge, no other studies have focused on the development of iEDDA-based hydrogel MPs by microfluidics technique. Because of inherent difficulties associated with the development of MPs via microfluidics, the use of an iEDDA-crosslinked system remains challenging due to its fast gelation kinetics. However, here we hypothesize that it is possible to use microfluidics to form iEDDA-crosslinked hydrogel MPs with suitable mechanical properties, swelling behaviour and biocompatibility.
The aim of this work was to develop a monodisperse in situ gellable iEDDA-crosslinked poly(ethylene glycol) (PEG)-based hydrogel38,39 MPs via water-in-oil emulsification (W/O)40 using glass microfluidics to form aqueous droplets where the corresponding polymers precursor solutions crosslink.4 For this, two different forms of PEG were used as precursor polymers and modified with either tetrazine or norbornene as functional groups. Using co-flow glass microfluidics,41 MPs were formed and, subsequently, their morphological, physicochemical, rheological and swelling properties were studied, as well as their use as potential drug delivery and cell encapsulation systems.
Experimental section
Materials
4-Arm 20 kDa polyethylene glycol-amine (PEG-amine, Creative PEGWorks USA), 5-[4-(1,2,4,5-tetrazin-3-yl)benzylamino]-5-oxopentanoic acid (Tz-COOH, Sigma Aldrich, USA), 1-methyl-2-pyrrolidinone (NMP, Sigma Aldrich, USA), triethylamine (Sigma Aldrich, USA), N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uranium hexafluorophosphate (HBTU, Sigma Aldrich, USA), diethyl ether (Sigma Aldrich, USA), 6 kDa linear hydroxyl polyethylene glycol (PEG-OH, Sigma-Aldrich, USA), 4-(dimethylamino) pyridine (Sigma-Aldrich, USA), pyridine (Tokyo Chemical Industry, Japan), mixture of endo and exo with predominant endo form of 5-norbornene-2-carboxylic acid (Sigma-Aldrich, USA), diisopropylcarbodiimide (Sigma-Aldrich, USA), deuterated dimethyl sulfoxide-d6 (DMSO, Sigma-Aldrich, USA), paraffin oil (Sigma-Aldrich, USA), Span 80 (Sigma-Aldrich, USA), Scigen O.C.T. compound cryostat embedding medium (Fisher Scientific, USA), 10× phosphate buffer solution (PBS, HyClone, GE Healthcare Lifesciences, USA), p-nitrophenyl acetate (p-NPA, Sigma-Aldrich, USA), Hank's balanced salt solution (HBSS, Termofisher Scientific, USA), sodium chloride (Sigma-Aldrich, USA), potassium chloride (Sigma-Aldrich, USA), calcium chloride (Sigma-Aldrich, USA), sodium bicarbonate (Sigma-Aldrich, USA), monosodium phosphate (Sigma-Aldrich, USA), magnesium chloride (Sigma-Aldrich, USA), 2-(N-Morpholino)ethanesulfonic acid (MES, Sigma-Aldrich, USA), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES, Sigma-Aldrich, USA), fetal bovine serum (FBS, Gibco, USA), penicillin and streptomycin (PEST, Gibco, USA), non-essential aminoacids (HyClone, GE Healthcare Lifesciences, USA), 1-glutamine 200 mm (HyClone, GE Healthcare Lifesciences, USA), trypsin (2.5%) (HyClone, GE Healthcare Lifesciences, USA), Triton X-100 (Merck Millopore, Germany), bovine serum albumin (BSA, Sigma-Aldrich, USA), bicinchoninic acid protein quantification assay (BCA, Sigma-Aldrich, USA), Dulbeccos's modified Eagle's medium (DMEM, Gibco, USA), CellTiter-Glo® luminescent assay (Promega, USA), and Live/Dead cell assay (Thermo Fisher Scientific, USA).PEG-tetrazine and PEG-norbornene synthesis
The 4-arm 20 kDa PEG-amine was functionalized with Tz-COOH to obtain 4-arm PEG-tetrazine (PEG-Tz) following previously described procedures with some modifications.42 Firstly, in a dry and argon purged round-bottom flask, 100 mg 4-arm 20 kDa PEG-amine (0.005 mmol, 0.02 mmol –NH2) were dissolved in 5 mL of NMP and mixed with 5.55 μL triethylamine (2× to –NH2, 0.04 mmol) during 15 min under argon atmosphere. Separately, 15 mg of Tz-COOH (2.5× to –NH2, 0.05 mmol) were dissolved in 2.5 mL NMP and activated with 18.96 mg of HBTU (2.5× to –NH2, 0.05 mmol) in another dry and argon purged container during 5 min. Subsequently, the activated Tz-COOH was added to the initial PEG-amine mixture and the functionalization was conducted during 15 h at room temperature under argon atmosphere. The obtained product was precipitated in 10-fold volume excess of cold diethyl ether (−20 °C), centrifuged, dried overnight under vacuum, dialyzed against ultrapure water for 48 h and lyophilized.6 kDa PEG was functionalized with norbornene acid to obtain PEG-norbornene (PEG-Nb) following previously described procedures with some modifications.42 Firstly, 5 g 6 kDa PEG (0.83 mmol, 0.83 mmol –OH), 50.91 mg 4-(dimethylamino) pyridine (0.5× to –OH, 0.4167 mmol) and 0.34 mL pyridine (5× to –OH, 4.17 mmol) were added in a dry and argon purged round-bottom flask and dissolved in 15 mL anhydrous dichloromethane. In another dry and argon purged vessel, 1.02 mL 5-norbornene-2-carboxylic acid (10 COOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 –OH, 8.33 mmol) were dissolved in 10 mL anhydrous dichloromethane and mixed with 0.65 mL diisopropylcarbodiimide (0.5× to Nb, 4.17 mmol) during 45 min at room temperature under argon atmosphere to obtain a dinorbornene anhydride. The obtained product was filtered to remove the resultant urea salts and, subsequently, added to the original mixture containing PEG. The functionalization was conducted overnight at room temperature and under argon atmosphere. The obtained product was precipitated in 10-fold volume excess of cold diethyl ether (−20 °C), centrifuged, dried overnight under vacuum, dialyzed against ultrapure water for 48 h and lyophilized.
:1 –OH, 8.33 mmol) were dissolved in 10 mL anhydrous dichloromethane and mixed with 0.65 mL diisopropylcarbodiimide (0.5× to Nb, 4.17 mmol) during 45 min at room temperature under argon atmosphere to obtain a dinorbornene anhydride. The obtained product was filtered to remove the resultant urea salts and, subsequently, added to the original mixture containing PEG. The functionalization was conducted overnight at room temperature and under argon atmosphere. The obtained product was precipitated in 10-fold volume excess of cold diethyl ether (−20 °C), centrifuged, dried overnight under vacuum, dialyzed against ultrapure water for 48 h and lyophilized.
Both obtained polymers were dissolved in deuterated DMSO and characterized by 1H-NMR to study the performed functionalization with an Ascend 400 MHz NMR spectrometer (Bruker, Switzerland). The functionalization degree of both polymers was calculated based on the area associated with specific protons of the added moieties.
Fabrication of polymeric hydrogel microspheres via microfluidic technique
MPs were fabricated by single emulsion in a glass capillary microfluidics chip with co-flow geometry.43 The microfluidics platform was designed based on two different glass capillaries aligned in a co-flow geometry. Firstly, the inner glass capillary (outer diameter 1.00 mm; 1B100-6, World Precision Instruments, USA) was shaped using a micropipette puller (P-97, Sutter Instrument, USA) and meticulously sanded obtaining a final inner diameter of approximately 100 μm. The inner capillary was inserted in the left side of an outer glass capillary (with an inner diameter of 1.10 mm; T150-6, World Precision Instruments, USA) and coaxially aligned.44For the preparation of the microspheres, different polymer concentrations were studied always maintaining a 1:1 ratio between the two reactive groups based on the obtained modification degrees. Firstly, two different aqueous inner phases between the two precursor polymers were prepared. PEG-Tz and PEG-Nb in 1× PBS precursor solutions were connected to a T-junction capillary, which was connected to the inner capillary of the glass microfluidics chip. Both precursor solutions were mixed and subsequently injected through the inner glass capillary, whereas a paraffin oil 1% Span 80 solution was simultaneously injected from the outer glass capillary. A 3D coaxial flow was obtained thanks to the injection of both phases in the same direction. Flow rates were set at 200 μL h−1 for both inner phases, at 20 mL h−1 for the outer phase and remained constant during the entire process. The spherically shaped droplets of polymer precursors immersed in an oil outer phase were obtained from the microfluidics platform and suspended in a recollection bath under 50 rpm shaking for their crosslinking. Subsequently, MPs were centrifuged at 95g (Hettich EBA 21, Germany) for 5 min, the oil and surfactant traces were aspirated, and the remaining MPs were immersed in 1× PBS. This washing and centrifuging procedure was repeated thrice to ensure the complete removal of oil and surfactant traces. Finally, the obtained MPs were stored at 4 °C until further use.
Morphological and physicochemical evaluation
MPs were morphologically studied using light microscopy (EVOS cell imaging system, Thermo Scientific, USA). For the measurements, MPs were initially immersed in 1× PBS. Characterization of each batch was based on the measurement of the diameter of 100 MPs using ImageJ software. Moreover, the coefficient of variation of each batch was subsequently calculated according to eqn (1): | (1) |
MPs external morphology and inner structure were also characterized via scanning electron microscopy (SEM Quanta FEG 250, FEI, USA) at an accelerating voltage of 4.0 kV. For the study of the external morphology, samples were dried overnight at room temperature under vacuum and, afterwards, placed over a silica substrate and coated with a 10 nm platinum layer. For the study of the inner structure without lyophilizing the samples, MPs were immersed in cryostat embedding medium and frozen in liquid nitrogen. Subsequently, samples were sectioned in 15 μm thin segments (Leica Cryostat CM1950, Leica Biosystems, Germany), placed over a silica substrate and coated with a 10 nm platinum layer.
Melting temperatures of both precursor polymers and freeze-dried MPs were also studied by differential scanning calorimetry (DSC, AG-DSC823E, Mettler Toledo, Switzerland) following a ramping up method at 1 °C min−1 from 0 to 200 °C under a nitrogen atmosphere.
Freeze-dried MPs were characterized by attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy (Bruker Vertex 70, Bruker, USA). FTIR spectra were recorded in the range of 5000–500 cm−1 with a resolution of 2 cm−1 using OPUS 8.1 software.
Rheological studies
The rheological behaviour of both bulk hydrogel and hydrogel MPs was studied (TA Instruments DHR-2rheometer, TA Instruments, USA) with a 20 mm diameter stainless steel plate-and-plate (parallel plate) geometry. In the case of the bulk hydrogels, prior monitoring the gelation, both precursor polymers solutions were quickly mixed and injected above the rheometer plate, allowing the gel to cure. The MPs were transferred to the rheometer plate by scooping. Different types of test methods were used: strain sweep, frequency sweep and time sweep. During the strain sweep, the moduli of the studied hydrogels were studied under a gradual strain variation from 0.1% to 1000% maintaining temperature and frequency constant at 25 °C and 1 Hz. During the frequency sweep, the variation of G′ and G′′ values of the studied hydrogels was studied modifying the applied oscillatory frequency from 20 to 0.1 rad s−1 under a constant strain (1%) within the linear viscoelastic range at 25 °C. Finally, for the time sweep, the measurement of precursor solutions of the hydrogel (PEG-TZ and PEG-NB) placed on the lower plate was started instantly and the variation of G′ and G′′ values was recorded under constant strain (1%), angular frequency (1 Hz) and temperature (25 °C).Swelling studies
MPs were immersed in synovial fluid mimicking buffer at 37 °C, as previous described.45 Briefly, synovial fluid mimicking buffer was composed by HBSS, 0.14 M of NaCl, 5.4 × 10−3 M of KCl, 1.62 × 10−3 M of CaCl2, 4.16 × 10−3 M of NaHCO3, 2.7 × 10−3 M of Na2HPO4·2H2O, 0.49 × 10−3 M of MgCl2·6H2O, and 4 mg mL−1 bovine serum albumin. 2 different buffers were prepared according to two different pH conditions, pH 5.5 and pH 7.4. MPs were immersed in the corresponding buffer and at specific time-points they were observed using optical microscopy (EVOS cell imaging system, Thermo Scientific, USA). Afterwards, their diameter was measured using ImageJ software and the mean diameter size and size distributions were subsequently calculated. Moreover, the swelling index was calculated according to eqn (2): | (2) |
Protein encapsulation, release studies and protein activity studies
BSA-loaded hydrogel MPs were prepared according to the protocol for the production of MPs described above. BSA was dissolved in both precursor polymer solutions and MPs were prepared as previously. The entrapment efficiency and loading degree of the developed MPs were determined by dissolving the loaded samples in a mildly acidic phosphate buffer with pH 5.8. After the complete degradation of the samples, the amount of BSA in the supernatant was determined using a BCA protein assay kit. The entrapment efficiency (EE) and loading degree (LD) values were calculated according to eqn (3) and (4): | (3) |
 | (4) |
In vitro release studies were performed at two different pH conditions, 5.5 and 7.4. BSA-loaded MPs were immersed in 5 mL of HBSS-HEPES (pH 7.4) and HBSS-MES (pH 5.5) at 37 °C. 200 μL of the release medium were removed at specific time points, from 5 min up to 48 h. After the removal of release medium at each time point, equal volume of fresh buffer was immediately added.
Subsequently, the release data was analysed according to different kinetic models including zero-order, first-order, Higuchi and Korsmeyer-Peppas models. Each of these models was calculated following eqn (5), (6), (7) and (8):
 | (5) |
 | (6) |
 | (7) |
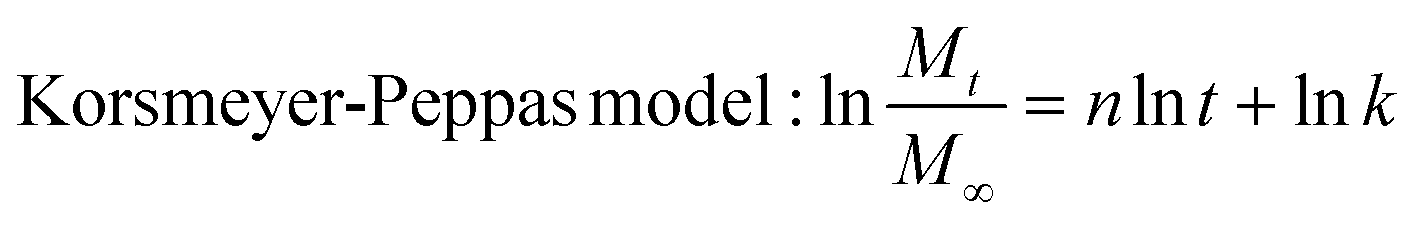 | (8) |
Esterase-like activity of loaded BSA was studied following previously described procedures.46 Briefly, p-nitrophenyl acetate (p-NPA) was dissolved in methanol and diluted with ultrapure water (1:100 ratio) with a final concentration of 3.5 mM. After determining the amount of BSA in the supernatant of each sample using a BCA protein assay kit, all released BSA samples immersed in HBSS–HEPES were diluted to obtain a final BSA concentration of 3 μM. Esterase-like activity was studied using 100 μL of 3 mM solution of p-NPA and 400 μL of sample with a BSA concentration of 3 μM. Afterwards, the absorption band at 400 nm was studied until no further changes could be observed with a Varioskan™ LUX multimode microplate reader (Thermo Scientific, USA). Esterase-like activity of unloaded BSA obtained from the original batch was used as control. The degree of esterase-like activity (EA) values were calculated, according to eqn (9):
 | (9) |
Cell culture
RAW 264.7 cells were obtained from the American Type Culture Collection (ATCC, USA). Cells were grown and maintained in DMEM supplemented with 10% (v/v) FBS, 1% (v/v) NEAA, 1% (v/v) L-glutamine and 1% (v/v) penicillin and streptomycin in a humidified incubator (BB16 gas incubator, Heraeus Instruments GmbH, Germany) at 37 °C, 5% CO2 and 95% relative humidity. Seeding density was 2 × 105 cells per mL. Cell medium was replaced every 2 days.Human primary tenocytes were enzymatically isolated from the human extensor indicis proprius, as previously described.88 Cells were grown and maintained in DMEM supplemented with 10% (v/v) FBS, 1% (v/v) penicillin and streptomycin and 200 μM of Ascorbic acid in a humidified incubator (BB16 gas incubator, Heraeus Instruments GmbH, Germany) at 37 °C, 5% CO2 and 95% relative humidity. Seeding density was 5 × 103 cells per cm2. Cell medium was replaced every 2 days. Patient’s recruitment, participation, and sample collection were obtained after receipt of a signed informed consent, approved by the Helsinki and Uusimaa Hospital District ethics committee (HUS/2785/2020) and by the institutional review board (HUS/234/2020)
Cytotoxicity studies
For cytotoxicity studies, RAW macrophages of passages #5–#10 and human tenocytes of passages #4 and #5 were seeded in a 96-well plate at 1 × 104 cells per well. After overnight attachment, the 10% PEG-TZ MPs suspended in triplicate in the appropriate cell medium were added at concentrations of 5, 25, 100, 250, 500 and 1000 μg mL−1, and complete medium and Triton X-100 were used as negative and positive controls. After 24 and 72 h, the cell viability was measured using CellTiter-Glo® luminescent assay and the luminescence was measured using a Varioskan™ LUX multimode microplate reader (Thermo Scientific, USA).Cell encapsulation
For cell encapsulation, all the used equipment was washed with 70% ethanol followed by UV sterilization for 24 h. Both aqueous inner phases between the two precursor polymers were prepared and sterilized by filtering through a 0.20 μm syringe filter. RAW cells of passages #5–#10 were suspended with a concentration of 20 × 106 cells per mL with both precursor polymers solutions in 1′ HBSS. The formation of the 10% PEG-TZ followed the same procedure, as previously described, and the formed MPs were collected in a 1% Span 80 paraffin oil recollection bath under gentle shaking. Subsequently, MPs were centrifuged at 95g for 5 min, the oil and surfactant traces were aspirated, and the remaining MPs were immersed in cell culture media. This procedure was repeated in triplicate to ensure the complete removal of oil and surfactant traces. Finally, the obtained MPs were maintained in an incubator at 37 °C and 5% CO2.Cell viability studies
The cell viability of encapsulated RAW after 1 and 7 days were determined by Live/Dead cell staining. The MPs were washed with PBS 1× and immersed in culture medium with 4 μM calcein AM and 2 μM EthD-1 without FBS. Subsequently, samples were incubated during 45 min at 37 °C and 5% CO2. MPs were observed by a Leica TCS SP8 STED confocal microscope (Leica Microsystems, Germany). Cell viability values were calculated based on the percentage of live cells observed in 15 different MPs per condition. Moreover, the total number of cells observed in each MP was counted and subsequently the difference between the total number of observed cells after 1 and 7 days was studied.Statistical analysis
The results of all the experiments are presented as the mean values (±SD) of triplicate samples made in three technical and independent experiments. For cell viability studies, a one-way ANOVA followed by a Turkey-Kramer post hoc test was used for the statistical analysis. The significance levels of the differences were set at the probabilities of *p < 0.05, **p < 0.001 and ***p < 0.0001 for comparison. For cell counting studies, a two-tailed Student's t-test was used for the statistical analysis. The significance levels of the differences were set at the probabilities of *p < 0.05, **p < 0.01, ***p < 0.0005 and ****p < 0.0001.Results and discussion
PEG-tetrazine and PEG-norbornene synthesis and fabrication of polymeric hydrogel microspheres
Both polymers used for the preparation of MPs (4-arm PEG-TZ and PEG-NB) were synthesized, following the previously described protocols in the literature32,42 The structures of both modified precursors were studied by 1H-NMR (Fig. S1 and S2).† Based on the obtained spectra and in accordance with literature, 4-arm PEG-TZ showed a modification degree of ∼85%, showing a series of peaks at δ 10.5 ppm, 8.4 ppm and 7.8 ppm associated with protons of the tetrazine group.35 Moreover, PEG-NB showed a modification degree of ∼76%, based on a series of peaks at δ 6.1 ppm and 5.9 ppm, corresponding to different protons of the norbornene group.47,48The effect of both inner and outer phases flow rates in the microfluidic setup (Fig. 1A) on the size and polydispersity of the obtained MPs was studied. In order to do so, we prepared different formulations maintaining fixed the mineral oil outer phase flow rate and varying the inner phase flow rate from 50 to 500 μL h−1. Different outer phase flow rates from 10 to 75 mL h−1 were studied. In each case, the obtained MPs were observed by the light EVOS microscope (Fig. 1B and C), the MPs diameter was measured using ImageJ software and the coefficient of variation was subsequently calculated.
 | ||
| Fig. 1 (A) Schematic representation of the production of the TZ-NB crosslinked MPs formed via W/O emulsification using microfluidics with an inner aqueous phase with the precursor polymers, and an outer oil phase. Two different forms of PEG were used as precursor polymers and modified with either tetrazine or norbornene as functional groups. The co-flow glass microfluidics device consisted on an inner capillary with an inner diameter of 1 mm and a 100 μm diameter tip, inserted in the left side of an outer glass capillary, with an inner diameter of 1.10 mm. (B) Optical images obtained by the light microscope during the MPs production process. Scale bar 1000 μm. (C) Optical images of the obtained MPs by the light microscope. Scale bar 1000 μm. | ||
Overall, higher inner phase flow rates under a fixed outer phase flow rate were correlated with MPs with higher diameter sizes without significant differences in their polydispersity index. Moreover, higher outer phase flow rates under a fixed inner phase flow rate were correlated with MPs with lower diameter sizes and also no statistically significant changes in their polydispersity was observed. A single microfluidics platform was used to obtain MPs with sizes ranging from 225 μm to 625 μm (Fig. 2 and S3†). For instance, for a fixed inner phase flow rate of 100 μL h−1, the diameter size of the obtained MPs decreased from 530 μm to 225 μm only by increasing the outer phase flow rate from 10 mL h−1 to 50 mL h−1. However, for a fixed outer phase flow rate of 20 mL h−1, the diameter size of the obtained MPs increased from 330 μm to 580 μm just by increasing the inner phase flow rate from 50 μL h−1 to 500 μL h−1.
 | ||
| Fig. 2 Optimization diagram including the MPs, size and the inner flow and the outer flow rates values. | ||
Morphological and physicochemical evaluation
SEM images of the MPs (Fig. 3A) showed an external uniform surface without the presence of visible pores. SEM images from the inner structure (Fig. 3B and C) of the MPs showed a highly porous structure with pore sizes ranging from 10 μm to 20 μm, in accordance with other similar reported systems.49 We hypothesize that the highly porous structure can be explained by the release of gaseous nitrogen as the inert byproduct of the iEDDA reaction between TZ and NB, as other iEDDA-crosslinked hydrogels have previously shown.50,51 Moreover, considering lyophilization is also responsible for the pore formation, non-lyophilized wet bulk hydrogels were observed by the light microscope and numerous bubbles located within the hydrogel matrix were observed, confirming that the production of bubbles was caused by the released nitrogen gas produced during the iEDDA reaction. | ||
| Fig. 3 (A) SEM image of the obtained MPs. Scale bar 100 μm. (B) SEM image of the inner structure of the MPs. Scale bar 100 μm. (C) SEM image of the porous inner structure of the MPs. Scale bar 50 μm. | ||
Characterization studies were conducted using DSC analysis. Fig. 4A shows a sharp endotherm peak at 46 °C in the case of 4-arm PEG-TZ and another peak at 70 °C in the case of PEG-NB, corresponding in both cases to the melting temperature of both precursor polymers.52 These sharp endotherm peaks were not observed in the formed MPs, confirming a change in polymer microstructure after the crosslinking of both precursor polymers. This effect is widely observed in chemically crosslinked hydrogels due to the inability of the precursor polymers to crystallize after their crosslinking due to their immobilization into the hydrogel structure, limiting its mobilization and crystallization.53–55
 | ||
| Fig. 4 (A) DSC spectra of 4-arm PEG-TZ, PEG-NB and the MPs. The measurement was conducted following a ramping up method at 1 °C min−1 from 0 to 200 °C under a nitrogen atmosphere. (B) ATR-FTIR spectra of 4-arm PEG-TZ, PEG-NB and the MPs. FTIR spectra were recorded in the range of 5000–500 cm−1 with a resolution of 2 cm−1. | ||
The chemical composition of the MPs was then analyzed using ATR-FTIR. Fig. 4B confirmed the presence of 4-arm PEG-TZ in the MPs, specifically the band observed at 1670 cm−1 corresponds to the double bound between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N from the tetrazine group.56 Moreover, the band around 2850 cm−1 corresponds to the C–H stretching and the band around 1080 cm−1 corresponds to the C–O stretching in accordance with the structure of both original polymers.57 In the case of PEG-NB, the band at 1480 cm−1 confirmed the presence of the CC bond from norbornene, apart from the previously mentioned bands associated with the structure of the original polymer.58 After crosslinking, a noticeable reduction of the band at 1670 cm−1 was observed, corresponding to the elimination of one CN bond associated with the coupling of both moieties and the release of gaseous nitrogen. Altogether, these results suggest the successful crosslinking within MPs’ structure.
N from the tetrazine group.56 Moreover, the band around 2850 cm−1 corresponds to the C–H stretching and the band around 1080 cm−1 corresponds to the C–O stretching in accordance with the structure of both original polymers.57 In the case of PEG-NB, the band at 1480 cm−1 confirmed the presence of the CC bond from norbornene, apart from the previously mentioned bands associated with the structure of the original polymer.58 After crosslinking, a noticeable reduction of the band at 1670 cm−1 was observed, corresponding to the elimination of one CN bond associated with the coupling of both moieties and the release of gaseous nitrogen. Altogether, these results suggest the successful crosslinking within MPs’ structure.
Rheological studies
The rheological properties of the MPs were studied and compared to the hydrogels formed by the bulk method. All the samples were prepared and allowed to swell in 1× PBS (pH of 7.4) for 24 h. Both types of samples were characterized through strain and frequency sweep measurements. Moreover, in the case of the bulk hydrogel, time sweep measurements were conducted to study the gelation kinetics of the system. In the bulk hydrogels, the strain sweeps (Fig. 5A) showed a stable behaviour up to strains of 10%. The values of the storage modulus G′ increased with increasing the precursor polymers concentration in the formulation. Subsequently, the frequency sweeps (Fig. 5B) were executed within the linear viscoelastic range. The observed G′ values remained constant for most part of the measurements, in agreement with the expected behaviour in covalently crosslinked hydrogel networks.59,60 Moreover, the obtained G′ values remained similar to other hydrogel systems formed by PEG-derivatives with the same type of crosslinking with G′ values around 1349 ± 345 Pa for 10% PEG-TZ and 2978 ± 217 Pa for 20% PEG-TZ.61,62 | ||
| Fig. 5 (A) Strain sweep of bulk hydrogel with 5% PEG-TZ, 10% PEG-TZ and 20% PEG-TZ. (B) Frequency sweep of bulk hydrogel with 5% PEG-TZ, 10% PEG-TZ and 20% PEG-TZ. | ||
Time sweep measurements (Fig. 6) of both precursor polymer solutions revealed that the gelation started to increase the storage modulus at around 120 ± 11 s after the precursor polymers were exposed to each other. The G′ values were observed to increase until 10 min later, corresponding to the complete gelation of the system and are in accordance with other bulk hydrogel systems with the same crosslinking method which describe the same behaviour after 144 s.63 Despite that the gelation kinetics can be tuned by modifying the substitution degree of both polymers, the observed gelation kinetics are highly favourable because they allow both precursor polymers to quickly mix in the microfluidic platform and form the MPs before the hydrogel matrix crosslinks.
 | ||
| Fig. 6 Time sweep of bulk hydrogel 10% PEG-TZ. | ||
Fig. 7A shows the rheological properties of the MPs. In the case of the strain sweep measurements, all the conditions were maintained in comparison with the measurements performed in bulk hydrogels. MPs also showed a stable behaviour for most part of the measurements with no statistically significant differences in terms of G′ values between both formulations with different precursor polymers concentrations. However, the obtained G′ values were considerably lower, in this case the G′ values were around 950 Pa, compared to the G′ values of the bulk hydrogels formed by the same precursor polymers. This can be explained by the different morphology of both samples, considering that the bulk system constitutes a homogeneously crosslinked polymer matrix, whereas MPs constitute a succession of individually formed micro-sized spheres. Moreover, frequency sweeps (Fig. 7B) showed constant G′ values and confirmed the differences previously observed between the storage modulus of the two types of samples. Overall, MPs showed the same rheological behaviour with lower storage modulus values in comparison to the bulk hydrogel system. The observed properties were comparable to other covalently crosslinked microspheres, such as vinylpiridine and divinylbenzene MPs.64
 | ||
| Fig. 7 (A) Strain sweep of MPs with 10% PEG-TZ and 20% PEG-TZ. (B) Frequency sweep of MPs with 10% PEG-TZ and 20% PEG-TZ. | ||
Swelling studies
Next, we evaluated the swelling behaviour of the developed systems. The swelling ratio and degradability remain highly important properties with a high influence in the mechanical properties and the loading and release profile of cargo molecules. We investigated how the developed formulations with different precursor polymer concentrations experienced volumetric swelling. For this, we exposed MPs to a synovial fluid mimicking buffer,45 as a physiologically mimicking model with high polyelectrolyte and protein content, and two different pH conditions, 5.5 and 7.4, a slightly acidic buffer mimicking the inflammatory environment, and the physiological environment respectively. Subsequently, we measured the MPs diameter at different times for 8 days and compared it to the initial diameter before immersion.Fig. 8A and B show that in both pH conditions tested the MPs had a fast increase in their swelling index during the first 5 min. At pH 7.4, both formulations of 10% and 20% 4-arm PEG-TZ showed mainly constant swelling index values between 0.2 and 0.3 during the 8 days. No apparent signs of volume changes or signs of degradation were observed based on the obtained images, following similar iEDDA-crosslinked PEG-based formulations with prolonged hydrolytic stability65,66 and similarly to poly-L-ornithine-coated alginate MPs, which own swelling index values below 0.75 under the same conditions.67–70 However, at pH 5.5, both formulations showed a different behaviour, characterized by a gradual increase in their swelling index until the complete degradation of the MPs, only observed after 8 days. Major differences were observed between formulations with either 10% or 20% of 4-arm PEG-TZ concentration, leading to an increase in the swelling index values for the formulations with lower polymer content. The different degradation behaviour may be explained by the hydrolysis of esters bonds of PEG-NB, which has been described to occur faster in acidic pH conditions by protonation of the ester bond at the carbonyl oxygen.42,71 This phenomenon might even be enhanced in MPs in comparison with the original bulk hydrogel system due to spherical morphology of the MPs, thus increasing the surface area of the system in comparison with the original bulk hydrogel.
 | ||
| Fig. 8 Swelling studies in MPs with 10% PEG-TZ and 20% PEG-TZ at 37 °C under pH 5.5 and 7.4. (A) Swelling index of 10% PEG-TZ and 20% PEG-TZ MPs at pH 5.5 and 7.7 for 8 days. (B) Swelling index of 10% PEG-TZ and 20% PEG-TZ MPs pH 5.5 and 7.4 for the initial 200 min. | ||
Overall, the developed MPs showed a low swelling behaviour and no degradation signs under the tested physiological conditions, with both a high and quick swelling profile under mildly acidic conditions followed by gradual degradation after 8 days.
Protein encapsulation, release studies and protein activity studies
The possibility to use the developed MPs as protein delivery carriers was also evaluated. We studied the loading and release of BSA as a model protein. The main advantage of MPs is based on their high drug loading potential compared to other microdevices. For the preparation of the BSA-loaded MPs, BSA was added and mixed with both precursor polymer solutions, and subsequently the MPs were prepared as usual. The calculated entrapment efficiency and loading degree values were 87.17% and 4.36%, respectively, with SD values of 2.41 and 0.248 respectively. These loading values were mainly attributed to the high hydrophilic nature of the loading cargo immersed in the hydrophilic matrix and the rapid crosslinking of the MPs after their formation via single emulsion.The BSA release profile was studied at two different pH conditions, 5.5 and 7.4, a slightly acidic buffer mimicking the inflammatory environment and physiological conditions, with two different precursor polymer concentrations. In both conditions, the MPs showed a burst release during the first 6 h (Fig. 9A), regardless of their polymer concentration. At pH 7.4, 10% and 20% of 4-arm PEG-TZ MPs already released 90% and 80%, respectively, of BSA after 1 day, followed by a sustained release of the remaining 10% and 20% during the following days. At pH 5.5, no statistically significant differences were observed compared with the previously tested conditions, as 10% and 20% 4-arm PEG-TZ MPs released a slightly higher amount of BSA, 95% and 87%, respectively, of the cargo after 1 day. Following the release studies, the obtained data during the first 6 hours was analysed based on different kinetic models, including zero order, first order, Higuchi and Korsmeyer-Peppas. Table S1† shows that, among the different models, the most fitting model with the highest linearity (R2) for both conditions and types of formulations was Korsmeyer-Peppas, with release exponent (n) values lower or equal to 0.45. Therefore, the analysis confirms that the initial release was controlled by a Fickian diffusion mechanism.72 These results show a similar release profile in comparison with other alginate and poly-L-lysine-alginate based MPs also used for protein encapsulation which similarly show predominant protein release during the initial 3 h.73–77
 | ||
| Fig. 9 (A) BSA release profiles from MPs with 10% PEG-TZ and 20% PEG-TZ at 37 °C under pH 5.5 and 7.4. (B) Esterase-like activity of released BSA using p-NPA, which is converted to p-NP, at 2, 4, 6, 24 and 48 hours in comparison with the initial esterase-like activity of unloaded BSA. The absorption of the nitrophenolate ion was measured in the UV-Vis band around 400 nm. | ||
Overall, the system showed a high burst release behaviour, which may be attributed to a high diffusion coefficient of the loaded carrier thanks to the highly porous inner structure, conditioned by the polymer molecular weight and crosslinking density, and the highly hydrophilic nature of the MPs, as it has been previously described in other similar systems which release more than the 80% of the protein content within 24 hours.78,79 Considering that the release of the encapsulated cargo was due to diffusion, the use of alternative PEG-based precursor polymers with different molecular weight and polymer concentration remains a different option to modify the mesh size of the hydrogel matrix from MPs and study any possible changes regarding protein encapsulation and release kinetics.80,81
After the release studies, esterase-like activity of loaded BSA was studied using p-NPA, which is converted to p-NP. This reaction can be investigated by studying the absorption of the nitrophenolate ion in the UV-Vis band around 400 nm. Nitrophenolate ion acquires a yellow tonality at pH values above its pKa = 7.08 at 22 °C.46,82 The absorption of released BSA samples at different time points was compared with the absorption of unloaded BSA from the original batch. Fig. 9B shows that released BSA at 2, 4, 6, 24 and 48 hours maintained a high percentage of its original esterase-like activity, around an 80%. This confirms that the loading of protein molecules into the hydrogel matrix of MPs does not cause major changes in their original activity, considering the non-cross-reactivity nature of iEDDA chemistry with biological moieties.
Cytotoxicity studies
Following the characterization studies above, we next studied the cytotoxicity of the developed MPs on RAW 264.7 macrophages and primary human tenocytes as model cell lines. The assay was performed using Cell-Titer Glo® luminescent assay after 24 h and 72 h of exposure to different concentrations of MPs, ranging from 5 μg mL−1 to 1000 μg mL−1 (Fig. 10A–B and S5†). No statistically significant differences in cytotoxicity were observed for any of the tested concentrations after 24 and 72 h exposure to the developed MPs. These results are in agreement with other PEG-based systems, which have been described as highly biocompatible, and with iEDDA-crosslinked hydrogels, considering that the two used chemical moieties do not own cross-reactive behaviour with other chemical moieties belonging to living cells,83 as well as other highly biocompatible common systems such as poly-L-lysine-alginate MPs.84 | ||
| Fig. 10 Cell proliferation of murine RAW 264.7 macrophages relative to control after the incubation with MPs’ concentrations from 5 μg mL−1 to 1000 μg mL−1 at time-points of 24 h (A) and 72 h (B). The results were plotted as mean ± standard deviation (n = 3 biological replicates in which each time three technical replicates have been used). A one-way ANOVA followed by a Turkey-Kramer post hoc test was used for the statistical analysis for comparison with the medium, which was used as a control in all tests. The significance levels of the differences were set at the probabilities of *p < 0.05, **p < 0.001 and ***p < 0.0001. | ||
Cell encapsulation studies
Next, we studied the use of the developed MPs for the encapsulation of living mammalian cells, such as RAW 264.7 macrophages. Fig. 11A–E and S6† show the cytotoxicity studies and the cell viability using a live/dead cell staining analysed by confocal microscopy. Based on the percentage of live cells observed in the images, a 74.0 ± 5.4% of cell viability was observed one day after the preparation of the MPs. After 7 days of culture, 70.0 ± 4.4% of viable cells were detected, and the cells maintained a rounded morphology. The observed cell morphology could be explained due to the lack of adhesion motifs in the polymer matrix and the extremely low degradation of the network, considering that many other similar systems own a faster degradation behaviour,85 which allows the encapsulated cells to spread and migrate through the hydrogel network.86 We hypothesize that the MPs fabrication process is responsible for the observed decrease of cell viability, probably due to the exposure to paraffin oil during the production process and the applied pressure associated with the used flow rates in the microfluidics platform. | ||
| Fig. 11 Representative Live/Dead z-stack confocal images of RAW cells encapsulated in 10% of PEG-TZ MPs at 37 °C. Calcein AM positive cells (green) represent live cells and Ethidium homodimer-1-stained cells (red) represent dead cells. (A and B) 1 day after encapsulation. Scale bar 50 μm (C) 3D z-stack image of RAW 264.7 macrophages encapsulated in 10% PEG-TZ MPs 1 day after encapsulation. Scale bar 100 μm. (D and E) 7 days after encapsulation. Scale bar 50 μm. (F) 3D z-stack image of RAW 264.7 macrophages encapsulated in 10% PEG-TZ MPs 7 days after encapsulation. Scale bar 100 μm. | ||
As observed in Fig. 12A, no statistically significant differences were observed between 1 and 7 days of culture, suggesting that the encapsulated cells remained alive without any decrease of cell viability, despite their impossibility to adhere through the polymer network. Furthermore, the number of observed cells per microsphere significantly increased between 1 and 7 days of culture, as observed in Fig. 12B. Considering that RAW 264.7 own a doubling time of approximately 12 hours or less under 2D in vitro culture,87 the observed increase of cells immersed in the hydrogel matrix after 7 days remains considerably shorter. Despite the lack of adhesion moieties, which we hypothesize to cause the observed cell proliferation decrease, RAW 264.7 macrophage cells were still capable of proliferating under encapsulation conditions. Future studies should focus on the development of different polymeric hydrogel MPs with naturally derived materials crosslinked with the same click-chemistry method with the aim to evaluate the proliferation and adherence of encapsulated cells in a more biologically suitable polymer matrix.
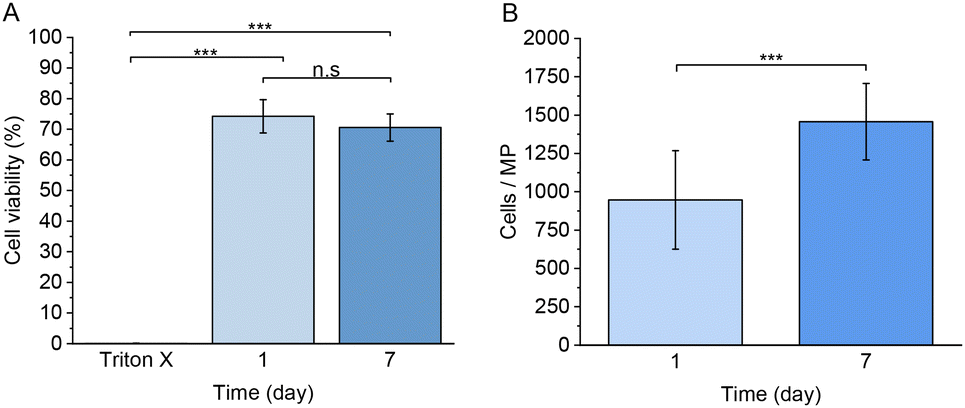 | ||
| Fig. 12 (A) Cell viability of RAW 264.7 macrophages 1 and 7 days after encapsulation. A one-way ANOVA followed by a Turkey-Kramer post hoc test was used for the statistical analysis. The significance levels of the differences were set at the probabilities of *p < 0.05, **p < 0.001 and ***p < 0.0001 for comparison of each test. (B) Average number of RAW 264.7 macrophage cells per microsphere 1 and 7 days after encapsulation. Values are expressed as the mean ± SD of 15 different microspheres per each time. A two-tailed Student t-test was used for the statistical analysis. The significance levels of the differences were set at the probabilities of *p < 0.05, **p < 0.01, ***p < 0.0005 and ****p < 0.0001. | ||
Conclusions
We developed monodisperse in situ gellable iEDDA-crosslinked PEG-based hydrogel MPs via W/O emulsification using a glass microfluidics platform. Both modified precursor polymers, 4-arm PEG-TZ and PEG-NB, owned fast crosslinking kinetics and allowed to obtain highly homogeneous MPs with diameter sizes within 200 μm and 600 μm range using a single microfluidic device. The morphological characterization results showed that the developed hydrogel MPs owned a uniform surface area and a highly porous inner structure. Moreover, the developed MPs showed the same rheological properties as similar covalently crosslinked hydrogel systems, maintaining a low swelling degree and slow degradation under physiological conditions, in contrast to mildly acidic conditions, which caused a high swelling degree and fast degradation. The hydrogel MPs also showed a high protein loading capacity and a predominant burst release behavior. Moreover, in vitro studies showed no cytotoxicity in RAW 264.7 macrophages of the MPs prepared and proved that the system is suitable for mammalian cells encapsulation. Overall, these results demonstrate that iEDDA-crosslinked MPs remain a potential platform for acting as drug carrier and cell encapsulation systems for biomedical applications.Author contributions
R. P. T. and H. A. S. designed the research. R. P. T, S. W., and A. C. performed the research. S. W., F. F., G. M., S. L. C., participated in the discussion of the results. S. H., J. H., G. B., and H. A. S. supervised the work and secured the funding for the research work. R. P. T. analyzed the data and wrote the first draft of the paper. All authors have revised the manuscript and given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Prof. H. A. Santos acknowledges financial support from the Academy of Finland (Grant No. 331151). S. Wang acknowledges Academy of Finland (decision no. 331106) for financial support. This project has received funding from the European Union's Horizon 2020 research and development programme under the Marie Skłodowska Curie grant agreement No. 955685. The authors thank the Light Microscopy Unit, Institute of Biotechnology, University of Helsinki (supported by HiLIFE and Biocenter Finland) for scanning electron microscope and confocal imaging.References
- Q. Feng, D. Li, Q. Li, X. Cao and H. Dong, Microgel assembly: Fabrication, characteristics and application in tissue engineering and regenerative medicine, Bioact. Mater., 2022, 9, 105–119 CrossRef CAS PubMed.
- E. M. Ahmed, Hydrogel: Preparation, characterization, and applications: A review, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed.
- M. Mahinroosta, Z. Jomeh Farsangi, A. Allahverdi and Z. Shakoori, Hydrogels as intelligent materials: A brief review of synthesis, properties and applications, Mater. Today Chem., 2018, 8, 42–55 CrossRef CAS.
- M. Chen, G. Bolognesi and G. T. Vladisavljević, Crosslinking Strategies for the Microfluidic Production of Microgels, Molecules, 2021, 26, 3752 CrossRef CAS PubMed.
- B. Li, et al., Injectable “nano-micron” combined gene-hydrogel microspheres for local treatment of osteoarthritis, NPG Asia Mater., 2022, 14, 1–15 CrossRef CAS.
- Y. C. Lu, et al., Designing compartmentalized hydrogel microparticles for cell encapsulation and scalable 3D cell culture, J. Mater. Chem. B, 2014, 3, 353–360 RSC.
- L. Yu, et al., Microfluidic formation of core-shell alginate microparticles for protein encapsulation and controlled release, J. Colloid Interface Sci., 2019, 539, 497–503 CrossRef CAS PubMed.
- S. S. Sivan, et al., Encapsulation of Human-Bone-Marrow-Derived Mesenchymal Stem Cells in Small Alginate Beads Using One-Step Emulsification by Internal Gelation: In Vitro, and In Vivo Evaluation in Degenerate Intervertebral Disc Model, Pharmaceutics, 2022, 14, 1179 CrossRef CAS PubMed.
- P. Liu, L. Jiang, L. Zhu and A. Wang, Novel approach for attapulgite/poly(acrylic acid) (ATP/PAA) nanocomposite microgels as selective adsorbent for Pb(II) Ion, React. Funct. Polym., 2014, 74, 72–80 CrossRef CAS.
- S. H. Ching, N. Bansal and B. Bhandari, Alginate gel particles–A review of production techniques and physical properties, Crit. Rev. Food Sci. Nutr., 2017, 57, 1133–1152 CrossRef CAS PubMed.
- F. A. Perrechil, A. C. K. Sato and R. L. Cunha, κ-Carrageenan–sodium caseinate microgel production by atomization: Critical analysis of the experimental procedure, J. Food Eng., 2011, 104, 123–133 CrossRef CAS.
- J. V. M. Benetti, J. T. do Prado Silva and V. R. Nicoletti, SPI microgels applied to Pickering stabilization of O/W emulsions by ultrasound and high-pressure homogenization: rheology and spray drying, Food Res. Int., 2019, 122, 383–391 CrossRef CAS PubMed.
- D. Liu, H. Zhang, F. Fontana, J. T. Hirvonen and H. A. Santos, Microfluidic-assisted fabrication of carriers for controlled drug delivery, Lab Chip, 2017, 17, 1856–1883 RSC.
- C. X. Zhao, Multiphase flow microfluidics for the production of single or multiple emulsions for drug delivery, Adv. Drug Delivery Rev., 2013, 65, 1420–1446 CrossRef CAS PubMed.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, The development of microgels/nanogels for drug delivery applications, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS.
- N. Chai, et al., Construction of 3D printed constructs based on microfluidic microgel for bone regeneration, Composites, Part B, 2021, 223, 109100 CrossRef CAS.
- Z. Wei, et al., Microfluidics Fabrication of Micrometer-Sized Hydrogels with Precisely Controlled Geometries for Biomedical Applications, Adv. Healthcare Mater., 2022, 11, 2200846 CrossRef CAS PubMed.
- C. Costa, et al., One-step microfluidics production of enzyme-loaded liposomes for the treatment of inflammatory diseases, Colloids Surf., B, 2021, 199, 111556 CrossRef CAS PubMed.
- C. Tramontano, et al., Microfluidic-Assisted Production of Gastro-Resistant Active-Targeted Diatomite Nanoparticles for the Local Release of Galunisertib in Metastatic Colorectal Cancer Cells, Adv. Healthc. Mater., 2022, 2202672 Search PubMed.
- I. Arduino, et al., Microfluidic preparation and in vitro evaluation of iRGD-functionalized solid lipid nanoparticles for targeted delivery of paclitaxel to tumor cells, Int. J. Pharm., 2021, 610, 121246 CrossRef CAS PubMed.
- T. Nardo, et al., Synthetic Biomaterial for Regenerative Medicine Applications, Kidney Transplantation, Bioengineering, and Regeneration: Kidney Transplantation in the Regenerative Medicine Era, 2017, pp. 901–921. DOI:10.1016/B978-0-12-801734-0.00065-5.
- S. Yang, et al., Fabricated technology of biomedical micro-nano hydrogel, Biomed. Technol., 2023, 2, 31–48 CrossRef.
- B. S. MohanKumar, et al., Hydrogels: potential aid in tissue engineering—a review, Polym. Bull., 2022, 79, 7009–7039 CrossRef CAS.
- T. Farjami and A. Madadlou, Fabrication methods of biopolymeric microgels and microgel-based hydrogels, Food Hydrocolloids, 2017, 62, 262–272 CrossRef CAS.
- V. L. Workman, S. B. Dunnett, P. Kille and D. D. Palmer, On-Chip Alginate Microencapsulation of Functional Cells, Macromol. Rapid Commun., 2008, 29, 165–170 CrossRef CAS.
- K. Enck, et al., Design of an Adhesive Film-Based Microfluidic Device for Alginate Hydrogel-Based Cell Encapsulation, Ann. Biomed. Eng., 2020, 48, 1103–1111 CrossRef PubMed.
- W. H. Tan and S. Takeuchi, Monodisperse Alginate Hydrogel Microbeads for Cell Encapsulation, Adv. Mater., 2007, 19, 2696–2701 CrossRef CAS.
- T. Braschler, et al., Link between alginate reaction front propagation and general reaction diffusion theory, Anal. Chem., 2011, 83, 2234–2242 CrossRef CAS PubMed.
- Z. Zhang, R. Zhang, L. Zou and D. J. McClements, Protein encapsulation in alginate hydrogel beads: Effect of pH on microgel stability, protein retention and protein release, Food Hydrocolloids, 2016, 58, 308–315 CrossRef CAS.
- J. Wu, W. Wei, L.-Y. Wang, Z.-G. Su and G.-H. Ma, Preparation of uniform-sized pH-sensitive quaternized chitosan microsphere by combining membrane emulsification technique and thermal-gelation method, Colloids Surf., B, 2008, 63, 164–175 CrossRef CAS PubMed.
- Y. Gao, et al., Covalently Crosslinked Hydrogels via Step-Growth Reactions: Crosslinking Chemistries, Polymers, and Clinical Impact, Adv. Mater., 2021, 33, 2006362 CrossRef CAS PubMed.
- D. L. Alge, M. A. Azagarsamy, D. F. Donohue and K. S. Anseth, Synthetically Tractable Click Hydrogels for Three-Dimensional Cell Culture Formed Using Tetrazine–Norbornene Chemistry, Biomacromolecules, 2013, 14, 949–953 CrossRef CAS PubMed.
- Y. Han, et al., Biomimetic injectable hydrogel microspheres with enhanced lubrication and controllable drug release for the treatment of osteoarthritis, Bioact. Mater., 2021, 6, 3596–3607 CrossRef CAS PubMed.
- W. Hu, Z. Wang, Y. Xiao, S. Zhang and J. Wang, Advances in crosslinking strategies of biomedical hydrogels, Biomater. Sci., 2019, 7, 843–855 RSC.
- F. Jivan, et al., Sequential Thiol–Ene and Tetrazine Click Reactions for the Polymerization and Functionalization of Hydrogel Microparticles, Biomacromolecules, 2016, 17, 3516–3523 CrossRef CAS PubMed.
- N. K. Devaraj and R. Weissleder, Biomedical applications of tetrazine cycloadditions, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS PubMed.
- C. F. Hansell, et al., Additive-free clicking for polymer functionalization and coupling by tetrazine-norbornene chemistry, J. Am. Chem. Soc., 2011, 133, 13828–13831 CrossRef CAS PubMed.
- C. C. Lin and K. S. Anseth, PEG hydrogels for the controlled release of biomolecules in regenerative medicine, Pharm. Res., 2009, 26, 631–643 CrossRef CAS PubMed.
- J. Zhu, Bioactive modification of poly(ethylene glycol) hydrogels for tissue engineering, Biomaterials, 2010, 31, 4639–4656 CrossRef CAS PubMed.
- A. A. Dias Meirelles, et al., Microfluidic approach to produce emulsion-filled alginate microgels, J. Food Eng., 2022, 315, 110812 CrossRef CAS.
- C. Costa, et al., One-step microfluidics production of enzyme-loaded liposomes for the treatment of inflammatory diseases, Colloids Surf., B, 2021, 199, 111556 CrossRef CAS PubMed.
- S. E. Holt, A. Rakoski, F. Jivan, L. M. Pérez and D. L. Alge, Hydrogel Synthesis and Stabilization via Tetrazine Click-Induced Secondary Interactions, Macromol. Rapid Commun., 2020, 41, 2000287 CrossRef CAS PubMed.
- J. P. Martins, et al., Microfluidic Nanoassembly of Bioengineered Chitosan-Modified FcRn-Targeted Porous Silicon Nanoparticles @ Hypromellose Acetate Succinate for Oral Delivery of Antidiabetic Peptides, ACS Appl. Mater. Interfaces, 2018, 10, 44354–44367 CrossRef CAS PubMed.
- D. Liu, et al., A Versatile and Robust Microfluidic Platform Toward High Throughput Synthesis of Homogeneous Nanoparticles with Tunable Properties, Adv. Mater., 2015, 27, 2298–2304 CrossRef CAS PubMed.
- F. Fontana, et al., Bioengineered Porous Silicon Nanoparticles@Macrophages Cell Membrane as Composite Platforms for Rheumatoid Arthritis, Adv. Funct. Mater., 2018, 28, 1801355 CrossRef.
- M. Kowacz and P. Warszyński, Beyond esterase-like activity of serum albumin. Histidine-(nitro)phenol radical formation in conversion cascade of p-nitrophenyl acetate and the role of infrared light, J. Mol. Recognit., 2019, 32, e2780 CrossRef PubMed.
- S. S. E. Michel, et al., Norbornene-Functionalized Chitosan Hydrogels and Microgels via Unprecedented Photoinitiated Self-Assembly for Potential Biomedical Applications, ACS Appl. Bio Mater., 2020, 3, 5253–5262 CrossRef CAS PubMed.
- J. van Hoorick, et al., Highly Reactive Thiol-Norbornene Photo-Click Hydrogels: Toward Improved Processability, Macromol. Rapid Commun., 2018, 39, 1800181 CrossRef PubMed.
- S. Li, et al., Injectable Click Chemistry-based Bioadhesives for Accelerated Wound Closure, Acta Biomater., 2020, 110, 95–104 CrossRef CAS PubMed.
- M. Gulfam, et al., Highly porous and injectable hydrogels derived from cartilage acellularized matrix exhibit reduction and NIR light dual-responsive drug release properties for application in antitumor therapy, NPG Asia Mater., 2022, 14, 1–17 CrossRef.
- S. b. Joo, et al., Fast Absorbent and Highly Bioorthogonal Hydrogels Developed by IEDDA Click Reaction for Drug Delivery Application, Materials, 2022, 15, 7128 CrossRef CAS PubMed.
- R. Paberit, et al., Cycling Stability of Poly(ethylene glycol) of Six Molecular Weights: Influence of Thermal Conditions for Energy Applications, ACS Appl. Energy Mater., 2020, 3, 10578–10589 CrossRef CAS.
- H. Tanuma, H. Kiuchi, W. Kai, K. Yazawa and Y. Inoue, Characterization and enzymatic degradation of PEG-cross-linked chitosan hydrogel films, J. Appl. Polym. Sci., 2009, 114, 1902–1907 CrossRef CAS.
- C. Zhou and Q. Wu, A novel polyacrylamide nanocomposite hydrogel reinforced with natural chitosan nanofibers, Colloids Surf., B, 2011, 84, 155–162 CrossRef CAS PubMed.
- N. A. Peppas and E. W. Merrill, Differential scanning calorimetry of crystallized PVA hydrogels, J. Appl. Polym. Sci., 1976, 20, 1457–1465 CrossRef CAS.
- S. Joshi, K. A. Raj, M. R. Rao and R. Ghosh, An electronic biosensor based on semiconducting tetrazine polymer immobilizing matrix coated on rGO for carcinoembryonic antigen, Sci. Rep., 2022, 12, 3006 CrossRef CAS PubMed.
- M. B. Browning, T. Wilems, M. Hahn and E. Cosgriff-Hernandez, Compositional control of poly(ethylene glycol) hydrogel modulus independent of mesh size, J. Biomed. Mater. Res., Part A, 2011, 98A, 268–273 CrossRef CAS PubMed.
- H. T. Hoang, et al., Dual pH-/thermo-responsive chitosan-based hydrogels prepared using ‘click’ chemistry for colon-targeted drug delivery applications, Carbohydr. Polym., 2021, 260, 117812 CrossRef CAS PubMed.
- Y. Qiao, et al., Gelatin Templated Polypeptide Co-Cross-Linked Hydrogel for Bone Regeneration, Adv. Healthc. Mater., 2020, 9, 1901239 CrossRef CAS PubMed.
- N. Yang, Y. Wang, Q. Zhang, L. Chen and Y. Zhao, In situ formation of poly (thiolated chitosan-co-alkylated β-cyclodextrin) hydrogels using click cross-linking for sustained drug release, J. Mater. Sci., 2019, 54, 1677–1691 CrossRef CAS.
- N. J. Darling, et al., Click by Click Microporous Annealed Particle (MAP) Scaffolds, Adv. Healthc. Mater., 2020, 9, 1901391 CrossRef CAS PubMed.
- N. H. Dimmitt, M. R. Arkenberg, M. M. de Lima Perini, J. Li and C. C. Lin, Hydrolytically Degradable PEG-Based Inverse Electron Demand Diels-Alder Click Hydrogels, ACS Biomater. Sci. Eng., 2022, 8, 4262–4273 CrossRef CAS PubMed.
- V. Delplace, et al., Nonswelling, Ultralow Content Inverse Electron-Demand Diels–Alder Hyaluronan Hydrogels with Tunable Gelation Time: Synthesis and In Vitro Evaluation, Adv. Funct. Mater., 2020, 30, 1903978 CrossRef CAS.
- J. J. Liétor-Santos, B. Sierra-Martín and A. Fernández-Nieves, Bulk and shear moduli of compressed microgel suspensions, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 060402 CrossRef PubMed.
- C. E. Ziegler, M. Graf, M. Nagaoka, H. Lehr and A. M. Goepferich, In Situ Forming iEDDA Hydrogels with Tunable Gelation Time Release High-Molecular Weight Proteins in a Controlled Manner over an Extended Time, Biomacromolecules, 2021, 22, 3223–3236 CrossRef CAS PubMed.
- H. Shih and C. C. Lin, Cross-linking and degradation of step-growth hydrogels formed by thiol-ene photoclick chemistry, Biomacromolecules, 2012, 13, 2003–2012 CrossRef CAS PubMed.
- M. D. Darrabie, W. F. Kendall and E. C. Opara, Characteristics of Poly-l-Ornithine-coated alginate microcapsules, Biomaterials, 2005, 26, 6846–6852 CrossRef CAS PubMed.
- A. Reyes Valenzuela, et al., Polymeric Microspheres Containing Human Vocal Fold Fibroblasts for Vocal Fold Regeneration, Laryngoscope, 2021, 131, 1828–1834 CrossRef CAS PubMed.
- H. Arimura, T. Ouchi, A. Kishida and Y. Ohya, Preparation of a hyaluronic acid hydrogel through polyion complex formation using cationic polylactide-based microspheres as a biodegradable cross-linking agent, J. Biomater. Sci., Polym. Ed., 2005, 16, 1347–1358 CrossRef CAS PubMed.
- K. Salma-Ancane, et al., Effect of crosslinking strategy on the biological, antibacterial and physicochemical performance of hyaluronic acid and ε-polylysine based hydrogels, Int. J. Biol. Macromol., 2022, 208, 995–1008 CrossRef CAS PubMed.
- K. Hori, et al., Theoretical study on the reaction mechanism for the hydrolysis of esters and amides under acidic conditions, Tetrahedron, 2007, 63, 1264–1269 CrossRef CAS.
- X. Zou, X. Zhao, L. Ye, Q. Wang and H. Li, Preparation and drug release behavior of pH-responsive bovine serum albumin-loaded chitosan microspheres, J. Ind. Eng. Chem., 2015, 21, 1389–1397 CrossRef CAS.
- K. T. L. Trinh, N. X. T. Le and N. Y. Lee, Microfluidic-based fabrication of alginate microparticles for protein delivery and its application in the in vitro chondrogenesis of mesenchymal stem cells, J. Drug Delivery Sci. Technol., 2021, 66, 102735 CrossRef CAS.
- J. S. Kjesbu, et al., Alginate and tunicate nanocellulose composite microbeads – Preparation, characterization and cell encapsulation, Carbohydr. Polym., 2022, 286, 119284 CrossRef CAS PubMed.
- L. Tang, et al., Microencapsulation of functional ovalbumin and bovine serum albumin with polylysine-alginate complex for sustained protein vehicle's development, Food Chem., 2022, 368, 130902 CrossRef CAS PubMed.
- B. Kupikowska-Stobba, M. Grzeczkowicz and D. Lewińska, A one-step in vitro continuous flow assessment of protein release from core-shell polymer microcapsules designed for therapeutic protein delivery, Probl. Biocybern. Biomed. Eng., 2021, 41, 1347–1364 CrossRef.
- S. Leick, A. Kemper and H. Rehage, Alginate/poly-l-lysine capsules: mechanical properties and drug release characteristics, Soft Matter, 2011, 7, 6684–6694 RSC.
- A. Famili and K. Rajagopal, Bio-Orthogonal Cross-Linking Chemistry Enables In Situ Protein Encapsulation and Provides Sustained Release from Hyaluronic Acid Based Hydrogels, Mol. Pharm., 2017, 14, 1961–1968 CrossRef CAS PubMed.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Click hydrogels, microgels and nanogels: Emerging platforms for drug delivery and tissue engineering, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- S. Lee, X. Tong and F. Yang, The effects of varying poly(ethylene glycol) hydrogel crosslinking density and the crosslinking mechanism on protein accumulation in three-dimensional hydrogels, Acta Biomater., 2014, 10, 4167–4174 CrossRef CAS PubMed.
- C.-C. Lin and K. S. Anseth, PEG Hydrogels for the Controlled Release of Biomolecules in Regenerative Medicine, Pharm. Res., 2009, 26, 631–643 CrossRef CAS PubMed.
- J. Pliego, et al., Monitoring Lipase/Esterase Activity by Stopped Flow in a Sequential Injection Analysis System Using p-Nitrophenyl Butyrate, Sensors, 2015, 15, 2798–2811 CrossRef CAS PubMed.
- N. H. Dimmitt, M. R. Arkenberg, M. M. de Lima Perini, J. Li and C.-C. Lin, Hydrolytically Degradable PEG-Based Inverse Electron Demand Diels–Alder Click Hydrogels, ACS Biomater. Sci. Eng., 2022, 8, 4262–4273 CrossRef CAS PubMed.
- S. H. Ching, N. Bansal and B. Bhandari, Alginate gel particles–A review of production techniques and physical properties, Crit. Rev. Food Sci. Nutr., 2017, 57, 1133–1152 CrossRef CAS PubMed.
- V. Delplace, et al., Inverse Electron-Demand Diels-Alder Methylcellulose Hydrogels Enable the Co-delivery of Chondroitinase ABC and Neural Progenitor Cells, Biomacromolecules, 2020, 21, 2421–2431 CrossRef CAS PubMed.
- S. T. Koshy, et al., Click-Crosslinked Injectable Gelatin Hydrogels, Adv. Healthc. Mater., 2016, 5, 541–547 CrossRef CAS PubMed.
- L. Kong, W. Smith and D. Hao, Overview of RAW264.7 for osteoclastogensis study: Phenotype and stimuli, J. Cell. Mol. Med., 2019, 23, 3077–3087 CrossRef PubMed.
- G. Molinaro, F. Fontana, R. P. Tello, S. Wang, S. L. Cérda, G. Torrieri, A. Correia, E. Waris, J. T. Hirvonen, G. Barreto and H. A. Santos, In Vitro Study of the Anti-inflammatory and Antifibrotic Activity of Tannic Acid-Coated Curcumin-Loaded Nanoparticles in Human Tenocytes, ACS Appl. Mater. Interfaces, 2023, 15(19), 23012–23023 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm00292f |
| This journal is © The Royal Society of Chemistry 2023 |