
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Subcellular localization and therapeutic efficacy of polymeric micellar nanoparticles encapsulating bedaquiline for tuberculosis treatment in zebrafish†
Madhavi
Bhandari‡
a,
Héctor
Soria-Carrera
 bcd,
Jens
Wohlmann
a,
Nils-Jørgen Knudsen
Dal
a,
Jesús M.
de la Fuente
bc,
Rafael
Martín-Rapún
*bcd,
Gareth
Griffiths
a and
Federico
Fenaroli
*ae
bcd,
Jens
Wohlmann
a,
Nils-Jørgen Knudsen
Dal
a,
Jesús M.
de la Fuente
bc,
Rafael
Martín-Rapún
*bcd,
Gareth
Griffiths
a and
Federico
Fenaroli
*ae
aDepartment of Biosciences, University of Oslo, Blindernveien 31, 0371 Oslo, Norway
bInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, C/Mariano Esquillor s/n, 50018 Zaragoza, Spain. E-mail: rmartin@unizar.es
cCIBER de Bioingeniería, Biomateriales y Nanomedicina, Instituto de Salud Carlos III, 28029 Madrid, Spain
dDepartamento de Química Orgánica, Facultad de Ciencias, Universidad de Zaragoza, c/Pedro Cerbuna 12, 50009 Zaragoza, Spain
eDepartment of Chemistry, Bioscience and Environmental Engineering, University of Stavanger, 4021 Stavanger, Norway. E-mail: federico.fenaroli@uis.no
First published on 19th January 2023
Abstract
The combination drug regimens that have long been used to treat tuberculosis (TB), caused by Mycobacterium tuberculosis, are fraught with problems such as frequent administration, long duration of treatment, and harsh adverse effects, leading to the emergence of multidrug resistance. Moreover, there is no effective preventive vaccine against TB infection. In this context, nanoparticles (NPs) have emerged as a potential alternative method for drug delivery. Encapsulating antibiotics in biodegradable NPs has been shown to provide effective therapy and reduced toxicity against M. tuberculosis in different mammalian models, when compared to conventional free drug administration. Here, we evaluate the localization, therapeutic efficacy and toxic effects of polymeric micellar NPs encapsulating a promising but highly hydrophobic and toxic antitubercular drug bedaquiline (BQ) in zebrafish embryos infected with Mycobacterium marinum. Our study shows that the NP formulation of BQ improves survival and reduces bacterial burden in the infected embryos after treatment when compared to its free form. The intravenously injected BQ NPs have short circulation times due to their rapid and efficient uptake into the endothelial cells, as observed by correlative light and electron microscopy (CLEM).
Introduction
Tuberculosis (TB), one of the oldest recorded diseases of mankind is caused by the bacterium Mycobacterium tuberculosis (M.tb).1 Until the unprecedented global pandemic of corona virus disease-2019 (COVID-19), TB was considered the most common cause of global mortality due to a single infectious agent.2 An estimated 10 million people develop active TB each year, and in 2020 approximately 1.5 million people succumbed to the disease. About one-fourth of the world's population harbors latent bacteria, making eradication of TB from countries where it is truly endemic even more difficult.3,4 The standard treatment regimen comprises a cocktail of the oral drugs, isoniazid,5 rifampicin (RIF), ethambutol (ETH) and pyrazinamide (PZA), effective when given for at least 6 months to patients with active TB harboring drug-sensitive mycobacteria.6 However, this treatment possesses intrinsic limitations such as the need for lengthy treatment, associated with harsh side effects.7 As a consequence, non-compliance of patients to the treatment has led to the emergence of drug resistant TB including multi-drug resistant tuberculosis (MDR-TB) where M.tb strains show resistance to two of the most effective first-line anti-TB drugs, isoniazid (INH) and rifampicin (RIF).8,9 Even more difficult to treat is extensively drug resistant TB (XDR-TB). XDR-TB bacterial strains fail to respond to more complex treatment with more toxic second-line drugs; a common protocol involves use of a fluoroquinolone, such as ciprofloxacin, levofloxacin, moxifloxacin or ofloxacin in conjunction with at least one of the injectable drugs: amikacin, capreomycin or kanamycin.10 Unfortunately, these second-line drugs (and even third-line drugs needed in extreme cases of drug resistance) are more expensive, more toxic, and have lower anti-mycobacterial activity which is why they require longer treatments.7,8 Since the highly resistant strains of M.tb continue to evolve, their further expansion would put at risk the effectiveness of our current antibiotics, therefore, we are in dire need of new treatment regimens.After over forty years since the last effective drug against TB was developed, bedaquiline (BQ)11,12 entered the market in 2012.13 Quite recently, a combined regimen of BQ along with pretomanid (another novel drug14) and linezolid (a repurposed drug for TB) (also called BPaL regimen) was approved by the United States Food and Drug Administration (U.S. FDA) for the treatment of highly drug-resistant adult pulmonary TB.15 This approval followed highly promising findings from a Phase III clinical trial (Nix-TB, 2015) which reported that a BPaL regimen achieved approximately 90% cure rates for MDR- and XDR-TB treatment in just 6 months. This is a huge improvement considering that 18 to 30 months is required by the current standard therapy showing a lower efficacy.16
BQ targets the adenosine triphosphate (ATP) synthase of mycobacteria and thus inhibits their ATP production, which is vital for energy metabolism and bacterial growth. BQ has the ability to kill both active and dormant forms of M.tb.10,17 The latter are responsible for latent infection, during which there is no apparent clinical manifestations of TB, a state that can last for decades, making it difficult to eradicate the disease.10,17 This makes BQ highly effective and theoretically more attractive than other antibiotics – such as protein-synthesis inhibitors – which only affect actively growing M.tb.10 However, BQ is associated with serious dose-dependent hepatic and cardiac toxicities, and poor oral bioavailability due to its hydrophobic nature, which has highly limited its use in clinical settings.18,19 Besides developing better and highly active BQ analogues, an alternative approach to enable oral drug delivery is to encapsulate drugs in nanoparticles (NPs < 1000 nm)20 which could minimize drug-induced toxicities,21 and further improve the treatment of MDR-TB.22
Relative to conventional free drug administration, the major advantages of NP-mediated drug delivery administration are the prolonged and targeted drug release with NPs, resulting in an increase in therapeutic efficacy and reduction of systemic toxicity of drugs by lowering their dosing frequency.22–24 Several studies have shown that encapsulation of anti-tubercular antibiotics into biodegradable NPs led to improved killing of mycobacteria – in mouse bone marrow primary macrophages,25 zebrafish embryo model26,27 and guinea pigs28 – when compared to the free drug. In a previous publication, we observed that the in vitro activity of BQ against Mycobacterium tuberculosis was improved when encapsulated in polypeptide-based micelles (2-fold increase) as compared with the free drug.29 However toxicity as well as in vivo cellular and subcellular localization of these NP had not been studied yet.
Here, we have investigated the therapeutic efficacy of NP formulation of BQ relative to the free drug in the treatment of zebrafish embryos infected with Mycobacterium marinum (M.m) as a model for M.tb infection. We have also studied in detail and at high resolution the biodistribution and subcellular localization of BQ encapsulated NPs by using a combination of fluorescence imaging and EM.
Materials and methods
Reagents
All reagents were purchased from Sigma-Aldrich unless otherwise specified. Nε-benzyloxycarbonyl-L-lysine N-carboxyanhydride (ZK NCA) and γ-benzyl-L-glutamate N-carboxyanhydride (BnE NCA) were prepared as previously reported,29 from Nε-benzyloxycarbonyl-L-lysine and L-glutamic acid γ-benzyl ester, which were purchased from Fluorochem. Dialysis was carried out in SnakeSkin™ Dialysis Tubing with 3.5 kDa molecular weight cut-off.NP preparation and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 12600g). The pellet was then washed thrice with water and freeze dried. ZK30-NH2 was obtained as a white solid (203 mg, 0.027 mmol) with 85% yield.
000 rpm, 12600g). The pellet was then washed thrice with water and freeze dried. ZK30-RhB was obtained as a pink solid (20 mg, 2.5 μmol) with 63% yield. Polymer composition was evaluated by 1H-NMR.
000 rpm, 12600g). The pellet was then washed thrice with water and freeze dried. ZK30-NH2 was obtained as a white solid (203 mg, 0.027 mmol) with 85% yield.
000 rpm, 12600g). The pellet was then washed thrice with water and freeze dried. ZK30-RhB was obtained as a pink solid (20 mg, 2.5 μmol) with 63% yield. Polymer composition was evaluated by 1H-NMR.
Polypeptides were characterized by gel permeation chromatography (GPC) with a setup that combines a Waters 2695 GPC with a tandem of detectors: multiangle laser light scattering (MALLS) detector at 50 °C and refractive index (RI) from Wyatt Tech. The system was equipped with one guard and two Phenogel narrow bore 5 μm, 300 × 4.6 mm columns (500 Å and 1E3 Å). Linear from Phenomenex. HPLC-grade N,N-dimethylformamide (DMF) containing 0.1 M LiBr was used as the mobile phase at a flow rate of 0.2 mL min−1 and 50 °C. ASTRA software from Wyatt Technology was used to collect and analyze the data.
Resazurin microtiter assay plate
The minimum inhibitory concentration (MIC) of free BQ required to inhibit 90% of M.m growth was determined using Resazurin Microtiter Assay Plate with a spectrophotometer.Zebrafish experiments
A high-resolution imaging of the cellular biodistribution of NPs was performed using the Zyla Spinning Disk Andor Dragonfly Confocal Microscope. Here, immediately after injection, a sedated zebrafish embryo was transferred to a glass-bottom dish (MatTek) and covered with 2% low melting point agarose (Promega, Madison) at about 35 °C. Upon solidification of the agarose, embryo water containing tricaine (0.02%) was added to keep the fish sedated.
Statistical analysis
Once the overall significance of the data set was verified, post hoc multiple comparisons tests was performed between the groups of interest. For data sets analyzed by column table, column statistics using D'Agostino & Pearson, Anderson–Darling, Shapiro–Wilk and Kolmogorov–Smirnov normality tests to check for Gaussian (normal) distribution of the data was used. When normally distributed according to all tests, data were analyzed by one-way ANOVA, followed by Dunnett's multiple comparisons test. In case of non-normal distribution, data were analyzed by Kruskal–Wallis test, followed by Dunn's multiple comparisons test. For data sets analyzed by survival table, the overall significance and pairwise comparison of groups was done based on the Kaplan–Meier estimate using the Log-rank (Mantel-Cox). Significance levels are indicated as *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.Results
NP preparation and characterization
We focused here on the same nanoparticles described in our previous report, consisting of micelles self-assembled from block copolypeptides. Block copolypeptide ZK30-BnE30 was synthesized through the ring opening polymerization of protected Nε-benzyloxycarbonyl-L-lysine N-carboxyanhydride (ZK NCA) and γ-benzyl-L-glutamate N-carboxyanhydride (BnE NCA).29 The block copolymer was obtained with low dispersion and good control of the molecular weight and monomer ratio as shown by gel permeation chromatography (GPC) and 1H-NMR spectroscopy (ESI Fig. 1 and 2†). The selective deprotection of the benzylic esters afforded the amphiphilic block copolypeptide ZK30-E30 (Fig. 1). We also prepared a polymer ZK30 consisting of only the hydrophobic block. The reaction of its N-terminus with rhodamine B (RhB) isothiocyanate allowed us to obtain the fluorescently labelled polypeptide ZK30-RhB. The 1H NMR spectra and GPC traces of these polymers are also shown in the ESI Fig. 1 and 2.†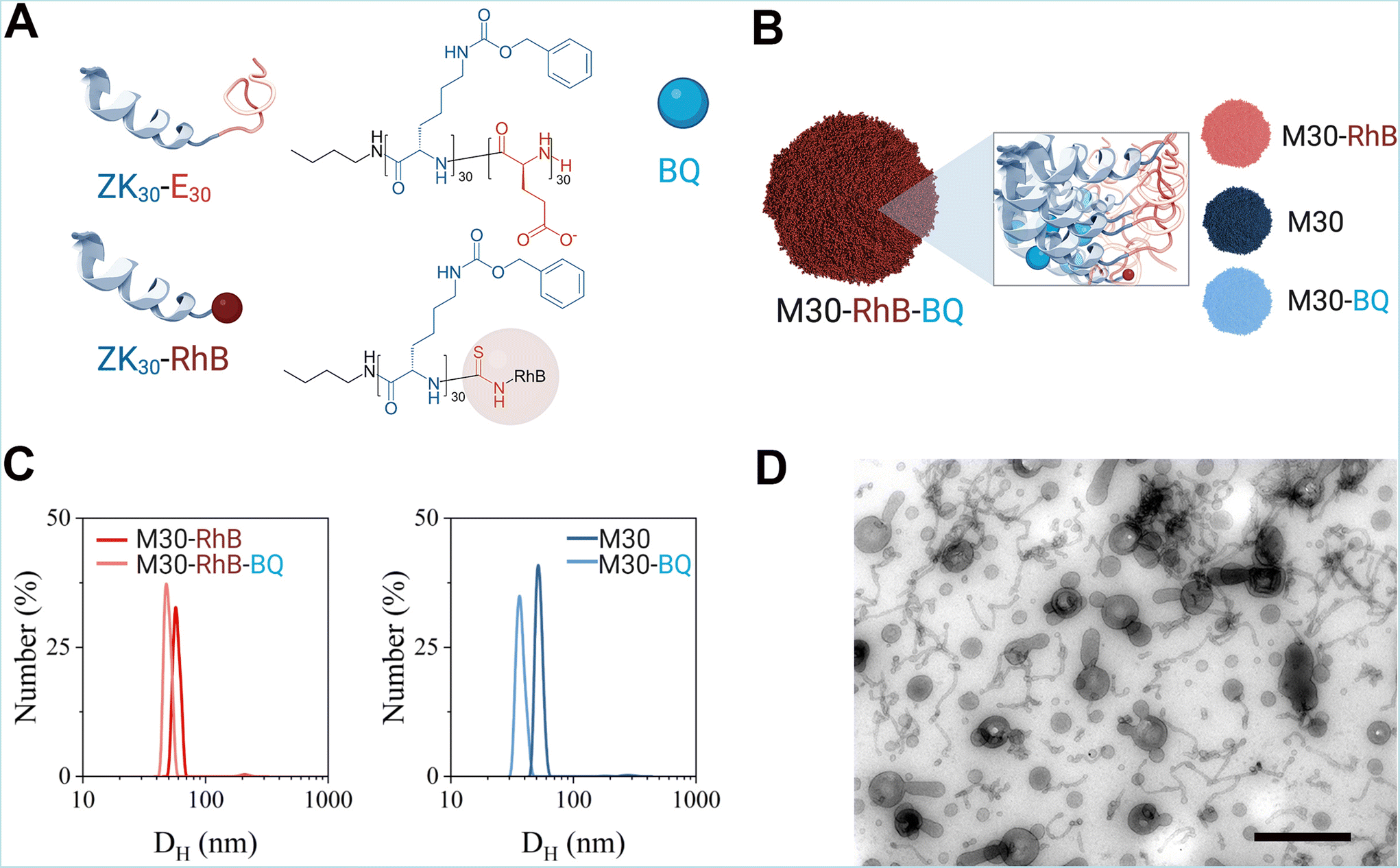 | ||
| Fig. 1 (A) Polymers used for the preparation of the micelles of this study. (B) Different types of micelles prepared for experiments. (C) DLS curves of RhB-labelled micelles (left) and DLS curves of non-labelled micelles (right). (D) Transmission electron micrographs of polymeric micellar BQ NPs, M30-RhB-BQ. Images were taken after subjecting the NPs to negative staining with 0.5% uranyl acetate/methylcellulose and methylcellulose embedding. Scale bar indicates and 500 nm (B). Part of this Figure was created with https://BioRender.com. | ||
The amphiphilic block copolypeptide ZK30-E30 was used to prepare nanoparticles, polypeptidic micelles, either empty (M30) or loaded with BQ (M30-BQ) as previously reported.29 For in vivo applications, we also developed fluorescent nanoparticles in which the fluorescent tag was covalently attached to the polymer molecules (ZK30-RhB), which allowed its incorporation into the hydrophobic block of empty (M30-RhB) or BQ-loaded micelles (Fig. 1A and B).
The nanoparticles (NPs) were characterized by their dynamic light scattering (DLS), ζ-potential, critical micellar concentration and transmission electron microscopy (TEM) images. Sizes measured by DLS were comparable for labelled and non-labelled nanoparticles ranging from 40 to 60 nm-with BQ-loaded NPs being slightly smaller than the empty (Fig. 1C and ESI Table 1†). All nanoparticles were found to be slightly polydisperse as PDI values were all between 0.1 and 0.2. However, PDI was higher for RhB-labelled NPs than for the unlabelled NPs, probably because of RhB destabilizing the NPs.
The different hydrophobic environment led to a decrease in fluorescence intensity for M30-RhB-BQ compared to M30-RhB (ESI Fig. 3†). However, the labelling process did not affect either the ζ-potential values or the BQ-loading capability of the micelle system, as shown by the measured drug loading and drug encapsulation efficiencies which were consistent with our previous report indicating the reproducibility of the system (ESI Table 1†). The hydrophobicity of BQ stabilized the NPs as shown by the lower CMC observed for BQ-loaded NPs, whereas RhB had the opposite effect (ESI Fig. 2†).29
We then observed the NPs using Transmission electron microscopy. In order to minimize drying artifact caused by the standard negative staining methodology we used methylcellulose that maintains the structure of NP when solidifying (see Materials and methods).36
Most M30-RhB-BQ were spherical in shape with long tubular or tentacular projections; similar appearance has been observed for M30-BQ, M30-RhB and M30 NPs although unlabelled NPs, M30-BQ and M30, appeared less heterogeneous, in line with the PDI observed by DLS (ESI Fig. 4†). NP size range varied between 20–100 nm thereby displaying an approximate correlation to DLS measurements (Fig. 1D).
NP characterization in zebrafish: biodistribution and accumulation analysis
We evaluated the circulation time of NPs by imaging them at different times after intravenous injection (directly after injection, 5 min, 1, 4, 8, 24, 48 and 72 h). Both BQ containing M30-RhB-BQ (NP-BQ), and empty NPs M30-RhB (NP-Blank) had short circulation times. After 5 min following intravenou administration into the PCV, more than 50% of NP-BQ as well as NP-Blank (the control group) were out of the circulation. In just 1 h post-injection, almost 80% of the NPs left the blood circulation. Both NP-BQ and NP-Blank were cleared from the circulation after 24 h following the injection (Fig. 2A).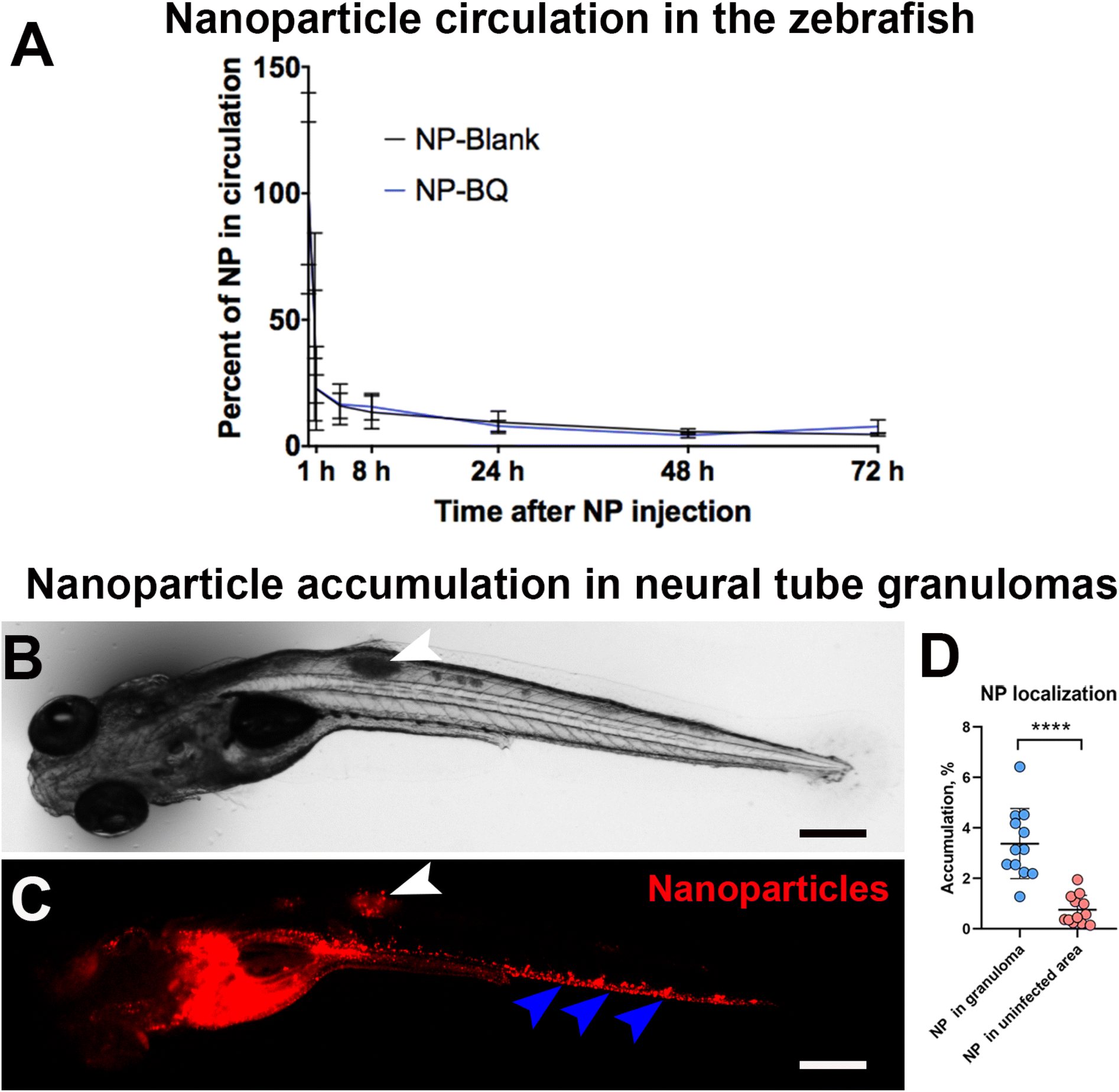 | ||
| Fig. 2 Zebrafish NP circulation and accumulation in granulomas. (A) Quantification of circulation time of NPs in the zebrafish embryos. The wild type zebrafish embryos were injected at 2 dpf with BQ containing (NP-BQ = M30-RhB-BQ) and empty (NP-Blank = M30-RhB) NPs and imaged at different time points, directly after injection, 5 min, 1, 4, 8, 24, 48 and 72 h respectively. N ≥ 5. (B) Transmission image of a neural tube infected embryo at day 5 post-infection. (C) Fluorescence image of the same embryo taken after 24 h post-injection of red fluorescent BQ NPs into the PCV. The white arrowheads in B and C indicate the granuloma region (B) and the relative accumulation of NP (C). The blue arrowhead in C indicates NP accumulation in the caudal region endothelium. (D) Quantification of fluorescence relative to NPs in the granuloma region and from an equivalent uninfected area (or background) at 24 h post-injection. N ≥ 12. Scale bars indicate 300 μm. | ||
In order to test whether the injected nanoparticles could reach mycobacteria infected areas we chose the neural tube model of zebrafish infection. As opposed to blood inoculation which results in multiple bacterial foci, neural tube injection of Mycobacterium marinum develops into a large and localized granuloma. These are assemblies of infected macrophages that often undergo necrosis, an important hallmark of human tuberculosis. Because of this feature, the neural tube model is considered a closer model to mimic human tuberculosis disease.33 For this experiment we injected M. marinum on day 3 post fertilization and 24 hours later we administered BQ NP intravenously. Using a fluorescence stereomicroscope, we observed a significant accumulation of BQ NPs (M30-RhB-BQ) in the neural tube granulomas (Fig. 2B and C, white arrowheads). On average, approximately 3.4% of the injected NPs localized in the granuloma area 24 h after injection (Fig. 2D). This value was almost 5 times higher than the equivalent uninfected control area adjacent to the granuloma, which had an average value of 0.75% (Fig. 2D). However, the majority of these NPs seemed to localize to the vascular wall in the caudal vein (Fig. 2C, blue arrowheads), indicating a likely accumulation in endothelial cells.
Having observed using low resolution microscopy NP accumulation in proximity to, and along the caudal vein we hypothesized that NP were actively taken up by endothelial cells. To verify this hypothesis, we used two different microscopy techniques: confocal microscopy and electron microscopy. We started by using a Zyla Spinning Disk Confocal where we imaged details of the endothelial lining of PCV following intravenous injection of NP. We found that 1 h following intravenous injection into the PCV, the lumen of the vessels was almost devoid of NPs (Fig. 3A) while, when looking at individual optical sections, there was a rapid and efficient uptake of NPs by the endothelial cells (Fig. 3B).
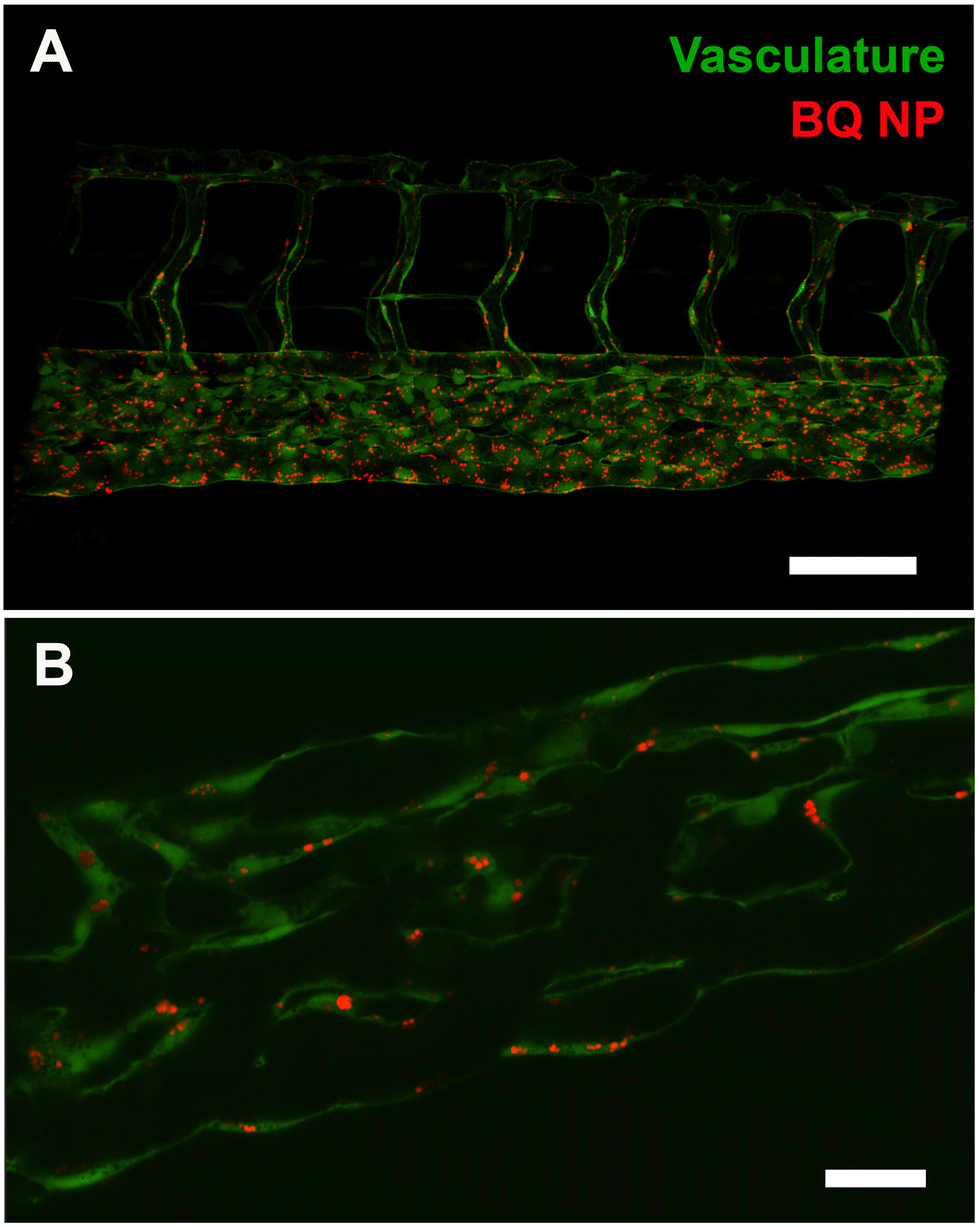 | ||
| Fig. 3 BQ NP (M30-RhB-BQ) accumulation in the zebrafish endothelium. (A) Confocal stack of the caudal region of zebrafish embryos, possessing green fluorescent vasculature, injected with red fluorescent NP and imaged 1 hour after injection. (B) Individual confocal plane showing NP (red) overlapping with the green vasculature. Scale bars: A, 100 μm; B, 20 μm. | ||
To validate this finding, we next performed higher resolution ultrastructural analysis after cellular uptake of NPs. For this we first injected intravenously 10 nm gold which is known to be rapidly taken up and to accumulate in late endosomes of endothelial cells.37,38 After 2 hours, we administered NP via the PCV allowing them to circulate for an additional 4 hours. All NPs (in red) were seen together with 10 nm gold inside large vesicles (green) into endothelial cells (blue), while none was found in the blood lumen (Yellow) (Fig. 4A). To further corroborate our findings, we performed correlative light and electron microscopy,23 which enabled correlation of red fluorescence of BQ NPs with the corresponding ultrastructure (Fig. 4B and C). Once again, BQ-NP were found inside late endosomes in the endothelium next to the blood vessels.
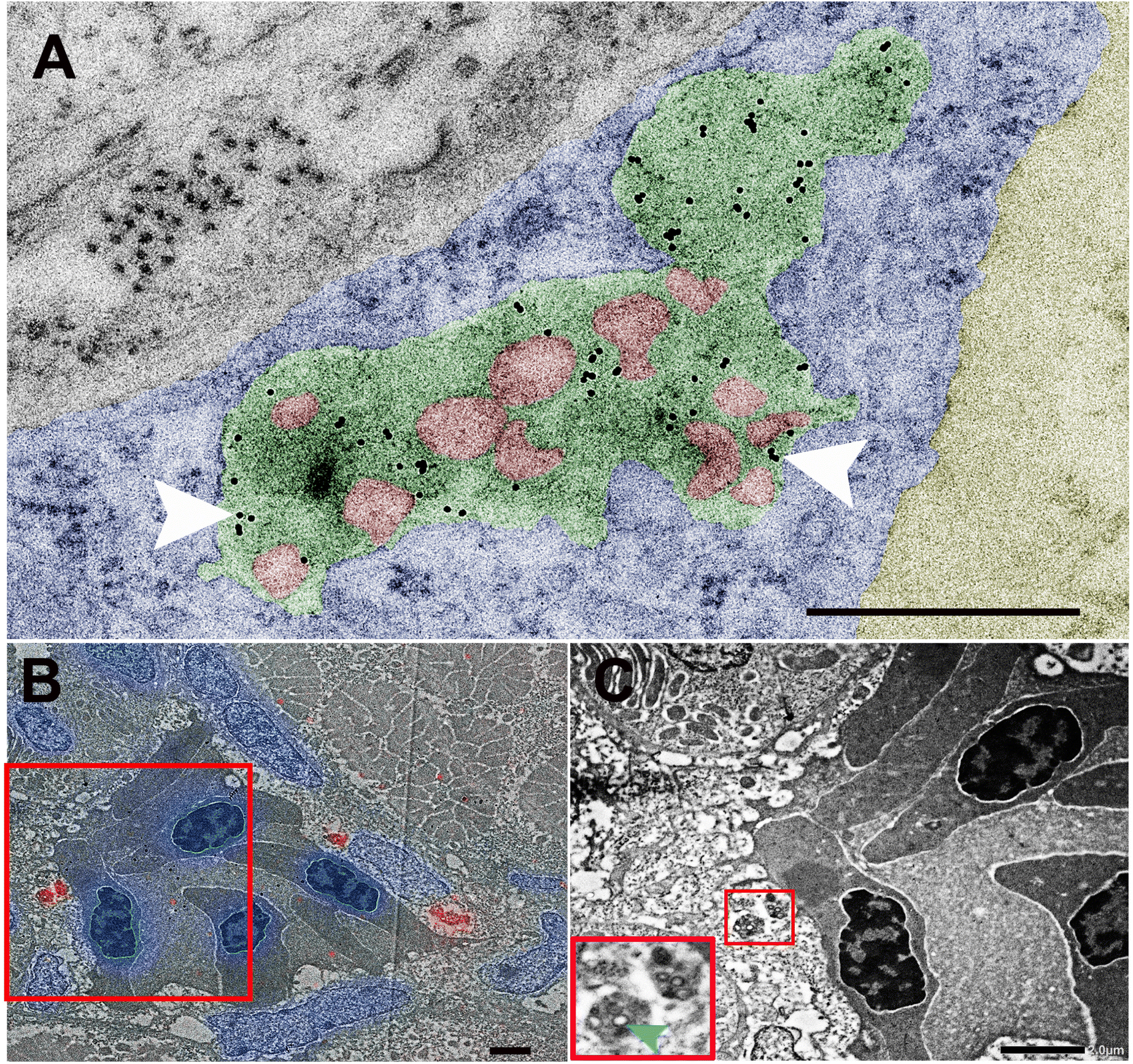 | ||
| Fig. 4 Ultrastructural analysis of biodistribution of BQ NPs (M30-RhB-BQ) in vivo. Representative electron micrographs of 60 nm epon sections taken across the transverse plane of the PCV of a zebrafish embryo injected with BQ NPs at 3 dpf. (A) At 4 h post-injection, BQ NPs (red) are seen inside late endosomes (green) in which 10 nm gold had been previously internalized (white arrowheads). Endothelial cell's cytoplasm and the blood lumen have been colored in blue and yellow, respectively. (B and C) CLEM in which a fluorescence microscopy image of NP (red) inside endothelial cells lining the PCV (DAPI in blue) overlapped with the correspondent transmission electron microscope image. In B the red box region is seen enlarged in C. In C the small red inset is seen enlarged in the left corner and shows at higher magnification NP inside late endosomes into endothelial cells near a blood vessel. Scale bars indicate 500 nm in A, 2 μm in B and C. | ||
To further corroborate our analysis and check whether our BQ NP break down in the endothelium over time. For this we checked by EM NP-containing endothelial endosomes at different time points after NP administration: 1 hour, 4 hours, 24 hours, 3 days(72 hours) and 7 days(168 hours). From our images it appears how NP are intact 1, 4 and 24 h after injection while they display degradation at 3 and 7 days after administration (Fig. 5).
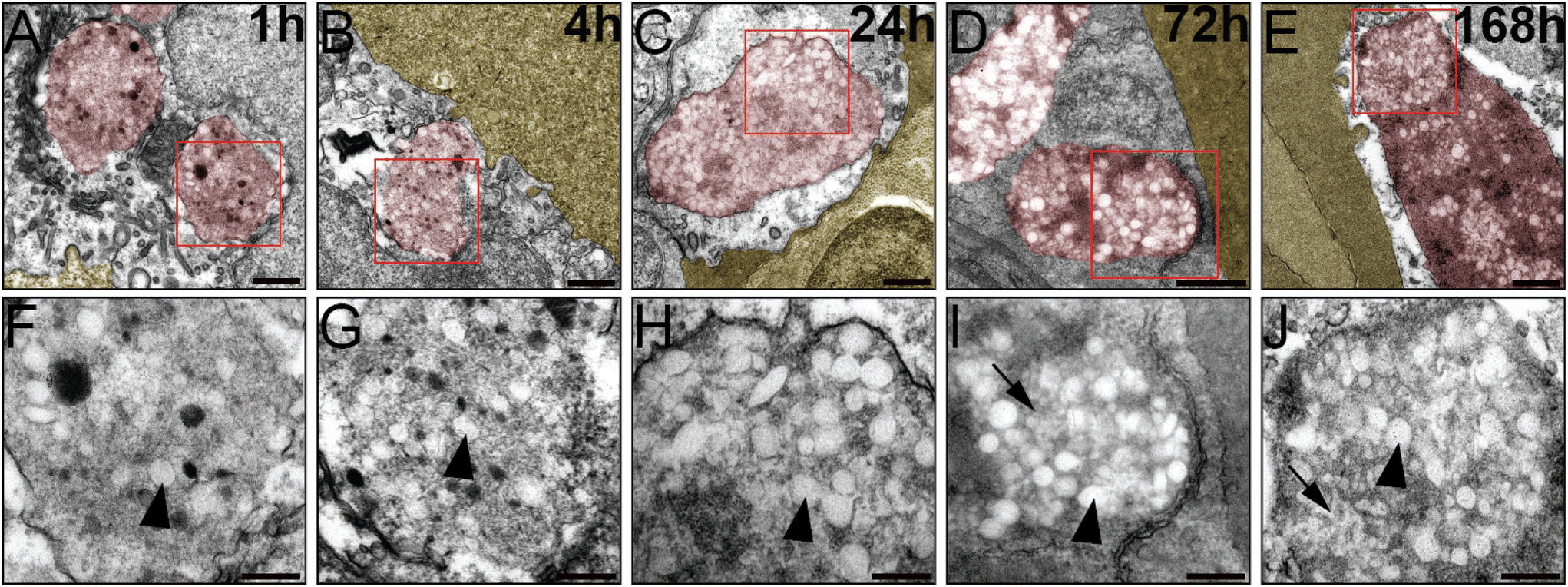 | ||
| Fig. 5 Ultrastructural analysis of BQ NPs (M30-RhB-BQ) in endothelial late endosomes (in red) near over time. Blood lumen neighboring the endothelium is shown in yellow. BQ NPs are observed at 1 h (A and F), 4 h (B and G), 24 h (C and H), 72 h (D and I) and 168 h (E and J) after injection. The images from the lower part (F–J) are enlarged imaged of the red box region above. Arrowheads at all time points indicate intact nanoparticles while arrows at 3 and 7 days indicate degraded NPs. Scale Bars indicate 500 nm in: (A–E); 200 nm in: (F–J). | ||
BQ activity against Mycobacterium marinum
After characterization of our NP in vivo biodistribution, we then focused on the potential of our NP containing BQ to kill M. marinum (M.m), the causative agent of Tuberculosis in ectotherms. For this, in vitro activity of BQ against M.m was determined before in vivo experimentation in the zebrafish embryos by using Resazurin Microtiter Assay Plate. The MIC value of BQ that killed 90% (MIC90) of M.m (as listed in Table 1) was found to be in the sub-micro molar amount (0.09 μM or 0.05 μg mL−1). Our result is in agreement with previous studies that had determined the MIC90 of BQ against M.tb (a close relative of M.m) to be in the range of 0.002 to 0.13 μg mL−1.5,19 The MIC90 for the control drugs Drug D39,40 and RIF is in agreement with our previous findings (Table 1).| Drugs | MIC90 for M.m (μM) |
|---|---|
| BQ | 0.09 μM |
| RIF | 0.23 μM |
| Drug D | 0.50 μM |
In vivo evaluation of toxicity associated with free and encapsulated BQ
The toxicity associated with both free BQ and its NP formulation was evaluated in vivo by monitoring the survival of wild type zebrafish embryos at 2 dpf after administration of 8.3 ng of BQ using NPs encapsulating BQ (NP-BQ = M30-BQ), empty NPs (M30), PBS (as a control), and free drug dissolved in DMSO (free BQ) into the PCV. We chose 8.3 ng as this was the highest injectable dose that could be administered (our drug concentration in the NP solution was 0.415 mg mL−1 and 20 nL is the maximum volume that could be injected without volume-related adverse effects). As a control, we injected 8.3 ng of BQ in 1 nL of pure DMSO, which is the maximum tolerated amount (previously established in the group) that could be injected into the embryos. The treated embryos exhibited a 10% better survival for NP-BQ compared to free BQ at a maximum used dose of 8.3 ng per embryo (ESI Fig. 5†).Therapeutic effects of encapsulated BQ against Mycobacterium marinum-infected zebrafish
In order to establish the therapeutic potential of BQ-containing NP (M30-BQ) we monitored zebrafish cumulative mortality and bacterial burden using fluorescence in different infection models: neural tube and blood infections. As described above, the neural tube infection forms large granulomas and is reminiscent of the most common form of human TB where mycobacteria are confined to the lungs.33 Blood infection is instead systemic causing numerous small and dispersed granulomas formed in proximity to the vasculature; this latter type of infection is also seen in humans (although less frequent) and is usually connected to poor immunity such as the case of individuals suffering from HIV co-infection.For these experiments we first tested different doses of encapsulated and free BQ in the blood infection model, where both M.m and treatment were administered via injections into the PCV. A dose of 1.1 ng per embryo showed an increase in survival of M.m infected embryos with the nanoparticle formulation (NP-BQ ∼ 70%) compared to the free drug (Free BQ ∼ 50%) at day 8 post-infection (Fig. 6B). Higher doses of BQ either free or inside NP resulted in complete eradication of M.m making all zebrafish survive the infection, (data not shown). A similar result, although with a higher concentration of BQ (2.5 ng), was found when using the neural tube model. Here zebrafish infected with M.m and treated with BQ-NP had a significantly higher survival versus all other groups (except uninfected zebrafish), including zebrafish treated with Free BQ (Fig. 6A).
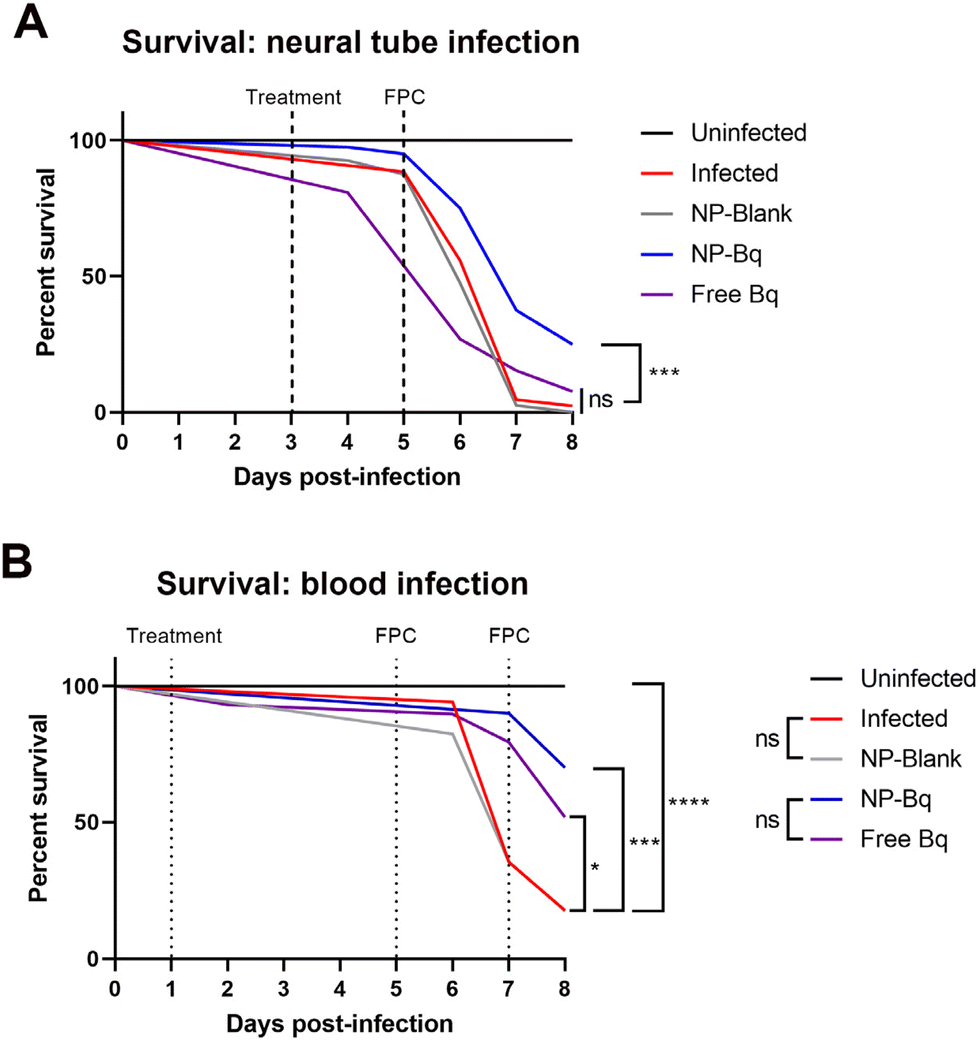 | ||
| Fig. 6 (A) Survival analysis comparing therapeutic efficacy of free (dissolved in DMSO) versus encapsulated BQ (NP-BQ = M30-BQ). N (embryos per group on the day of infection) ≥ 40. (B) FPC analysis to measure bacterial burden associated with red fluorescence of M.m used for the infection of zebrafish embryos in the neural tube. N (embryos per group on the day of imaging) in A ≥ 26 while in B ≥ 16. Statistical analysis was performed using a Log-rank (Mantel-Cox) test. | ||
In addition to survival, the relative bacterial burden was determined in all the groups by measuring fluorescence pixel count (FPC) of the red fluorescent bacteria (ESI Fig. 6†). The FPC method quantifies the total fluorescence associated with the red fluorescent protein expressed by the dsRed M.m.26 Based on our experience, FPC analysis is a different readout that could therefore complement our survival test results. The FPC results correlated well with the survival analysis and in both the blood model and the neural tube model the NP-BQ group had significantly lower bacterial burden compared to controls.
Discussion
NPs reduce drug-related toxicity and improve treatment efficacy
We have shown that the free drug BQ is highly effective in inhibiting mycobacterial growth in vitro as its MIC required to effectively kill M.m (0.09 μM) was approximately 6 times more potent than the Pretomanid derivative (Drug D, 0.5 μM) and 3 times more potent than RIF (0.23 μM). Moreover, also after encapsulation into polymeric micelles, BQ has shown to be effective against M.tb (H37Rv strain) growth and its antimycobacterial activity was higher compared to the free drug according to the MIC assay.29In addition, we have tested for potential drug-related toxicity of encapsulated BQ in vivo, which, when compared to its free form showed an improvement in survival. Even upon injection of the highest possible dose, restricted due to injectable volume constraints, a higher survival was observed in BQ NPs group compared to those receiving the free drug. Although the difference was only 10%, the deaths in the group receiving free BQ increased over time while deaths in the group injected with BQ NPs occurred only within the first 24 h post injection, after which survival remained stable. One explanation for this observation could be that the mortality observed in the group receiving NPs were the result of injection injury. However, the difference in toxicity between the encapsulated BQ and the free drug group could also be due to the toxicity of the DMSO used to dissolve the drug. No toxic solvent was used in the NP formulations and dimethyl sulfoxide (DMSO) was needed for injections of the free hydrophobic drug. In this regard, BQ NPs provided a much safer alternative, as NPs without drug were non-toxic. Besides, edemas were seen in most of the zebrafish embryos injected with the Free BQ, most likely due to the high permeability of DMSO that causes it to leak into tissues upon injection into the vasculature.
Apart from toxicity, we evaluated therapeutic effects of different doses of BQ NPs, which showed an improved survival against fish TB relative to the free drug. In our view this can be ascribed to two main reasons: 1 While free drug mixed with DMSO likely distributes evenly in the zebrafish following administration, NP-BQ accumulate rapidly into granulomas (about 3.4% of the injected dose within 24 h) before being taken up by the endothelium (see below). 2 Nanoparticles degrade slowly into endothelial cells and this contributes, unlike free drug administered with DMSO, to sustained release of bedaquiline that can extert its antitubercular activity over several days.
We evaluated NP activity in comparison with free drug via two analyses: survival assay – measuring number of deaths post-treatment in infected zebrafish embryos and Fluorescence pixel count (FPC) analysis that quantified bacterial growth in the treated embryos. Both analyses showed an improvement of survival and a reduction in bacterial growth (Fig. 5 and ESI Fig. 4†) of M.m infected zebrafish embryos post-treatment with BQ NPs. Although FPC is an important assay, we consider the results less reliable to assess drug potency than survival analysis. The reason is that, as the first embryos started to die due to infection, these were removed before imaging took place for FPC and only the live ones were considered for the analysis. Since the dead fish displayed the highest FPC, choosing the day for analysis where embryos have already died weakens the analysis of the surviving embryos.
Behavior of NPs in vivo
The characterization of NPs, especially their biological interactions and fate after in vivo administration is crucial for optimization of NP design to increase their efficacy and reduce toxicity associated with the encapsulated drugs for better therapeutic applications. To study the biodistribution of intravenously injected BQ NPs in vivo, we used a combination of live imaging and electron microscopy that enabled a more detailed and a higher-resolution study of biological interactions of NPs.We found that BQ NPs (M30-RhB-BQ) were short circulating in the embryos, since within minutes of injection into the vasculature, almost half of them disappeared from the circulation. This could possibly indicate their uptake by either blood-resident macrophages,26 endothelial cells lining the vasculature, or both.41 In a previous study in the blood infection model, following injection of poly (lactide co-glycolide) (PLGA) NPs, all of them were rapidly engulfed by the blood-resident macrophages that delivered the NPs to the blood associated granulomas.26 However, in the case of BQ NPs, most of them were found to localize inside the endothelial cells lining the vasculature instead to macrophages in blood-circulation. Surprisingly, when we assessed the accumulation of BQ NPs in neural tube granulomas, a significant amount was localized in the granuloma region compared to the nearby and equivalent uninfected region (Fig. 3C). A similar degree of accumulation has been shown for other short-circulating NPs, for example, PLGA NPs had approximately 3% accumulation in the granulomas at 12 h post-injection.33 However, compared to the long circulating NPs, BQ NPs seemed to accumulate less in the granulomas. For instance, 6.5% of PEGylated liposomes were found to accumulate in neural tube granulomas in just 4 h after injection33 while only about 3.4% of BQ NPs localized in the granulomas in 24 h post-injection. Again, the possible explanation could be the massive uptake of BQ NPs by endothelial cells rather than blood-resident macrophages ignoring “passive” extravasation as potential mechanism.
The endothelial cells lining the vasculature have been found to be active in the uptake of short circulating NPs,34,41 which was also the case for BQ NPs. Our initial investigation of biodistribution of BQ NPs was performed by live-imaging, using an epi-fluorescence stereomicroscope, on wild type zebrafish embryos that were injected with red fluorescent NPs. Already 5 minutes following intravenous injection, almost half of the BQ NP had left the blood circulation, and after 1 hour, almost all of them were found to be cleared from the circulation. The NPs seemed to be in close association with the cells lining the vasculature (especially those lining the PCV), suggesting possible interactions with those cells, which were again analyzed with high-resolution imaging using a spinning disk confocal microscope. Upon analysis of optical sections from the PCV, a massive number of BQ NP appeared to enter the endothelial cells.
To differentiate between stable binding to the cell surface and uptake, a higher resolution method, namely EM was used that can better identify the localization of NPs. EM analysis showed selective NP uptake into the cells of the vascular endothelium, mainly of the PCV but also in the dorsal aorta or intersegmental vessels during 4 h of the circulation. Ultrastructural analysis alone lacks specific markers to establish unequivocally the compartments where the NP are localized within the vasculature endothelium. Therefore, we proceeded with two different EM methodologies that would allow us to identify NP location. We first injected colloidal gold (easily identifiable due to its high electron density) which is known to be internalized in endothelial cells and reach late endosomes and lysosomes (which we classify as ‘lysosomes’ for simplicity.42,43 Four hours after the intravenous injection of BQ NP, particles within the size range of our BQ NP were found to selectively accumulate in lysosomes together with the gold. A further confirmation of this finding was obtained using a combined light and electron microscopy method, CLEM method, where a section of zebrafish embryo previously injected with fluorescent BQ NP was imaged first by light microscopy and then by EM.
Endothelial uptake of NP
According to Rességuier and colleagues, one reason for endothelial uptake of NPs could be associated with the charge of NPs. The negatively charged PLA NPs used in their study were rapidly internalized by endothelial cells – scattered along the entire vasculature – as well as by macrophages in circulation.41 Similarly, in a study by Campbell et al., the internalization of a wide range of anionic NPs was found to be inside the endothelial cells, although restricted to specific venous regions of the zebrafish embryos.44While NP uptake into endothelial cells may not be desirable for treatment against TB, evidence for potential benefits of endothelial uptake of NPs was shown recently, where a new and striking mechanism of NP transfer i.e., from endothelial cells to macrophages was observed.41 This new insight into NP interaction with the vascular endothelium has the potential to open new possibilities of macrophage targeting for anti-tubercular nanotherapeutics.
Conclusion
In this study, we took advantage of the optically transparent vertebrate fish model, the zebrafish embryo to understand the biodistribution and biological interactions of BQ NPs. Using high-resolution non-invasive live imaging, as well as EM, NP interactions at the tissue, cellular and ultrastructural level was analyzed. BQ NPs had short circulation times, because of their rapid and efficient uptake by vascular endothelial cells. Our findings demonstrate the importance of testing NP-based therapies in vivo, particularly in the field of nanomedicine, which requires extensive studies on both the vehicle and the drug along with their combined formulations to be of clinical relevance. To our knowledge, this is the first study assessing the in vivo activity and distribution of a NP formulation of BQ. Encapsulating BQ in polymeric micellar NPs not only reduced drug-related toxicity, but also improved survival and reduced bacterial burden post-treatment in M.m infected zebrafish embryos. Taken together, we propose an improved in vivo testing regime for NP-based drugs. Our findings open possibilities for increasing clinical application of this novel and powerful drug BQ against both drug-susceptible and MDR-TB.Abbreviations
| TB | Tuberculosis |
| NPs | Nanoparticles |
| BQ | Bedaquiline. |
Author contributions
Madhavi Bhandari has carried out all the zebrafish experiments under the supervision of Federico Fenaroli and Gareth Griffiths. Héctor Soria-Carrera prepared and characterized nanoparticles under the supervision of Rafael Martín-Rapún and Jesús M. de la Fuente. Jens Wohlmann supervised with electron microscopy experiments. Nils-Jørgen Knudsen Dal assisted with zebrafish experiments and performed the in vitro analysis of Bedaquiline activity. All authors have contributed and given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors report no conflict of interest. This work was supported by the Norwegian Research Council grants – Frimedbio (275873) and Bedrehelse (273319). In addition, this work was funded through the grant PID2019–109333RB-I00 funded by MCIN/AEI/10.13039/501100011033 (Ministerio de Ciencia e Innovación/Agencia Estatal de Investigación, Spain), through project LINKA20270 i-Link+ 2019 funded by CSIC and through Fondo Social de la DGA (grupo DGA E15_20R). HSC is grateful for a predoctoral fellowship FPU2016/02456 funded by Ministerio de Universidades (Spain). We acknowledge the generous support of the EM facility (head: Norbert Roos) and the Imaging Platform (head: Oddmund Bakke) at the Department of Biosciences of the University of Oslo. We also gratefully acknowledge the assistance of Eugenio Vispe and the Chromatography and Spectroscopy Service of the Instituto de Síntesis Química y Catálisis Homogénea (CSIC-Universidad de Zaragoza). Part of Fig. 1 was created with https://BioRender.com.References
- T. M. Daniel, Respir. Med., 2006, 100, 1862–1870 CrossRef PubMed.
- World Health Organization, Global tuberculosis report, WHO, 2021, ISBN 978-92-4-003702-1 Search PubMed.
- J. Chakaya, M. Khan, F. Ntoumi, E. Aklillu, R. Fatima, P. Mwaba, N. Kapata, S. Mfinanga, S. E. Hasnain, P. Katoto, A. N. H. Bulabula, N. A. Sam-Agudu, J. B. Nachega, S. Tiberi, T. D. McHugh, I. Abubakar and A. Zumla, Int. J. Infect. Dis., 2021, 113(Suppl 1), S7–S12 CrossRef CAS PubMed.
- R. M. Houben and P. J. Dodd, PLoS Med., 2016, 13(10), e1002152 CrossRef PubMed.
- L. De Matteis, D. Jary, A. Lucía, S. García-Embid, I. Serrano-Sevilla, D. Pérez, J. Ainsa, F. Navarro and J. De la Fuente, Chem. Eng. J., 2018, 340, 181–191 CrossRef CAS.
- E. L. Nuermberger, M. K. Spigelman and W. W. Yew, Respirology, 2010, 15, 764–778 CrossRef PubMed.
- A. Zumla, P. Nahid and S. T. Cole, Nat. Rev. Drug Discovery, 2013, 12, 388–404 CrossRef CAS PubMed.
- W. W. Yew, C. Lange and C. C. Leung, Eur. Respir. J., 2011, 37, 441–462 CrossRef CAS PubMed.
- E. D. Chan and M. D. Iseman, Curr. Opin. Infect. Dis., 2008, 21, 587–595 CrossRef CAS PubMed.
- A. Bahuguna and D. S. Rawat, Med. Res. Rev., 2020, 40, 263–292 CrossRef PubMed.
- A. H. Diacon, A. Pym, M. Grobusch, R. Patientia, R. Rustomjee, L. Page-Shipp, C. Pistorius, R. Krause, M. Bogoshi, G. Churchyard, A. Venter, J. Allen, J. C. Palomino, T. De Marez, R. P. van Heeswijk, N. Lounis, P. Meyvisch, J. Verbeeck, W. Parys, K. de Beule, K. Andries and D. F. Mc Neeley, N. Engl. J. Med., 2009, 360, 2397–2405 CrossRef CAS PubMed.
- A. H. Diacon, P. R. Donald, A. Pym, M. Grobusch, R. F. Patientia, R. Mahanyele, N. Bantubani, R. Narasimooloo, T. De Marez, R. van Heeswijk, N. Lounis, P. Meyvisch, K. Andries and D. F. McNeeley, Antimicrob. Agents Chemother., 2012, 56, 3271–3276 CrossRef CAS PubMed.
- E. Cox, FDA accelerated approval letter to Janssen Research and Development, Food and Drug Administration, Washington, DC, 2012, Available at: http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2012/204384Orig1s000ltr.pdf Search PubMed.
- R. Singh, U. Manjunatha, H. I. Boshoff, Y. H. Ha, P. Niyomrattanakit, R. Ledwidge, C. S. Dowd, I. Y. Lee, P. Kim and L. Zhang, Science, 2008, 322, 1392–1395 CrossRef CAS PubMed.
- W. World Health Organization, 2020, Licence: CC BY-NC-SA 3.0 IGO.
- F. Conradie, A. H. Diacon, N. Ngubane, P. Howell, D. Everitt, A. M. Crook, C. M. Mendel, E. Egizi, J. Moreira, J. Timm, T. D. McHugh, G. H. Wills, A. Bateson, R. Hunt, C. Van Niekerk, M. Li, M. Olugbosi, M. Spigelman and T. B. T. T. Nix, N. Engl. J. Med., 2020, 382, 893–902 CrossRef CAS PubMed.
- H. Patel, R. Pawara, K. Pawara, F. Ahmed, A. Shirkhedkar and S. Surana, Tuberculosis, 2019, 117, 79–84 CrossRef CAS PubMed.
- R. P. van Heeswijk, B. Dannemann and R. M. Hoetelmans, J. Antimicrob. Chemother., 2014, 69, 2310–2318 CrossRef CAS PubMed.
- E. B. Chahine, L. R. Karaoui and H. Mansour, Ann. Pharmacother., 2014, 48, 107–115 CrossRef PubMed.
- J. Kreuter, Eur. J. Drug Metab. Pharmacokinet., 1994, 19, 253–256 CrossRef CAS PubMed.
- J. Trousil, Z. Syrová, N. K. Dal, D. Rak, R. Konefał, E. Pavlova, J. Matějková, D. Cmarko, P. Kubíčková, O. Pavliš, T. Urbánek, M. Sedlák, F. Fenaroli, I. Raška, P. Štěpánek and M. Hrubý, Biomacromolecules, 2019, 20, 1798–1815 CrossRef CAS PubMed.
- Z. Baranyai, H. Soria-Carrera, M. Alleva, A. C. Millán-Placer, A. Lucía, R. Martín-Rapún, J. A. Aínsa and J. M. de la Fuente, Adv. Ther., 2021, 4, 2000113 CrossRef.
- D. L. Clemens, B. Y. Lee, M. Xue, C. R. Thomas, H. Meng, D. Ferris, A. E. Nel, J. I. Zink and M. A. Horwitz, Antimicrob. Agents Chemother., 2012, 56, 2535–2545 CrossRef CAS PubMed.
- A. Sharma, R. Pandey, S. Sharma and G. Khuller, Int. J. Antimicrob. Agents, 2004, 24, 599–604 CrossRef CAS PubMed.
- R. Kalluru, F. Fenaroli, D. Westmoreland, L. Ulanova, A. Maleki, N. Roos, M. Paulsen Madsen, G. Koster, W. Egge-Jacobsen, S. Wilson, H. Roberg-Larsen, G. K. Khuller, A. Singh, B. Nyström and G. Griffiths, J. Cell Sci., 2013, 126, 3043–3054 CAS.
- F. Fenaroli, D. Westmoreland, J. Benjaminsen, T. Kolstad, F. M. Skjeldal, A. H. Meijer, M. van der Vaart, L. Ulanova, N. Roos, B. Nyström, J. Hildahl and G. Griffiths, ACS Nano, 2014, 8, 7014–7026 CrossRef CAS PubMed.
- J. Trousil, Z. Syrová, N.-J. K. Dal, D. Rak, R. Konefał, E. Pavlova, J. Matějková, D. Cmarko, P. Kubíčková, O. Pavliš, T. Urbánek, M. Sedlák, F. Fenaroli, I. Raška, P. Štěpánek and M. Hrubý, Biomacromolecules, 2019, 20, 1798–1815 CrossRef CAS PubMed.
- R. Pandey, A. Sharma, A. Zahoor, S. Sharma, G. Khuller and B. Prasad, J. Antimicrob. Chemother., 2003, 52, 981–986 CrossRef CAS PubMed.
- H. Soria-Carrera, A. Lucía, L. De Matteis, J. A. Aínsa, J. M. de la Fuente and R. Martín-Rapún, Macromol. Biosci., 2019, 19, 1800397 CrossRef PubMed.
- N. D. Lawson and B. M. Weinstein, Dev. Biol., 2002, 248, 307–318 CrossRef CAS PubMed.
- N.-J. K. Dal, M. Speth, K. Johann, M. Barz, C. Beauvineau, J. Wohlmann, F. Fenaroli, B. Gicquel, G. Griffiths and N. Alonso-Rodriguez, Disease Models & Mechanisms, 2021, 15(1), dmm049147 Search PubMed.
- K. N. Adams, K. Takaki, L. E. Connolly, H. Wiedenhoft, K. Winglee, O. Humbert, P. H. Edelstein, C. L. Cosma and L. Ramakrishnan, Cell, 2011, 145, 39–53 CrossRef CAS PubMed.
- F. Fenaroli, U. Repnik, Y. Xu, K. Johann, S. Van Herck, P. Dey, F. M. Skjeldal, D. M. Frei, S. Bagherifam, A. Kocere, R. Haag, B. G. De Geest, M. Barz, D. G. Russell and G. Griffiths, ACS Nano, 2018, 12, 8646–8661 CrossRef CAS PubMed.
- N. K. Dal, A. Kocere, J. Wohlmann, S. Van Herck, T. A. Bauer, J. Resseguier, S. Bagherifam, H. Hyldmo, M. Barz, B. G. De Geest and F. Fenaroli, Small, 2020, 16, e1906719 CrossRef PubMed.
- E. Carlemalm, W. Villiger, J. A. Hobot, J. D. Acetarin and E. Kellenberger, J. Microsc., 1985, 140(Pt 1), 55–63 CrossRef CAS PubMed.
- G. Griffiths, A. McDowall, R. Back and J. Dubochet, J. Ultrastruct. Res., 1984, 89, 65–78 CrossRef CAS PubMed.
- L. Ghitescu, A. Fixman, M. Simionescu and N. Simionescu, J. Cell Biol., 1986, 102, 1304–1311 CrossRef CAS PubMed.
- G. E. Palade, Circulation, 1961, 24, 368–388 CrossRef CAS PubMed.
- I. Kmentova, H. S. Sutherland, B. D. Palmer, A. Blaser, S. G. Franzblau, B. Wan, Y. Wang, Z. Ma, W. A. Denny and A. M. Thompson, J. Med. Chem., 2010, 53, 8421–8439 CrossRef CAS PubMed.
- N.-J. K. Dal, G. Schäfer, A. M. Thompson, S. Schmitt, N. Redinger, N. Alonso-Rodriguez, K. Johann, J. Ojong, J. Wohlmann, A. Best, K. Koynov, R. Zentel, U. E. Schaible, G. Griffiths, M. Barz and F. Fenaroli, J. Controlled Release, 2023 DOI:10.1016/j.jconrel.2023.01.037.
- J. Rességuier, J. P. Levraud, N. K. Dal, F. Fenaroli, C. Primard, J. Wohlmann, G. Carron, G. W. Griffiths, D. Le Guellec and B. Verrier, J. Controlled Release, 2021, 331, 228–245 CrossRef PubMed.
- G. Griffiths and J. Gruenberg, Trends Cell Biol., 1991, 1, 5–9 CrossRef CAS PubMed.
- G. Griffiths, B. Hoflack, K. Simons, I. Mellman and S. Kornfeld, Cell, 1988, 52, 329–341 CrossRef CAS PubMed.
- F. Campbell, F. L. Bos, S. Sieber, G. Arias-Alpizar, B. E. Koch, J. Huwyler, A. Kros and J. Bussmann, ACS Nano, 2018, 12, 2138–2150 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01835g |
| ‡ Present address: Faculty of Health Sciences, Department of Life Sciences and Health Pharmacy, Oslo Metropolitan University, Pilestredet 50, 0167 Oslo, Norway. |
| This journal is © The Royal Society of Chemistry 2023 |