
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReaching new lights: a review on photo-controlled nanomedicines and their in vivo evaluation
Rik C. P. A.
Remmers
and
Kevin
Neumann
 *
*
Institute for Molecules and Materials, Radboud University, Nijmegen, Heyendaalseweg 135, 6525 AJ Nijmegen, the Netherlands. E-mail: kevin.neumann@ru.nl
First published on 12th January 2023
Abstract
The selective and efficient delivery of bioactive molecules to sites of interest remains a formidable challenge in medicine. In recent years, it has been shown that stimuli-responsive drug delivery systems display several advantages over traditional drug administration such as an improved pharmacokinetic profile and the desirable ability to gain control over release. Light emerged as one of the most powerful stimuli due to its high biocompatibility, spatio-temporal control, and non-invasiveness. On the road to clinical translation, various chemical systems of high complexity have been reported with the aim to improve efficacy, safety, and versatility of drug delivery under complex biological conditions. For future research on the chemical design of such photo-controlled nanomedicines, it is essential to gain an understanding of their in vivo translation and efficiency. Here, we discuss photo-controlled nanomedicines that have been evaluated in vivo and provide an overview of the state-of-the-art that should guide future research design.
1. Introduction
Drug delivery systems (DDS) hold great promise for the development of new treatment opportunities for several diseases. Chiefly, a drug that is formulated by a carrier vehicle presents several advantages over a free drug such as reduced clearance and systemic toxicity. Various structures have been employed as drug carriers, including liposomes, micelles, metal-nanoparticles, and hydrogel drug depots.1–5 Notably, several DDS formulations of clinically used drugs have been approved by the American Food and Drug Administration.6 Examples include DOXIL® (Janssen), a liposome-encapsulated form of the chemotherapeutic doxorubicin, and Estrasob™ (Novavax), a micellar DDS for the delivery of estradiol. More recently, a small interfering RNA (siRNA) delivering nanoparticle ONPATTRO® (Alnylam) has been approved for clinical usage.7 These approvals highlight that there is a strong clinical interest in advanced DDS, and that such systems could improve drug performance, mainly by enhancing drug pharmacokinetics and reducing side-effects.However, these traditional DDS still suffer from major drawbacks. Drug release cannot be actively controlled, which potentially leads to premature leakage of the encapsulated drug in the bloodstream. Indeed, many DDS suffer from a so-called burst release, leaking a portion of their encapsulated drugs upon intravenous injection.8,9 This makes traditional DDS design somewhat contradictory; increasing carrier stability can prevent burst release, but then drug liberation at the desired site becomes a challenge. Additionally, just like a free drug, DDS that deliver chemotherapy cargos are still susceptible to the formation of drug-resistant tumors, diminishing their usefulness. Furthermore, the clinical value of higher tumor drug concentration resulting from nanomedicines has been contested.10 It has been suggested that tumor drug accumulation is not the main driver for clinical success, with delivery of combination therapy and better on-site/off-side drug balance being of greater importance. While traditional DDS are able to prolong drug blood circulation, enhance the bioavailability, and reduce systemic toxicity, they have unfortunately not resulted in significant improvement of the overall survival rate of patients.11–14
Consequently, the next generation of so-called smart DDS addresses the challenges displayed by traditional DDS and instead of the “inject and forget” mode of action, have been designed to be stimulus responsive. Stimuli can be divided into internal stimuli such as pH, enzymes and redox environment, or external stimuli including light, magnetic fields, and acoustic stimuli.15,16 Advantageously, external stimuli offer active control over drug release, while internal stimuli do not. Particularly light is attractive as an external stimulus due to its good biocompatibility, excellent spatio-temporal control, and non-invasiveness. Furthermore, it allows for facile combination with photothermal therapy (PTT) and/or photodynamic therapy (PDT).
Still, light also suffers from a considerable drawback: ultraviolet (UV, 200–400 nm) and visible light (400–700 nm) do not penetrate deeply into tissue, a few millimeters at most.17,18 Furthermore, UV light can cause damage upon elongated periods of exposure.19 While magnetic fields and acoustic stimuli penetrate deeper, these approaches have their own drawbacks. For example, both magnetic fields and acoustic stimuli require exotic equipment, meaning a visit to a dedicated facility would be necessary to trigger the DDS.20,21 Near-infrared (NIR, 700–1400 nm) light seems the most promising, owing to its superior penetration depth and improved safety profile.21–23 Others have opted to employ deeply penetrating radio waves or X-rays to achieve controlled drug release. We would like to refer to timely literature that discusses drug release with these stimuli.24–28
Major steps have been taken in the usage of NIR light for DDS, with the majority of recently reported DDS being sensitized to the deeper penetrating NIR light and demonstrating great in vivo therapeutic effect.29–33 Additionally, advancements have been made in the clinical efficacy of light-sensitive DDS through combination therapy and diagnostics delivery.34–38 Clinical safety has also been addressed, with some reported DDS capable of payload liberation under irradiation with low power density light.39–43 Furthermore, active targeting of DDS and in situ (refillable) drug implants have been leveraged to localize drug concentrations at diseased sites and reduce them elsewhere, avoiding systemic toxicity.44–48 Photosensitive DDS have also been extended beyond just small molecules, with some capable of biopharmaceutical delivery (Fig. 1).49,50 Notably, some of these advances have been made with body-native cells, employing them as tumor-targeted drug delivery vectors or as a circulatory dynamic drug depot.51,52
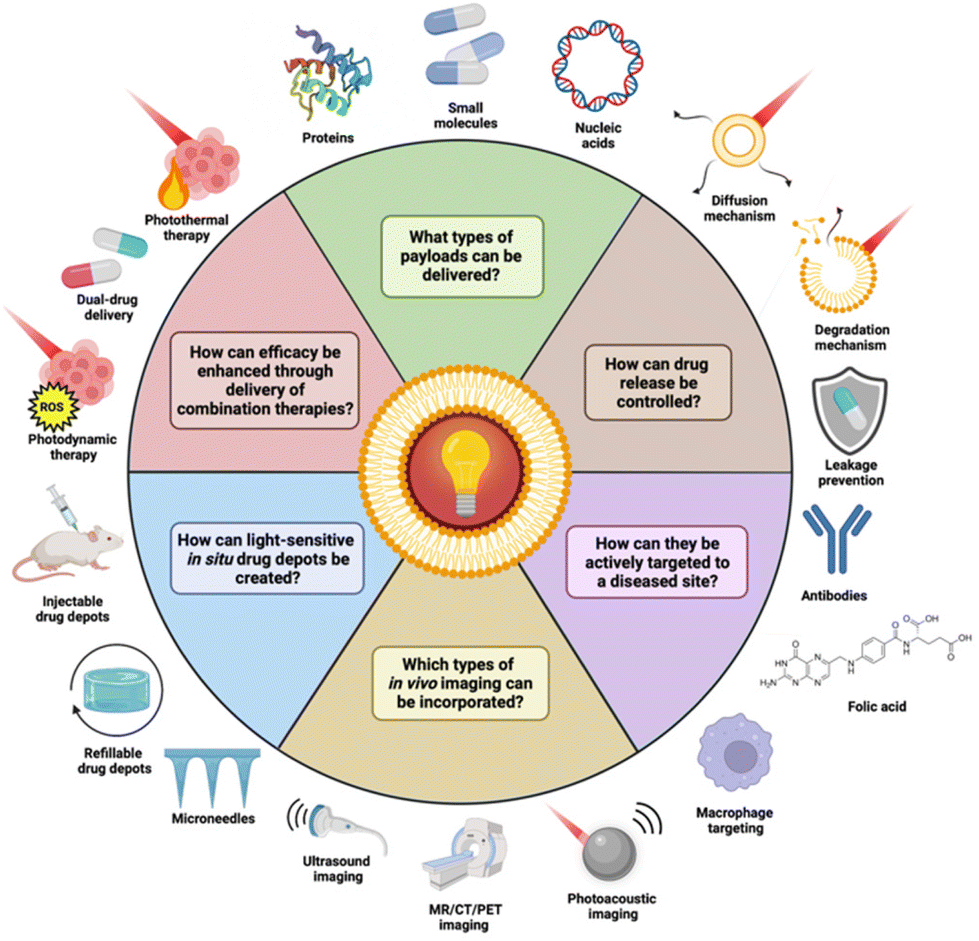 | ||
| Fig. 1 Recent advances surrounding light-sensitive DDS. In this review, we will provide an overview of these applications and the current state-of-the-art. Created with BioRender.com. | ||
In this review, we will discuss these recent advancements in the development of light-responsive DDS. As one of the major promises of DDS formulations entails improved pharmacokinetics and reduced systemic toxicity, we feel that a focus on photo-controllable DDS that have been evaluated in vivo is beneficial to provide a comprehensive overview. For a comprehensive overview of in vitro evaluated systems, we would like to refer to others.53,54 Additionally, a review discussing the medical applications of nanomaterials from a broader perspective can be found.55
2. Combination therapy DDS
While DDS serve to improve the pharmacokinetic profile of a drug, they do not amend one of the most persistent issues in cancer treatment, namely drug resistance to chemotherapy. A monotherapy often fails to fully destroy the tumor, and instead induces drug resistance.56 In recent years, much work has focused on not only the enhancement of pharmacokinetic properties by DDS, but also on increasing the efficacy provided by treatment with a DDS. Ideally, a singular DDS must be capable of enabling combination therapy. This combination therapy should be orthogonal, meaning that the individual constituent therapies do not target the same pathway. Otherwise, drug resistance could still arise.57Combination therapy delivered by DDS can be accomplished in a variety of forms (Table 1). For example, instead of loading a single chemotherapeutic drug, multiple anti-cancer drugs can be packed in a DDS for synergistic chemotherapy. In a recent example, Wu et al. synthesized a photo-responsive polymeric nanoparticle carrying both a cisplatin prodrug in the polymer backbone and doxorubicin, which could be released under green light irradiation.58In vivo, these nanoparticles demonstrated stronger tumor growth inhibition in Skov3 tumor bearing mice compared to nanoparticles without DOX and free cisplatin. In other work, Sun et al. designed metallopolymer micelles that carried a Ru-chlorambucil drug conjugate. The metallopolymer employed an amphiphilic PEG-b-Poly(6-(4-cyanophenoxy)hexylmethacrylate) block copolymer as the backbone to which the Ru-chlorambucil drug conjugate was grafted through a photocleavable Ru-N coordination bond. This grafted metallopolymer self-assembled to form micelles with a hydrodynamic diameter of ∼22 nm.59 Upon exposure to 660 nm light, the Ru–N bond in the polymer is cleaved and the drug conjugate is liberated from the micelle. In HeLa tumor-bearing mice, these micelles effectively inhibited tumor growth upon light irradiation.
| DDS | Payload | Model | Admin. | Irrad. | |
|---|---|---|---|---|---|
| Polym. NP | DOX/cis-platin | Skov3/RFP mouse xenograft | Intrav. | 500 nm, 20 mW cm−2, 10 min | 58 |
| Micelle | Chlorambucil | HeLa mouse xenograft | Intratum. | 660 nm, 360 J cm−2 | 59 |
| Liposome | DOX/sunitinib | B16F10 mouse xenograft | Intrav. | 660 nm, 300 mW cm−2, 10 min | 60 |
| Polym. NP | siRNA/DOX | MCF-7/ADR mouse xenograft | Intratum. | 365 nm, 1 W cm−2, 10 min | 50 |
| Micelle | DOX/PDT | CT-26 mouse xenograft | Intrav. | 670 nm, 100 mW cm−2, 10 min | 38 |
| Micelle | Sparfloxacin/PDT | 4T1 mouse xenograft | Intrav. | 980 nm, 1 W cm−2, 5 min | 33 |
| Micelle | DOX/PDT | HT-29 mouse xenograft | Intrav. | 808 nm, 1 W cm−2, 5 × 30 s | 35 |
| Cerasome | DOX/PTT | HT-29 mouse xenograft | Intrav. | 808 nm, 2 W cm−2, 10 min | 29 |
| Nanorods | Cinnamaldehyde/PTT | S. aureus infect.mice | Topical gel | 808 nm, 1 W cm−2, 10 min | 75 |
| Polym. NP | DOX/PDT/PTT | 4T1 mouse xenograft | Intrav. | 785 nm, 650 mW cm−2, 4 min | 32 |
| Liposome | TPZ/PDT/PTT | A549 mouse xenograft | Intrav. | 660 nm, 0.1 W cm−2, 10 min | 77 |
| Polym. Nano-capsule | DOX/PDT/PTT | KB mouse xenograft | Intrav. | 808 nm, 0.3 W cm−2, 5 min | 78 |
Various authors have reported light-controllable DDS capable of sequential drug release. Lai et al. designed photosensitized liposomes from DOX-loaded polymer nanoparticles (40 nm) and the angiogenic inhibitor sunitinib.60 Additionally, the liposomes were hybridized with red blood cell (RBC) membranes in an effort to extend circulation time. RBC membranes were selected specifically to avoid the accelerated blood clearance observed when only polyethylene glycol (PEG) is used, owing to the formation of anti-PEG antibodies in the blood stream.61 Typically, the co-delivery of chemotherapeutic and antiangiogenic agent would be somewhat contradictory, as angiogenesis inhibition can hinder chemotherapy from accumulating at the tumor site.62 However, when exposed to 660 nm light, the sunitinib was quickly released from the liposomes (100% after 5 min), after which the liberated DOX-loaded nanoparticles could internalize into the tumor cells. In mice, this sequential delivery system showed better performance for tumor growth inhibition than liposomes loaded with only DOX-nanoparticles or sunitinib. Additionally, the combinatorial delivery of these drugs strengthened the antitumor immune response. Others have explored sequential siRNA – DOX liberation to improve the efficacy of chemotherapy.50 Photoresponsive polymeric nanoparticles were coated with a cationic redox-sensitive polyethyleneimine shell that absorbed P-glycoprotein (P-gp) siRNA onto its surface. P-gp is part of the ABC transporter facility of proteins, which are known to facilitate (multi)drug resistance in cancer.63 Upon exposure to the reductive tumor microenvironment, the increased neoplastic glutathione concentration facilitated the cleavage of disulfide bonds in the cationic polymer shell, eroding it and, subsequently, releasing the P-gp siRNA.64 Irradiation with light (365 nm) triggered release of the DOX from the nanoparticles (Fig. 2). The benefit of this therapeutic strategy was demonstrated in vivo on mice bearing multidrug-resistant MCF-7/ADR subcutaneous tumors. When DOX-loaded nanoparticles carrying scrambled siRNA were injected intratumorally, only weak tumor growth inhibition resulted. In contrast, when the nanoparticles were decorated with P-gp siRNA, a strong antitumor effect was observed. This difference in therapeutic efficacy highlighted the value of tandem P-gp siRNA – chemotherapy delivery for the treatment of multidrug-resistant tumors. However, the use of high energy light (365 nm) does not seem promising for future translation to clinical settings. Adaption of this DDS to NIR light could be worthwhile future work.
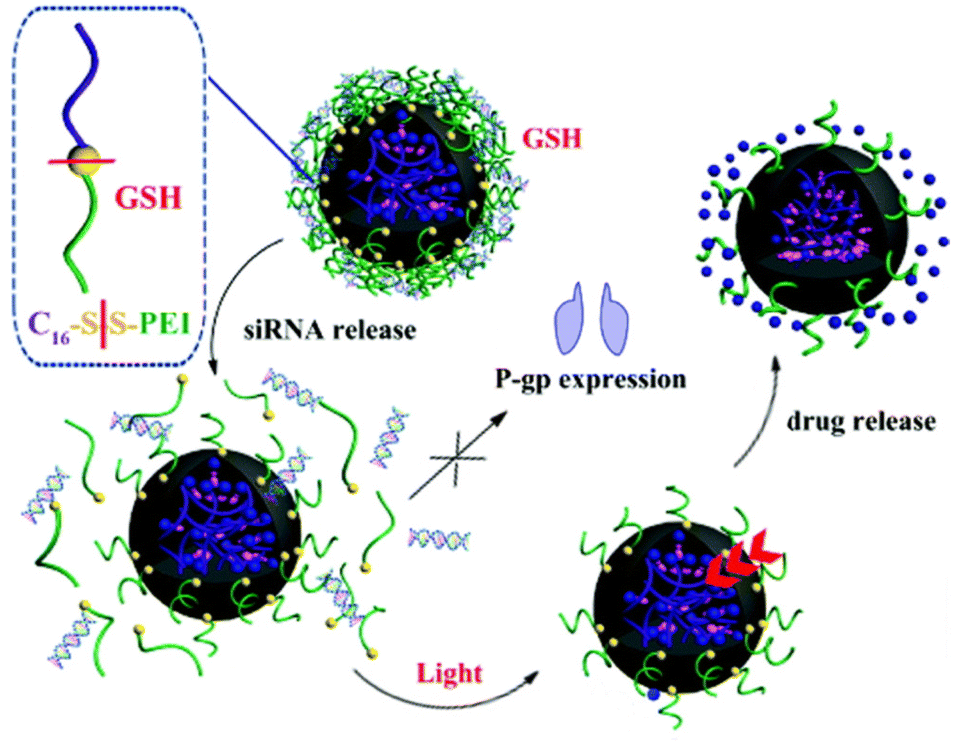 | ||
| Fig. 2 Sequential release of DOX and siRNA triggered by light irradiation and redox environment from dual-responsive polymeric prodrug nanoparticles. Reproduced from ref. 50 with permission from Royal Society of Chemistry, copyright 2018. | ||
Instead of the delivery of two agents, DDS that co-incorporate therapies with a completely different mode of action have gained significant attention. Photodynamic therapy (PDT) does not directly rely on the delivery of a drug molecule, but rather on the in situ generation of reactive oxygen species (ROS) such as hydroxyl radicals (˙OH), singlet oxygen (1O2), and superoxide anions (O2˙−) from molecular oxygen by a photosensitizer.65 Besides causing damage to the tumor via ROS production, PDT also vitalizes the immune system by inducing the presentation of tumor antigens to immune cells.66,67 This makes PDT an attractive partner for chemotherapy DDS. Indeed, various chemophotodynamic DDS have been reported. For example, Uthaman et al. prepared photoactivatable thioketal nanomicelles that co-encapsulated DOX and pheophorbide A, a photosensitizer.38 When internalized into a tumor cell, the increased ROS levels present in tumors would passively cleave the thioketal backbone, resulting in loss of structural integrity and DOX leakage. Irradiation with 670 nm light and subsequent ROS production by the loaded pheophorbide A further compromised the nanomicelle, inducing even more drug release. Furthermore, the authors noted a micelle-invoked antitumor immune response that was not present in any of their control experiments. In other work, Chu et al. reported sparfloaxin loaded micelles in which sparfloaxin acted as both the chemotherapeutic agent and the photosensitizer.33,68 Exposure to a 980 NIR laser induced thermal release of the sparfloxacin, which resulted in strong in vivo murine tumor suppression owing to the combined chemophotodynamic therapy. Additionally, the micelles suppressed tumor metastasis, which was attributed to the PDT induced immune response.
Despite its clear usefulness as cancer therapy, the nature of cancer also inherently limits the maximum effectiveness of PDT. Namely, PDT relies on molecular oxygen for the generation of ROS. However, tumor microenvironments are often hypoxic, stunting the effectiveness of PDT.69 To amend this unwanted dampening of efficacy, Yang et al. designed a NIR-responsive micelle for chemophotodynamic therapy that carried its own supply of oxygen.35 The micelles employed a perfluoroalkane core capable of carrying molecular oxygen linked to a hydrophilic PEG outer surface via a IR780 scaffold, which served both as the photosensitizer and a light-triggered degradable linker. In vivo, the oxygen-enriched micelles demonstrated a significantly stronger antitumor effect than the control micelles. When the micelles were additionally loaded with DOX, the MCF-7 tumor was nearly fully eliminated. Furthermore, micelle treatment resulted in downregulation of hypoxia-inducible factor 1-alpha (HIF-1α), demonstrating in vivo tumor hypoxia relief. Crucially, HIF-1α is known to upregulate the expression of P-gp.70 Potentially, these oxygen-carrying micelles could not only provide more efficient photodynamic therapy but could also increase the efficacy of chemotherapy by virtue of their indirect knockdown of tumor P-gp expression. The work conducted by Yang and coworkers clearly highlights the value of resolving local hypoxia in tumors for PDT.
Similar to photodynamic therapy, photothermal therapy (PTT) has also been investigated for synergistic therapy by light-sensitive DDS. PTT is mediated by photothermal conversion agents that absorb NIR light and convert it into heat.69 Besides the ability of PTT to not only prompt hyperthermia-induced cancer cell death in tumor tissue, PTT makes DDS more available to cancer cells by increasing tumor blood flow and decreasing tumor interstitial fluid pressure.71,72 Recently, Zhang et al. reported light-sensitive DOX-loaded cerasomes, a lipid bilayer vesicle with an external silica coating for combined chemophotothermal therapy of colon cancer.29,73 The cerasome membrane was grafted with the photothermal conversion agent cyanine 7, which upon NIR exposure would generate heat. The generated heat destabilized the cerasome membrane and subsequently triggered DOX release and induced hyperthermia in the tumor tissue. In a HT-29 xenograft mouse model, the cerasomes effectively ablated HT-29 tumors upon 808 nm light irradiation for 10 minutes while preserving body weight, with some mice only showing black scars at the tumor site which fell off after 15 days. No tumor recurrence was noted during this time. Others designed polymeric micelles loaded with both DOX and IR780 that released their payload through dissociation at their upper critical solution temperature.74 This release mechanism provided quick and thorough DOX release from the micelles upon exposure to 808 nm light. In vivo, the micelles strongly stunted growth of MCF-7 tumors, displaying ∼90% growth inhibition. Interestingly, hyperthermia induced downregulation of P-gp, with a 72% decrease in expression levels. Possibly, this could be explained by the fact tumor hyperthermia can alleviate hypoxia, knocking down HIF-1α and thus resulting in decreased P-gp expression.70,71 Nevertheless, the clear reduction in P-gp levels undoubtedly increased the micelle antitumor efficacy in vivo.
DDS capable of delivering drugs and PTT have also seen applications outside the field of cancer treatment. Notably, Sun et al. designed copper-gallic acid-cinnamaldehyde-polydopamine nanorods for photo-controllable antibacterial therapy.75 When exposed to 808 nm light, the cinnamaldehyde payload was released, alongside heat via photothermal conversion. The cinnamaldehyde acted as the antibacterial agent, while the generated heat compromised the structural integrity of the bacterial cells, providing synergistic sterilization. In mice, these nanorods efficiently combatted S. aureus infection, with wound healing being significantly faster in the nanorod + NIR treated group. Furthermore, no signs of animal cell or tissue toxicity were observed, hinting at the biocompatibility of the design.
Considering the increased efficacy of combination treatment, some groups designed trimodal DDS, capable of delivering small molecule drugs, PDT, and PTT synergistically. For cancer treatment, the combination of PDT and PTT is especially powerful. Under normal circumstances, the efficacy of PDT is limited by the hypoxic tumor microenvironment, but -as previously mentioned- PTT is able to relieve tumor hypoxia, which in turn could increase PDT-mediated ROS generation.71 In this view, Liu et al. reported a photo-controllable docetaxel-loaded nanoparticle capable of trimodal therapy that demonstrated both high ROS quantum yield (62%) and photothermal conversion efficiency (38%).32 This was achieved by incorporating a novel thiophene-conjugated photosensitizer based on boron dipyrromethene into the nanoparticles. In addition to the high photo-efficiency of the dye, conjugating thiophene to the dye scaffold increased the absorption coefficient of the dye and redshifted the wavelength of maximum absorbance from 738 nm to 752 nm, which has marginally better tissue penetration.76 Furthermore, the nanoparticles had their surface equipped with folic acid for active targeting of cancer cells (please note that active targeting will be discussed in more detail in paragraph 5). In a 4T1 tumor xenograft mouse model, these nanoparticles effectively ablated tumor growth under 785 nm light irradiation, demonstrating the high antitumor efficacy of their trimodal therapy delivery. In other work, Dai et al. synthesized trimodal liposomes that encapsulated a hypoxia-activated tirapazamine (TPZ) prodrug, which was converted to its active radical form under PDT conditions.77 Notably, this system features cascade activation; the liposomes release their content under 808 nm light exposure via photothermal conversion agent-mediated heating, which also provides the PTT. The tumor is then further irradiated with 660 nm light to induce PDT and activate the TPZ prodrug for chemotherapy (Fig. 3).
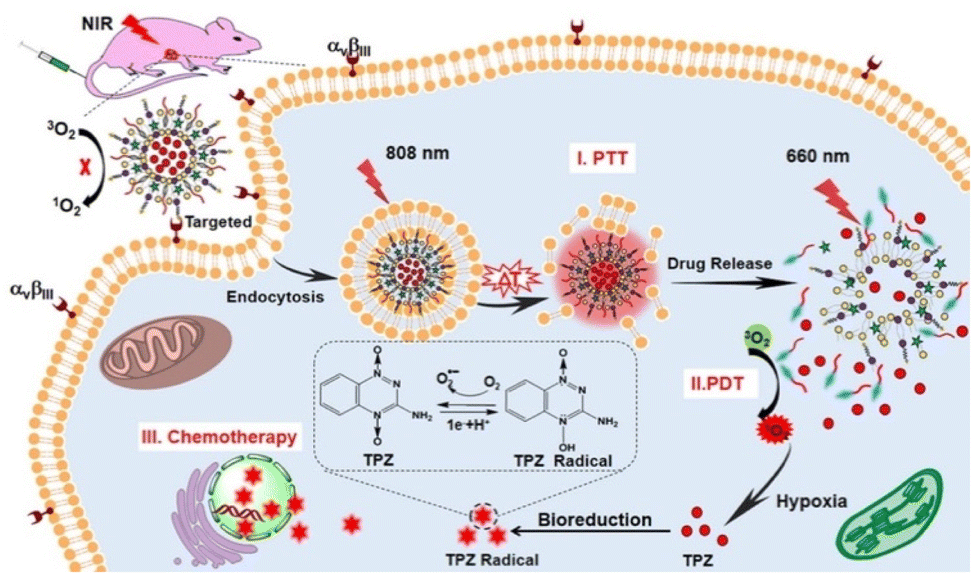 | ||
| Fig. 3 Combination Therapy Triggered by NIR Light. Reproduced from ref. 77 with permission from American Chemical Society, copyright 2019. | ||
The anti-cancer efficacy of the liposomes was demonstrated in A549 tumor-bearing nude mice, where they achieved roughly 88% tumor growth inhibition upon 808/660 nm light exposure. Interestingly, irradiating the tumor with 808 nm light followed by 660 nm showed much better efficacy than irradiation with only 808 nm light, demonstrating the increased efficacy of the trimodal therapy over mere PTT alone. Furthermore, this system offers increased controllability over therapy delivery.
In order to further improve the efficacy of trimodal therapy DDS, Wang et al. fabricated DOX-loaded size-switchable nanocapsules that were sensitized to both the lower pH of the tumor microenvironment and externally applied NIR light (Fig. 4).78 Importantly, controlled size-switching behavior provides active control over the pharmacokinetic and pharmacodynamic properties of nanoparticles. Larger size (100–200 nm) endows nanoparticles with a better half-life in circulation but limits their ability to penetrate the tumor. In contrast, smaller size (4–20 nm) facilitates superior tumor penetration, but also makes nanoparticles prone to drug leakages and renal clearance.79,80 The reported nanoparticles shrink their diameter from 220 nm to 55 nm upon 808 nm laser irradiation in a moderately acidic environment with a pH of 6.5. Since the tumor microenvironment is also of acidic nature, this means the nanocapsules could be capable of this size-switching in more complex biological environment; the reported nano-devices possess good circulatory stability with a size around 200 nm, and when irradiated at the tumor site, they shrink to facilitate drug release and deep tumor penetration.81 The authors hypothesize that this size-shrinking is mediated via a change in radial stress caused by heating of the polymer shell above its Tc.
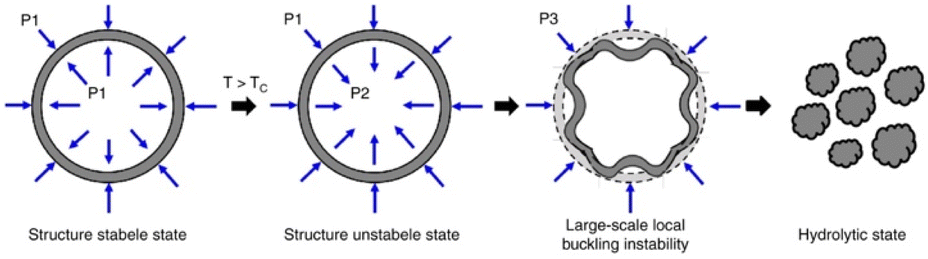 | ||
| Fig. 4 Radial stress analysis of nanocapsules that display size-switchable behaviour (radial stress is represented for the nanocapsules at different stages with P1, P2 and P3). Reproduced from ref. 78 with permission from Springer Nature, copyright 2019. | ||
The nanoparticles strongly inhibited the growth of KB tumors in mice upon NIR light exposure for 5 minutes through their trimodal therapy delivery, presenting a decrease in tumor weight during the 18-day experiment while showing favorable biosafety (Table 1).
Excellent progress has been made adapting photo-controllable DDS for multimodal therapy. Notably, recently reported photo-controllable DDS can synergistically deliver chemotherapy, PDT, and PTT under deeper penetrating NIR light irradiation, demonstrating good in vivo efficacy even after only short exposure times. However, most of these DDS still require light with a high-power density (1 W cm−2), which could hinder their clinical translation due to possible safety concerns. Indeed, clinical studies often utilize light with a lower power density (<150 mW cm−2).82–85 Sensitizing these multimodal DDS to light with a lower power density could be a valuable next step on their path towards the clinic.
3. Control over drug release
Active control over drug release is one of the core principles of stimuli responsive DDS. Current research has not only focused on improving treatment efficacy, but also on enhanced controllability. The ideal DDS should feature robust on/off states, in which the drug is released during the “on” state (stimulus present), and the drug is retained during the “off” state (stimulus absent). In an ideal delivery system, switching between these states should also be near instantaneous, where removal of the stimulus immediately induces a transition from “on” to “off”. Furthermore, in the “on” state, drug release can be fast for immediate total release, or slower for repeated in situ dosing. Drug release from DDS could be mediated via two different mechanisms, namely (i) a diffusion mediated mechanism in which the drug diffuses out from the nanoparticle under stimulus-induced structure changes of the carrier, or (ii) a degradation mediated mechanism in which the drug escapes the carrier because of stimulus-induced carrier degradation.86 The diffusion mechanism could be preferred when greater control is needed, while the degradation mechanism could be preferred when quick drug release is required.Regarding degradation-mediated release nanoparticles, recent work has investigated photo-controllable nanoparticles sensitive to ROS generation (Table 2) Seah et al. reported a paclitaxel-loaded micelle that also relied on ROS cleavage of a thioketal for drug liberation (Fig. 5A).42,87 Upon ROS formation in the micelle via a photosensitizer, the polythioketal polymer was cleaved and the structural integrity of the micelle was compromised, leading to drug release. These micelles efficiently released their entire payload over a course of six hours upon 20-minute 808 nm light exposure. When this test was repeated with a tissue-like phantom, total drug release was still achieved, although it was somewhat slower (roughly 10 hours for 80% drug release). In mice carrying a A549 tumor, the micelles effectively destroyed the tumor after 20 days. Strangely, the control group injected with PBS buffer initially demonstrated strong tumor growth, followed by a period of full tumor collapse (no more tumor present after 30 days). The authors speculate that this could be due to the ability of the mice to produce mature T-lymphocytes as they enter adulthood.88 Alternatively, the authors postulate other functional immune cells could also contribute to the observed antitumor effect.89,90 Still, the group treated with the micelles undeniably showed a stronger therapeutic effect, with no tumor growth being observed in the first place. Motivated by a need to reduce unwanted drug leakage, Pei et al. designed a thioketal-linked conjugate nanoparticle co-loaded with DOX and chlorin e6 (Ce6), a photosensitizer.43 In contrast with the work of Seah et al., the drug was directly conjugated to a thioketal moiety which was itself grafted to the random copolymer backbone making up the nanoparticle. Upon irradiation with 660 nm light, Ce6-mediated ROS generation would cleave the thioketal linkage between the DOX and the polyphosphoester polymer backbone, releasing the drug. Notably, this design resulted in excellent drug retention (less than 10% leakage after 80 hours in PBS buffer) in the dark, and good controllable drug release (20% total DOX release after each NIR light exposure, three cycles). The nanoparticles were tested in a MDA-MB-231 murine xenograft tumor model, where they significantly slowed down tumor growth. Additionally, no significant weight loss or toxic effects were observed in the mice.
| DDS | Payload | Model | Admin. | Irrad. | |
|---|---|---|---|---|---|
| Polym. NP | DOX | MDA-MB-231 mouse xenograft | Intrav. | 660 nm, 0.1 W cm−2, 30 min | 43 |
| Polym. NP | Paclitaxel | A549 mouse xenograft | Intrav. | 808 nm, 300 mWcm−2, 20min | 42 |
| Polym. NP | DOX | WERI-Rb-1 mouse xenograft | Intraocular | 505 nm, 50 mW cm−2, 5min | 39 |
| Micelle | Chlorambucil | HeLa mouse xenograft | Intratum. | 635 nm, 200 mW cm−2,5 min | 40 |
| Silica NP | Naproxen | Wistar albino rat | Intrav. | 254 nm, 40 W, 30 min | 94 |
| Polym. NP | DOX | H22 mouse xenograft | Intrav. | 808 nm, 1.0Wcm−2, 2 min | 96 |
| Polym. NP | Pramipexole | C57BL/6 mouse | Intramuscular | 808 nm, 1.0Wcm−2, 2min | 97 |
 | ||
| Fig. 5 Chemical structure and reactivity of photo-controllable linkages utilized for the design of DDS discussed in this paragraph. | ||
Comparing the two different designs of Seah et al. and Pei et al., directly attaching the drug to the nanoparticle via a thioketal linker endowed the system with slower but more controllable drug release which could be repeatedly triggered. In contrast, encapsulating the free drug inside a polythioketal nanoparticle resulted in full drug release in a short amount of time, creating an “all or nothing” design. Interestingly, both studies discussed the use of low power density light (0.1 and 0.3 W cm−2 respectively) combined with long irradiation times (30 and 20 min respectively) to achieve in vivo drug release. The use of lower power density light could be attractive from a clinical perspective, highlighting another potential advantage of ROS-induced degradative drug release.
Naturally, degradation-mediated release can also be achieved without reliance on ROS. Long et al. designed a DOX-loaded polymer nanoparticle which released its payload through direct photocleavage of its coumarin-based trigonal conjugate building blocks by green light (Fig. 5B).39,91 Because the nanoparticle was designed for intravenous chemotherapy of retinoblastoma, light depth penetration wasn't an issue and shorter wavelength green light could be used for photocleavage instead of the longer wavelength NIR light. In a retinoblastoma mouse model, these polymer nanoparticles inhibited tumor growth while demonstrating good biosafety, showing no signs of weight loss or systemic toxicity after intraocular injection. Again, these results were achieved using low power density light (50 mW cm−2, 5 min).
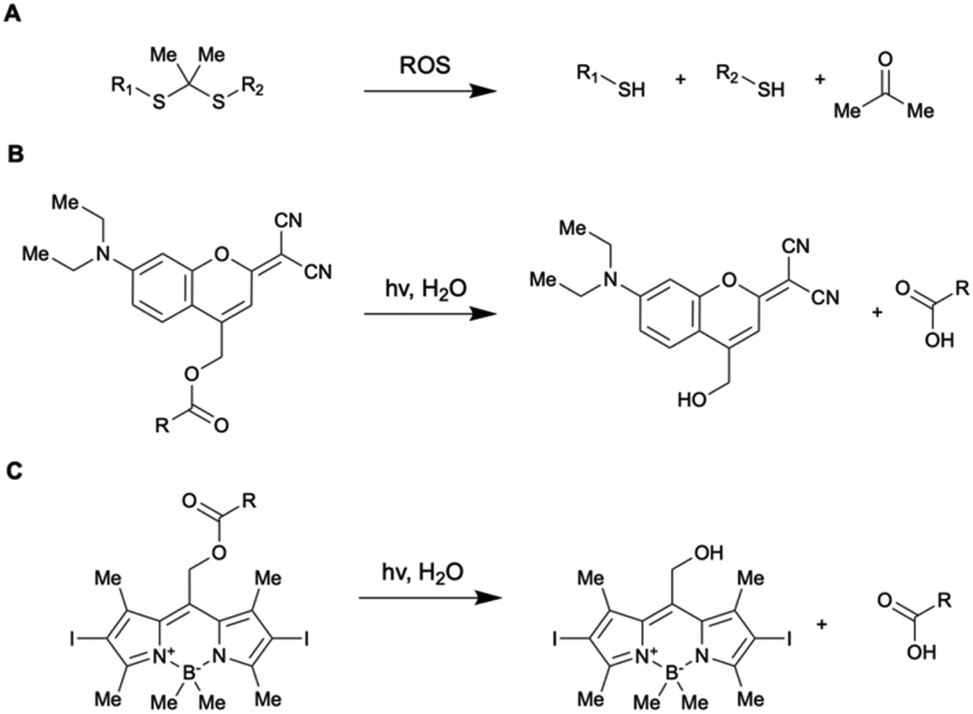
Others have opted to control their drug release through the design of nanocarriers that release their payload through a diffusion-mediated mechanism. Several aspects of photo-controllable diffusion-release nanocarrier design have been recently investigated. Lv et al. reported a micelle capable of one-photon upconversion-like photolysis through a triplet–triplet energy transfer process for the release of the chemotherapy drug chlorambucil.40 This was motivated by a desire to use more biocompatible NIR light to cleave UV-sensitive prodrugs, and the limited theoretical quantum yield of previously reported two-photon upconversion systems (at most 0.5).92,93 The chlorambucil was conjugated to boron-dipyrromethene to afford a photocleavable prodrug which was co-loaded with a Pt-based photosensitizer. Using low power density 635 nm light (200 mW cm−2), these micelles strongly suppressed tumor growth in HeLa tumor-bearing mice. This demonstrated the use of one-photon upconversion in vivo, which conceivably has better biocompatibility than two-photon upconversion processes, which requires more photons and thus more energy-dense light. In other work, Almáši et al. set out to investigate “zero-premature release” mesoporous silica nanoparticles for naproxen sodium salt (NAP) delivery.94 The particles had their pores gated with a UV-photo-controllable switch for active control over drug diffusion (Fig. 6).
 | ||
| Fig. 6 Pore closing and pore opening triggered via UV light irradiation. Reproduced from ref. 94 with permission from Springer Nature, copyright 2021. | ||
To verify the robustness of the system, in vivo pharmacokinetic experiments were performed in Wistar rats. The drug concentration in the blood serum was measured. In the absence of UV light, little drug was detected (1.17 ± 0.39 μg mL−1). However, when the rats were exposed to UV light, the serum drug concentration was measured to be much greater (36.05 ± 8.17 μg mL−1). This confirmed that the photogate was active in vivo and that the nanoparticles only presented minimal burst release upon injection. This concept has also been extended to NIR light-sensitive mesoporous silica nanoparticles by Li et al., utilizing base-paired nucleotides as gatekeepers to prevent DOX leakage.95
Further probing the aspects of nanocarrier design, He et al. conducted an investigation into the effect of polymer chain length for NIR-induced combinatorial drug release and PTT from polymeric nanocarriers via the diffusion mechanism.96 They constructed a thermosensitive poly(etheramine)-based nanocarrier co-loaded with DOX and a photosensitizer while varying the ratio of hydrophobic to hydrophilic blocks in the polymer. Two designs were synthesized, one with a higher ratio hydrophobic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hydrophilic (PEA81) and one with a lower ratio (PEA11). Because PEA81 had a lower critical solution temperature (LCST) compared to PEA11, PEA81 more readily released its payload under NIR irradiation to 45 °C. In contrast, PEA11 had a LCST of 60.8 °C, and thus released little DOX under the same conditions. Perhaps unsurprisingly, PEA11 did suffer from less ambient drug leakage than PEA81. During in vivo trials, PEA81 demonstrated stronger antitumor efficacy upon intravenous injection followed by NIR irradiation in mice than PEA11. Crucially, PEA81 and PEA11 showed comparable DOX biodistribution, meaning the reduced leakage of PEA11 turned out to make little impact in vivo.
:hydrophilic (PEA81) and one with a lower ratio (PEA11). Because PEA81 had a lower critical solution temperature (LCST) compared to PEA11, PEA81 more readily released its payload under NIR irradiation to 45 °C. In contrast, PEA11 had a LCST of 60.8 °C, and thus released little DOX under the same conditions. Perhaps unsurprisingly, PEA11 did suffer from less ambient drug leakage than PEA81. During in vivo trials, PEA81 demonstrated stronger antitumor efficacy upon intravenous injection followed by NIR irradiation in mice than PEA11. Crucially, PEA81 and PEA11 showed comparable DOX biodistribution, meaning the reduced leakage of PEA11 turned out to make little impact in vivo.
While the previously discussed work focused on nanocarrier design, Li et al. showed that not only is the design important, the fabrication method can be consequential as well.97 They synthesized poly(D, L-lactide-co-glycolide) (PLGA) microspheres co-loaded with hollow gold nanoparticles (HGNP) and pramipexole for treatment against Parkinson's disease. Upon NIR-light exposure, the HGNP heat up and facilitate the release of pramipexole from the microspheres. The microspheres were synthesized either by a water-in-oil-in-water (W/O/W) method, or a solid-in-oil-in-water (S/O/W) method. Interestingly, the carriers made with the S/O/W method showed better drug release under NIR light and presented a better area-under-the-curve parameter in in vivo pharmacokinetic tests. Furthermore, mice that had received the S/O/W microspheres performed better in a rotarod test, a test that screens for neuromuscular impairment, than mice that had received the W/O/W microspheres. These improved properties likely stemmed from the higher HGNP encapsulation efficiency of the S/O/W microspheres (65%) as compared to the W/O/W microspheres (29%).
Conclusively, recent work has shown that the structure and fabrication method of a photo-controllable DDS can be adapted to tune its drug releasing properties, allowing for better control over pharmacodynamics and pharmacokinetics. Importantly, in vivo experiments must be used to determine the optimal balance between drug retention and drug release. As demonstrated by He et al., an in vitro benefit (less drug leakage) does not always translate to an in vivo benefit (toxicity comparable to a leakier system). To fill knowledge gaps around these topics, future work could center around systematically investigating the in vivo effect of drug retention, drug release kinetics and the design choices that alter these parameters.
4. Delivery of biomolecules
In recent years, biomolecules have established themselves as a superb form of highly selective therapeutics. Importantly, biomolecule-based therapeutics are readily available with the potential to personalize, and target previously undruggable pathways.98–100 Additionally, they possess unrivaled substrate specificity compared to small molecules. In the clinic, DDS that can deliver mRNA or entire proteins could find applications in protein replacement therapy, while delivery of siRNA could have applications in anti-cancer therapy. DDS are perhaps a natural fit for biomolecules, as their free forms are often sensitive to clearance and metabolism.101,102 In the same vein, great care must be taken when attempting to combine biomolecule drugs with PDT, as they are known to be sensitive to oxidation.103 As already shown by the previously discussed siRNA-delivering nanoparticles of Wu et al., light sensitive DDS have also been adapted to the delivery of such modalities.Indeed, several groups have reported dedicated systems for biomolecule delivery (Table 3). Chu et al. reported a NIR light-activated singe-strand DNA (ssDNA) delivery system based on upconversion nanoparticles (UCNP) and photocaged CpG oligonucleotides (PCpG) to selectively activate antitumor immunity.104 When exposed to NIR light, the UCNP facilitated the conversion of NIR light to UV light, cleaving off the complementary photocage strand and liberating the CpG oligonucleotides. In 4T1 tumor bearing mice, the nanoparticles were taken up by the immune system, notably macrophages, B cells, and dendritic cells, resulting in the intratumoral concentration of the UCNP peaking after 3 hours. Upon one-minute NIR light irradiation, the murine tumors were nearly fully eliminated through activation of the immune system. The CpG-loaded nanoparticles lacking the photocleavable complementary strand showed greater antitumor efficacy, but also demonstrated severe systemic toxicity. Notably, despite inferior antitumor activity, the photocaged CpG-loaded nanoparticles extended mouse survival more than the non-photocaged CpG-loaded nanoparticles, further hinting at the severity of the systemic toxicity.
In other work, Huo et al. designed light-sensitive Gold-DNA nanosunflowers for targeted silencing of the c-myc oncogene.105 Triplex-forming POY2T oligonucleotides were attached to ultrasmall (∼2 nm) gold nanoparticles, and the resulting conjugates were loaded into complementary DNA-mediated self-assembled gold-DNA nanocarriers. Upon irradiation with light, the nanocarrier disassembled through melting dissociation of the carrier DNA strands, liberating the DNA-nanoparticle conjugates. The DNA-tail of the nanoparticles could then insert itself in the c-myc oncogene, down-regulating its expression.
In MCF-7 tumor xenograft mice, the nanosunflowers demonstrated significant suppression of tumor growth upon intravenous injection followed by 10-minute NIR light exposure. Of the mice irradiated after 1, 3, 6 and 12 hours, only the last two groups showed significant tumor suppression, with the best tumor growth inhibition (∼90%) observed in the 12-hour group. Additionally, the nanosunflowers showed excellent blood compatibility and induced no damage to healthy tissues. The authors further speculated that the “nanoparticle in nanoparticle” design helped significantly with achieving long time blood circulation, effective tumor accumulation and penetration, and cellular uptake.
Biomolecule delivery by light-sensitive DDS has not limited itself to the delivery of oligonucleotides. Recently, Qu et al. succeeded in photo-controllable delivery of a full protein. Through a NIR-responsive NDA-mediated nanotetrahedron carrier, they were able to deliver the pro-apoptotic protein granzyme B for the clearance of senescent cells, which play a pivotal role in various age-related diseases.49,106 Granzyme B was coupled to a UNCP, which was then caged inside a DNA tetrahedron. The tetrahedron was fitted gold nanoparticles coated with anti-B2MG to enable selective targeting of senescent cells. Upon NIR exposure, a boronic ester linkage between the DNA and gold nanoparticles cleaved and the granzyme B-doped UNCP was liberated, resulting in subsequent apoptosis via the granzyme B-mediated activation of caspase-3.
To validate their design in vivo, the nanotetrahedrons were evaluated in senescence-accelerated SAMP8 mice by intravenous injection followed by 30-minute irradiation with 980 nm light. One month after the start of nanotetrahedron treatment, the mice showed good clearance of senescent cells, and senescent indicators normally in present in these mice at elevated levels decreased to the same concentration as found in healthy mice. Furthermore, the density of their fur coat improved and the distance they ran daily on a running wheel increased, hinting at their newfound vitality.
The distinctive nature of photo-controllable biomolecule DDS endows them with unique opportunities for disease treatment, as they can directly intervene at the nuclear DNA or modulate the immune system. Yet, regarding clinical translation, this could also be the very thing troubling them. Due to their distinctive properties, biomolecules could be at greater risk of undesirably interfering with cellular processes or eliciting an unwanted response from the immune system, making (long-term) toxicity a major concern. Care must be taken to not overcomplicate the DDS in the name of efficacy, where the focus instead should be on their biocompatibility and safety profile. As the work by Chu et al. encouragingly shows, good efficacy can be achieved with relatively minimalistic photo-controllable biomolecule DDS. With this in mind, we believe that further development of simple photo-controllable biomolecule DDS could therefore be a big step on their path towards clinical translation.
5. Light-responsive DDS after molecular targeting
While in cancer treatment circulatory DDS are generally already passively targeted to solid tumors via the EPR effect, tumor accumulation can be enhanced through active targeting.107 Perhaps more importantly, smart targeting of a DDS could reduce off-site drug levels, something which has been suggested to be crucial for clinical translation, even more so than the increased tumor accumulation.10 In this view, the benefit of active targeting is not so much the increased drug concentration at a diseased site, but a favorable shift in the balance between off-site and on-site drug levels. Clinically, this could translate to lower dosage levels and less side-effects, improving the quality of life for a patient and fulfilling the promises that DDS offered.To accomplish active targeting, site-specific molecular recognition is leveraged to steer DDS towards the diseased site. This site-specific recognition can for example be achieved by conjugation of NPPS with antibodies for surface proteins overexpressed in the diseased cells, or by attachment of small molecules that bind to overexpressed receptors at the diseased site. Naturally, light-controllable DDS would benefit greatly from active targeting, as both the active targeting and the external controllability of the DDS work synergistically to increase drug delivery to the appropriate site and reduce unwanted drug leakage elsewhere, fostering a better on/off-site drug balance.
Indeed, several groups have recently published in vivo studies that validate this idea (Table 4). Ma et al. reported on DOX-loaded host–guest poly(pyrrole) nanocomplexes that leveraged anti-gelactin-3 antibodies to actively target the nanocomplexes towards differentiated thyroid cancer (DTC) cells.45 This strategy was motivated by the observation that galectin-3 is highly overexpressed in DTC patients but is undetectable in a healthy thyroid. In mice bearing TPC-1 subcutaneous tumors, the nanocomplexes effectively destroyed the neoplasm after intravenous injection followed by 10-minute 808 nm light irradiation, leaving only scars that fell of two weeks later. Critically, pharmacokinetic studies were also performed. It was found that the nanocomplexes decorated with the anti-galectin antibodies demonstrated not only a higher DOX concentration in the tumor, but also a lower DOX concentration in the heart and muscle tissue of mice. This is especially noteworthy since cardiotoxicity is most significant side effect of DOX, limiting its usage in the clinic.108,109 Nanocomplexes equipped with dummy IgG-antibodies also showed a better on/off-site drug balance, but only to a lesser extent.
| DDS | Payload | Model | Admin. | Irrad. | |
|---|---|---|---|---|---|
| Hybrid nanocomplex | DOX/PTT | TPC-1 mouse xenograft | Intrav. | 808 nm, 2 W m−2, 10 min | 45 |
| Lipsome | DOX/PDT | Various mouse xenografts | Intrav. | 808 nm, 1 W m−2, 5 min | 44 |
| Micelle | DOX | MCF-7/ADR mouse xenograft | Intrav. | 808 nm, 1 W m−2, 5 min | 31 |
| Macrophage nanoparticle carrier | Oxaliplatin/PDT | 4T1/EMT6 mouse xenograft | Intrav. | 671 nm, 250 mW cm−2, 10min | 52 |
In another study by Li et al., a DOX-loaded photosensitive liposome fitted with HER2 antibodies for synergistic chemophotodynamic therapy was reported.44In vivo biodistribution experiments were performed in both MCF-7 and A549 murine tumor xenograft models. Not only did the liposomes accumulate significantly more in the MCF-7 tumor compared to the A549 tumor due to the overexpression of human epidermal growth factor receptor 2, but they also showed relatively less DOX presence in peripheral organs, especially the liver. In pharmacodynamic trials, the liposomes proved very effective for stunting MCF-7 tumor growth upon NIR light irradiation.
As previously mentioned, the usage of active targeting moieties has not limited itself to just antibodies. Recent work has also highlighted the potential of FA as an active-targeting agent for photosensitive DDS. Zhang et al. reported a FA-doped DOX-loaded micelles sensitized to 808 nm light for targeted chemotherapy delivery.31 Additionally, for biodistribution experiments, the micelles were loaded with a fluorescent dye. In mice, these micelles quickly accumulated at an MCF-7 tumor 4 h after intravenous injection, demonstrating little nonspecific DOX fluorescence in the liver and the spleen. In contrast, the non-targeted micelles lacking the FA doping demonstrated not only decreased tumor fluorescence, but also stronger off-site hepatic and splenic fluorescence. In conjunction, the micelles proved potent therapy in the mice, significantly ablating tumor growth upon 808 nm light irradiation while not affecting mouse body weight or inducing tissue abnormalities in major organs as observed by histological analysis.
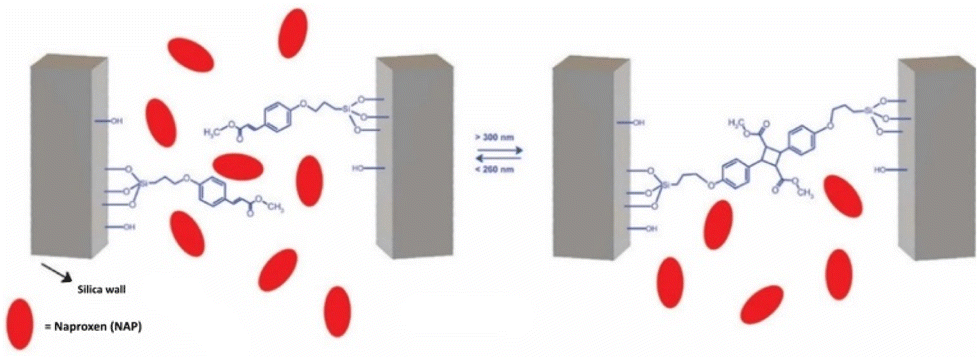
Notably, the development of active-delivery photosensitive DDS has also been extended beyond molecular recognition. Instead of leveraging singular receptor–ligand interactions for their molecular recognition, Huang et al. instead chose to harness entire macrophages.52 Indeed, macrophages do possess several traits that make them excellently suited as DDS: they can actively follow cancer-related chemokine and cytokine signals, overcome biological barriers and transmigrate into tumor tissues.110 When carrying a drug load, macrophages could act like Trojan horses for the tumors they target. To accomplish macrophage-mediated chemophotodynamic therapy delivery, the authors endowed macrophages with liposomes loaded with an oxaliplatin prodrug and a zinc phthalocyanide photosensitizer (Oxa(IV)@ZnPc@M). Intriguingly, the authors note that the macrophages became polarized to the anti-cancer M1 phenotype upon drug loading. In vivo, these drug-loaded light-controllable macrophages combined with anti-PD-L1 antibodies effectively eliminated both primary 4T1 tumors and secondary bone metastatic tumors after irradiation with 671 nm light for 10 minutes (Fig. 7). Interestingly, the treatment was also able to suppress unexpected lung metastasis during the murine experiments. Furthermore, the immunogenic tumor cell death induced by the therapy served to release tumor-associated antigens that acted as in situ vaccines. The authors demonstrated this by first treating 4T1 tumor bearing mice with their macrophages, then subsequently surgically removing the remaining tumor tissue. Upon reintroduction of 4T1 tumor cells, the mice showed slower primary tumor growth and inhibited bone and lung metastasis. The mice that had received previous treatment demonstrated prolonged survival over the mice that had not. However, the authors mention that the macrophages die upon irradiative drug liberation, potentially hampering the potential efficiency of their proposed system. In future work, Huang et al. want to further investigate the potential side-effects and long-term toxicity of their macrophages.
 | ||
| Fig. 7 Oxa(IV)@ZnPc@M mediated chemo-photodynamic therapy is triggering immune responses applicable for anti-primary and bone metastatic tumors. Photodynamics therapy (PDT), Dendritic cells (DCs) and immunogenic cell death (ICD). Reproduced from ref. 52 with permission from Springer Nature, copyright 2021. | ||
These recent reports highlight the potential of active targeting in combination with photo-controllable DDS. In vivo, active targeting of light-sensitive DDS increased drug concentration at the diseased site, and importantly, reduced it elsewhere. Still, active targeting leveraging only a single receptor could be vulnerable to acquired resistance when that receptor is downregulated. A targeted DDS employing multiple different recognition elements could be an interesting design strategy to amend this. Indeed, body-native macrophages have also shown to be viable photo-controllable DDS, capable of smartly seeking out tumor sites and even succeeding in unexpected lung metastasis suppression. With the potential side-effects and long-term toxicity unknown, further exploration of cell-based photo-controllable DDS remains of great interest owing to their potential for superior efficacy and biocompatibility.
6. Photo-controlled drug release from drug depots
The majority of DDS discussed in this review are nanostructures that carry their payload to a site of interest via the circulatory system. In contrast, drug depots are structures that remain in place, releasing their drug directly at the site of implantation. This endows drug depots with a couple of crucial advantages over circulatory DDS. Firstly, they directly deliver their payload to the diseased locus, increasing drug concentration at the relevant site and reducing systemic toxicity. Secondly, patient compliance for long-term treatment will potentially be much better for drug implants. Instead of having to get repeated injections for long-term dosing, a singular placement of a drug depot suffices, resulting in less burden to the patient.111 Thirdly, implants are capable of directly bypassing physical barriers that would otherwise hinder drugs, such as the blood–brain barrier.112Recent developments of light-sensitive drug depots have centered around the so-called in situ implant type. Traditional implants have to be pre-formed before being introduced to the body, often requiring invasive surgery to place at the desired location. In contrast, in situ implants can be injected into the body, and will only take their final shape when allowed to harden. Importantly, several in situ drug implants have already obtained clinical approval.113 However, in situ drug depots also come with some downsides. Mainly, they suffer from irregular release patterns, often demonstrating high burst release shortly after injection, followed by a period of strongly decreased drug release which could fail to meet the therapeutic threshold.114–116 As a consequence, light-controllable in situ drug implants have risen as a prominent method of ameliorating the problems associated with their non-controllable counterparts (Table 5).
| DDS | Payload | Model | Admin. | Irrad. | |
|---|---|---|---|---|---|
| Polym. gel implant | Tetracaine | Spara-gue Dawley rat | — | — | 48 |
| Hydrogel implant | mHNK | BALB/c nude mouse | — | 680 nm, 150 mW cm−2, 5min | 117 |
| Liposome-loaded capsule implant | Methotrexate | New Zealand white rabbit | Intravitreal | 1064 nm, 1.1 Wcm−2, 30 × 20s | 118 |
| Nano-composit. Hydrogel implant | DOX/PTT | 4T1 mouse Xenogr. | Intratum. | 808 nm, 0.3 W cm−2, 3min | 30 |
| Hydrogel implant | DOX/PTT | 4T1 mouse Xenogr. | Intratum. | 808 nm, 2 W cm−2, 150s | 119 |
| Nano-composite hydrogel | DOX | MDA-MB-231 xenogr. | Intratum. | 808 nm, 1 W cm−2, 5min | 120 |
| Hydrogel micro-needle implant | DOX/PTT | B16F10 xenogr. | Intrav. | 808 nm, 0.4 W cm−2, 1min | 121 |
| Hydrogel implant | DOX/PTT | 4T1 mouse xenograft | Intratum. | 808 nm, 0.15 W cm−2, 5min | 41 |
| Hydrogel implant | Cisplatin/PTT | MDA-MB-231 xenogr. | Intratum. | 1064 nm, 0.5 Wcm−2, 5min | 47 |
| Red blood cells | Dexamethasone | DBA1/J mouse | Intrav. | 635 nm, 3 mW, 5 min | 51 |
| Refillable drug depot | DOX | CD1 mouse | Intraderm./intrav. | 405 nm, 10 mW cm−2, 30 min | 46 |
For example, Zhang et al. created a light-triggerable polymer–drug conjugate for on-demand local anesthesia.48 Tetracaine, a clinically used local anesthetic was directly bound to a polymer backbone via a photocleavable carbamate bond. Upon irradiation with 400 nm UV light, the tetracaine is liberated from the polymer backbone, resulting in nerve block. Crucially, adjusting the intensity of the light allowed for complete control over the amount of released tetracaine. In mouse trials, it was validated that UV irradiation resulted in in vivo local anesthesia, and that this anesthesia could be induced multiple times by interval irradiation. Interestingly, the duration of the nerve block was linearly correlated with the power density of the UV light, endowing the system with good controllability. However, while suited for surface level skin implants, the usage of UV light to trigger drug release from other implants is less than desirable due to its shallow penetration. Hence, various implants sensitive to NIR light have been designed. Very recently, a NIR light-triggered implant for the treatment of insomnia was reported.117 This hydrogel-based implant could effectively induce non-rapid eye movement sleep in mice via the NIR-induced release of GABAa receptor agonists. In another example, He et al. propsed a size-exclusive PLGA capsule that contains NIR-activated liposomes.118 Irradiation with NIR resulted in on-demand in vivo ocular release of methotrexate, an anti-inflammatory drug. Notably, these implants were very resistant to burst release.
Recent developments have also focused on improving the anti-cancer efficacy of light-controlled in situ drug depots by combining drug release with photothermal therapy for synergized cancer treatment. Zheng et al. created a DOX-loaded in situ CuS nanocomposite hydrogel that was able to synergistically deliver both chemotherapy and photothermal therapy to tumors when exposed to NIR light.30 In 4T1 tumor bearing mice, the two-way treatment was able to fully eradicate the tumor after irradiation with 808 nm light for 150 seconds. Hou et al. reported a NIR-sensitized agarose gel incorporating DOX that incorporated sodium humate, a saponificated derivative of the natural product humic acid, as the photothermal conversion agent.119 This implant was able to effectively inhibit tumor growth, but did not result in complete tumor ablation. In other work, Qiu et al. used black phosphorous as a photothermal conversion agent for the delivery of synchronous chemo- and photothermal therapy by a DOX-loaded hydrogel.120 Upon NIR light exposure, the generated heat triggers hydrolysis, releasing the DOX payload. In a murine MDA-MB-231 tumor model, complete inhibition of tumor growth was achieved. Furthermore, Song et al. developed a specialized implant for the treatment of superficial skin tumors consisting of indocyanine green-doped hydrogel microneedles for combined chemo-photothermal.121 While not technically an in situ implant, the microneedles are implanted in the skin quickly (5 minutes) and non-invasively, thereby retaining the characteristic advantages of in situ implants. Upon irradiation with NIR light, the microneedles are heated due to photothermal conversion by the indocyanine green molecules, resulting in their ablation and liberation of DOX. While the microneedles succeeded in locally releasing DOX and inducing tumor hyperthermia, they were not able to fully stop tumor growth, only stunting growth progression.
Efforts have also been made to improve the efficiency and safety of NIR-responsive in situ drug implants. Importantly, nanocomposite hydrogels suffer from possible leakage of the physically mixed photothermal conversion agent, and often present only limited photothermal conversion efficiency. This can diminish the in vivo photothermal conversion efficiency of nanocomposite hydrogels over time.122 To facilitate the development of more efficient and safe NIR-sensitized drug implants, Liu et al. designed a DOX-loaded hydrogel based on poly(N-phenylglycine) (PLNG) and α-cyclodextrin (α-CD) where the polymer backbone itself acted as the photothermal conversion agent, reaching a photothermal conversion efficiency of 52.62%.41 The implant was able to ablate 4T1 tumors in mice after exposure to 808 nm light with an ultralow power density (0.15 W cm−2), highlighting the much improved efficiency of this system in vivo. In other work, Ruan et al. applied this concept to cisplatin-releasing hydrogels sensitive to the less commonly utilized NIR-II region (1000 to 1400 nm) instead of the NIR-I region (750–1000 nm) to deliver hydrogel-based chemo-photothermal therapy.47 The NIR-II region has strategic advantages over the NIR-I region, mainly better tissue penetration depth and a higher maximum permissible exposure.76,123 Comparable to Liu et al., the authors also used PLNG and α-CD as the polymer backbone for their in situ drug implant. When the implant was irradiated with a 1064 nm laser, it was able to fully suppress tumor growth in MDA-MB-231 tumor bearing mice using a 0.5 W cm−2 laser intensity.
Innovatively, Zywot et al. succeeded in using the circulatory system as a light-controllable drug depot for the release of the anti-inflammatory drug dexamethasone.51 The authors conjugated both Dex and the infrared dye cyanine 5 to vitamin B12. The vitamin B12 served to suspend the Dex inside RBC, as vitamin B12 is membrane impermeable, while cyanine 5 served to tune the photocleavable Dex-B12 bond to 650 nm light. Upon light exposure, the Dex was liberated from the B12 anchor and was released from the RBC owing to its membrane permeability. This approach was validated for human RBC in vitro and murine RBC in vivo. In an arthritic mouse model, the Dex-loaded murine RBC effectively reduced arthritic severity after 635 nm light irradiation for 5 minutes. This was achieved in a similar manner to IV dosed Dex, but notably the RBC drug depot needed 78% less Dex to achieve comparable clinical remission. In follow-up studies, the authors wish to further enhance the potential of this system by loading different drugs sensitized to different wavelengths of light for separate drug delivery to designated sites.
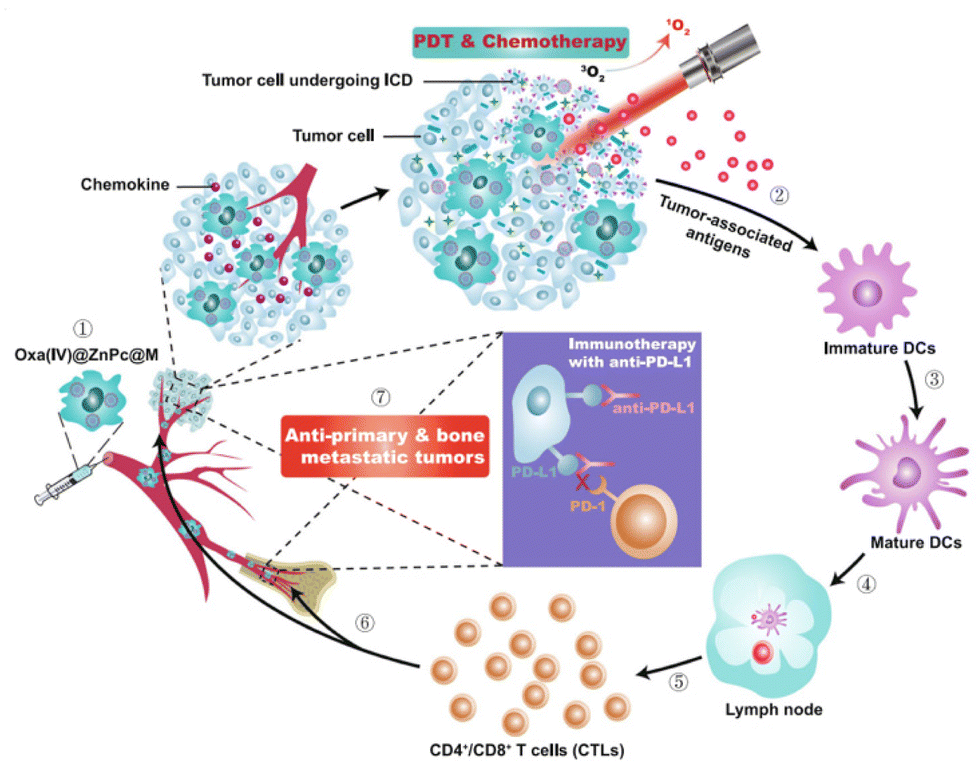
Still, despite undeniably impressive progress, all drug implants discussed so far will exhaust their drug content over time, resulting in a diminished therapeutic effect. Palvai et al. recently reported a refillable in situ drug depot that leverages strain-promoted alkyne–azide cycloaddion (SPAAC) chemistry for drug rechargeability (Fig. 8).46 Tissues are first labelled with azide groups through direct local injection of the labeling agent 3-azidopropionic acid sulfo-NHS ester (step 1), followed by introduction of a photocleavable dibenzocyclooctyne-drug conjugate (step 2). The resulting SPAAC reaction fixes the drug to the local tissue, and drug liberation can be induced by irradiation with 405 nm visible light (step 3). After drug depletion, step 2 can be repeated to recharge the drug depot. Elegantly, this system supports drug switching without the need for removal or modification of the implant, allowing for quick adaptation to a changed clinical situation. The rechargeability of this drug depot was confirmed in vivo in CD-1 mice. The authors demonstrate successful DOX loading of the depot and release of DOX upon 30-minute light irradiation, followed by DOX recharge and further drug decaging. While certainly possessing desirable attributes, the authors themselves also note the limitations of the current system. After a certain amount of drug recharges, the azide groups become depleted and require replenishment. Additionally, adaption of the system to a deeper penetrating wavelength of light is desirable, and the long-term biocompatibility of the currently proposed refillable drug depot remains as of yet unexplored.
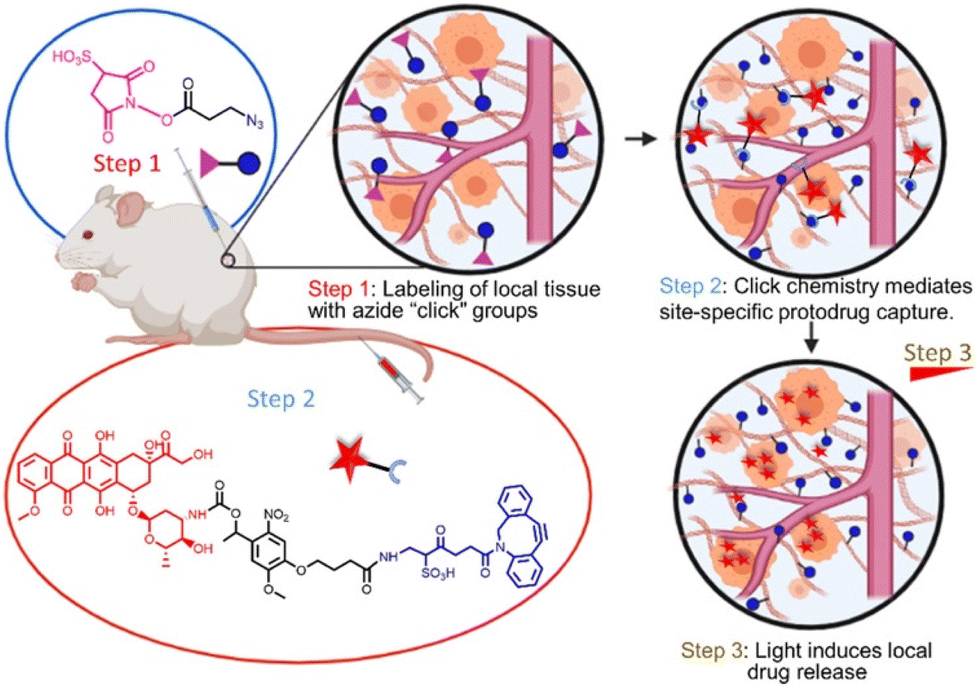 | ||
| Fig. 8 Pre-targeting of tissues is achieved using click-chemistry and allows subsequent photo-controlled release of DOX. Reproduced from ref. 46 with permission from American Chemical Society, copyright 2021. | ||
The recently reported light-sensitive drug implants have made major steps on the path to clinical viability, as almost all are sensitized to more deeply penetrating NIR light. Additionally, incorporation of combination therapy has proven to be a viable option for in situ drug implants. Advantageously, the RBC based drug depot reported by Zywot et al. can be injected intravenously, and thus could also be applicable to hard-to-reach disease sites where in situ injection is not an option. Owing to its natural biocompatibility, we feel that further investigation of this system will be of great interest to the community. Moreover, the first steps have been taken towards the development of refillable photocontrollable drug depots. Future work could focus on extending this concept to a contained implant, as aselectively labeling tissues with azide handles is at risk of creating a poorly defined drug depot with unclear long-term biocompatibility.
7. Towards photo-controllable theranostics
Apart from delivering therapeutic action, some DDS also incorporate diagnostic imaging modalities. These “theranostics” (a portmanteau of therapeutics and diagnostics) allows for non-invasive image-guided treatment of disease in a fully integrated manner. They enable real-time pharmacokinetic tracking of administered medication, endowing them with applications in personalized medicine.124 A theranostic nanomedicine can be administered to a diseased patient and the fate of the drug in de body could be monitored. If a drug accumulates at the diseased site, a patient is likely to respond to the therapy. Further long-term monitoring could take place to confirm sustained treatment effectiveness. If a drug does not accumulate at the diseased site, a patient is likely to not respond to the therapy. In this case, the theranostic agent can be switched, or alternatively, conventional therapy could be employed. Disregarding their application in personalized medicine, theranostic DDS offer another major advantage, namely the potential to quickly reveal newly formed cancer metastasis, allowing for rapid medical response. The potential impact of this capability is huge, as the vast majority of cancer-related deaths are due to cancer metastasis.125Considering the potential game-changing nature of theranostics, it isn't surprising that many light-activated theranostic DDS have been reported in the literature with in vivo experiments. For the purposes of this review, we will only discuss theranostics that carry an integrated separate imaging modality (Table 6).126,127 Fluorescence imaging by encapsulating fluorescent drugs such as DOX will not be accounted for, as such modalities are severely restricted in their imaging capability when switching to a non-fluorescent drug, limiting their potential for widespread clinical translation.
| DDS | Payload | Model | Admin. | Irrad. | |
|---|---|---|---|---|---|
| Iron oxide – carbon NP | DOX/PTT | 4T1 mouse xenogr. | Intrav. | 808 nm, 1 W cm−2, 5min | 128 |
| Liposome | DOX | MCF-7 mouse xenogr. | Intrav. | 365 nm, 60 Wcm−2, 4 × 2min | 36 |
| Bi2S3 micro-sphere | DOX/PTT | MDA-MB-231 | Intratum. | 808 nm, 0.35 W cm−2, 10min | 130 |
| Polymeric NP | DOX/CPT/PTT | 4T1 mouse Xenogr. | Intrav. | 808 nm, 2 W cm−2, 5min | 34 |
| MoS2 nano-sheet | DOX/PTT | MCF-7/ADR mouse xenogr. | Intrav. | 808 nm, 0.6 W cm−2, 10min | 133 |
| Si-NP | Cisplatin/PTT | A549 mouse xenogr. | Intrav. | 808 nm, 1 W cm−2, 5min | 37 |
| Yolk–shell NP | DOX/PTT | HepG-2 mouse xenogr. | Intrav. | 808 nm, 1 W cm−2, 5min | 135 |
| Janus NP | DOX/Dtxl | H-22 mouse xenogr. | Intrav. | 808 nm, 1 W cm−2, 5min | 138 |
Magnetic resonance (MR) imaging is non-invasive and boasts superior tissue penetration, making it potentially very suitable for theranostics. Recently, DOX-loaded Fe3O4-coated carbon nanoparticles with MR imaging capabilities have been reported.128 These nanoparticles delivered chemophotothermal therapy upon NIR irradiation, and additionally served as a T2-contrast agent. In vivo, they enhanced the MR signal intensity in the tumors of 4T1 xenografted mice and were able to efficiently inhibit the growth of these tumors. Apart from carbon nanoparticles, liposomes have also been used for MR theranostics.36 Liu et al. synthesized a multifunctional lipid by conjoining Gd3+-chelating gadopentic acid to a lipid tail via a UV-sensitive o-nitrobenzyl ester. The lipid was then used to produce DOX-encapsulating liposomes that were both pH and NIR responsive. The direct conjugation design was selected to promote MR contrast without interfering with liposome drug encapsulation or release. Crucially, the liposomes proved superior MR T1-contrast agents for tumor imaging both in vitro and in MCF-7 tumor bearing mice when compared to free gadopentic acid. Furthermore, they strongly inhibited mouse tumor growth under 365 nm light exposure.
Not exclusively keeping to MR imaging, recent research has also focused on the development of photoacoustic (PA) theranostic DDS. Like MR imaging, PA imaging is a non-invasive and non-ionizing imaging technique perfectly suited for the visualization of soft tissue, including tumors.129 Zhang et al. designed urchin-like DOX-loaded Bi2S3 hollow microspheres for chemo-photothermal tumor therapy and PA diagnostic imaging.130 Notably, they employed ZnS composite microspheres as a sacrificial template, allowing for quick and facile Bi2S3 hollow nanoparticle synthesis. Owing to the nanorod spikes on their surface, the nanoparticles proved effective photoacoustic contrast agents in vivo. Additionally, they stunted tumor progression in MDA-MB-231 mice under 808 nm light, although only to a moderate degree. In other work, Yang et al. reported DOX and camptothecin-loaded polypyrole nanoparticles capable of delivering chemophotothermal therapy and serving as a PA contrast agent.34 Interestingly, these nanoparticles release their DOX payload by swelling up (diameter: 200 nm) due to NIR light-induced heating, and shrinking again (diameter: 40 nm) due to cooling when the light source is removed, creating “breathing” nanoparticles. They proved excellent in vivo PA contrast agents, increasing the tumor PA signal 10.6-fold 4 hours after intravenous injection in a 4T1 tumor xenograft mice model as compared to the pre-injection signal. Irradiating the mice with 808 nm NIR light for 5 minutes also resulted in significant tumor growth suppression and complete mouse survival after a period of 50 days.
Like MR and PA, positron emission tomography (PET) imaging has also been used for non-invasive real time diagnostics. Notably, PET offers unlimited penetration depth and is extremely sensitive to contrast agents (10−11 to 10−12 M).131,132 Recently, Dong et al. reported MoS2 nanosheets for PET theranostics.133 The nanosheets were sensitized to release DOX under exposure to acidic pH, NIR light, and the enzyme hyaluronidase, owing to their incorporation of hyaluronic acid. Additionally, the hyaluronic acid actively targeted the nanosheets to the CD44 receptor, which is commonly overexpressed in drug-resistant cancer cells.134 The nanosheets were able to achieve 96% tumor growth inhibition in MCF-7-ADR tumor bearing mice when irradiated with 808 nm NIR light for 10 minutes. In contrast, tumor growth inhibition was only 76% without the NIR laser, and tumor regrowth was observed. Upon labeling the nanosheets with a 64Cu radioactive tracer, the nanosheets proved very effective contrast agents for in vivo tumor diagnostics, with the actively targeted nanosheets achieving a twofold PET signal over nanosheets without hyaluronic acid, highlighting the potential impact of active targeting in theranostics.
However, not all DDS theranostics have stuck with a singular imaging technique. Multiple systems with multimodal visualization capabilities have been reported. For example, Xiao et al. described a cisplatin-loaded silica-coated nanovesicle that possessed MR and ultrasound (US) imaging capacity in addition to furnishing synergistic NIR-triggered chemo-photothermal therapy.37 Ultrasmall superparamagnetic iron oxide nanoparticles served as a T2-weighted MR contrast agent, while perfluorohexane acted as the US contrast agent. Not only were these nanovesicles able to nearly fully eliminate A549 tumors in mice owing, but they also proved efficient theranostic tools by virtue of their MR and US imaging ability. Ding et al. reported bowl-shaped DOX-loaded Au@PDA nanoparticles that enhanced both PA and computational tomography (CT) imaging (Fig. 9).135 CT and PA were chosen to complement each other; CT is excellent for visualizing hard tissue, while PA possesses high soft-tissue sensitivity.136,137 This combination neatly demonstrates the value of incorporating multimodal imaging in theranostics. Notably, the unusual bowl-shape of the nanoparticles much enhanced their DOX loading capacity through the enlarged PDA surface area. These nanoparticles delivered chemo-photothermal therapy when exposed to 808 nm light, and passively released moderate amounts of DOX when exposed to acidic pH. In vivo, they were successful in inhibiting tumor growth under NIR light and visualizing the tumor via both CT and PA imaging.
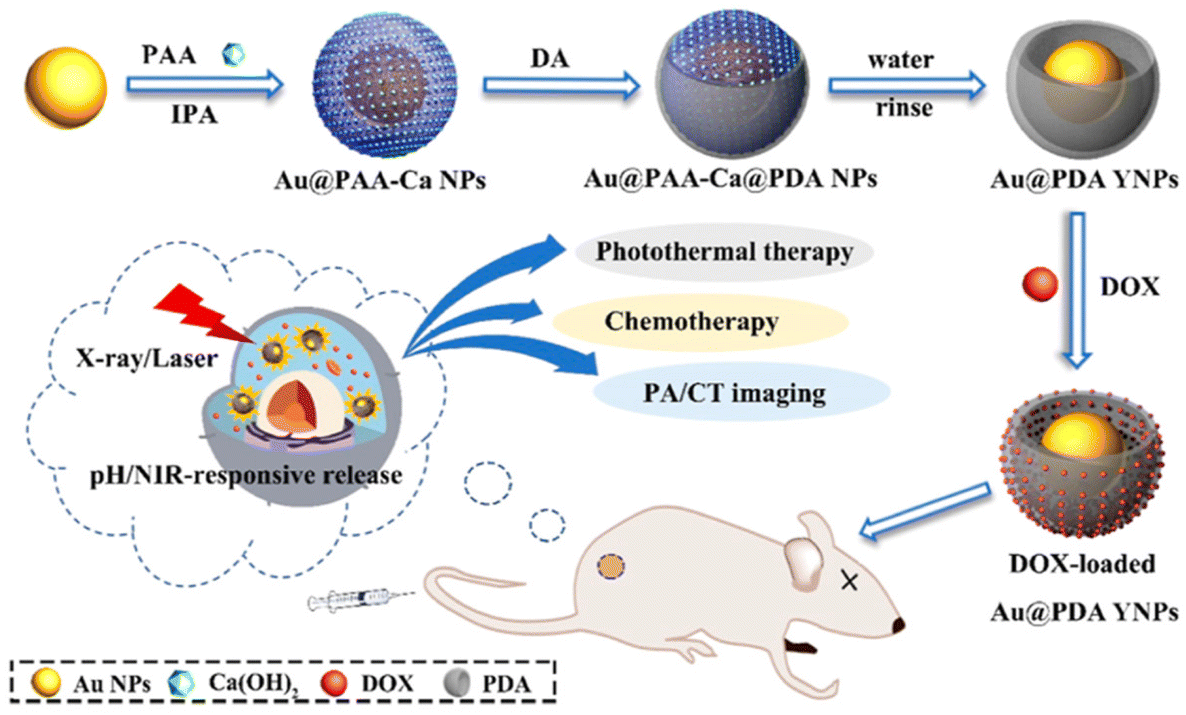 | ||
| Fig. 9 Controlled drug release from bowl-shaped Au@PDA YNP triggered by pH. NIR-responsiveness allows imaging alongside synergistic chemotherapy. Reproduced from ref. 135 with permission from American Chemical Society, copyright 2022. | ||
The concept of light-sensitive theranostics has also been applied to Janus nanoparticles (JNP). A recent publications highlights a nanocube-nanosphere amphiphilic JNP capable of encapsulating both hydrophobic and hydrophilic drugs in separate compartments.138 At low pH, DOX is released from the nanosphere, and under NIR light, Dtxl is liberated. Simultaneously, the Au nanocube and the Fe3+ coated nanosphere serve as the CT and MR imaging contrast agent, respectively. The JNPs were able to more than double both the CT and MR signal intensity. Much like the bowl-shaped nanoparticles designed by Ding et al., the combination of these imaging techniques in one theranostic agents enables synergistic visualization of both hard and soft tissues. Importantly, these JNPs achieved ∼90% tumor growth inhibition in a H-22 liver cancer xenograft mice model upon intravenous injection followed by 5-minute NIR irradiation.
Going even further, light-controllable theranostics have also been extended to trimodal imaging capabilities by Li et al. They reported surface-engineered 5-fluorouracil-loaded polymer nanoparticles with an internal air pocket (dubbed “nanobubbles”) with NIR, MR and US diagnostic applications.139 Additionally, when exposed to the lower pH of the tumor microenvironment or endosomes/lysosomes the nanobubbles were capable of switching to a positive surface charge, facilitating better cellular uptake and drug release. Complementary irradiation with NIR for 5 minutes resulted in better drug release than lower pH alone. The nanobubbles employed Gd3+ ions as T1-weighted MR contrast agents, and IR-780 was used as both the NIR contrast agent and the photothermal converter. The nanobubbles proved to be an effective in vivo trimodal imaging system in MGC-803 tumor bearing mice, with the tumor site clearly being visible by fluorescence imaging, MR imaging, and US imaging.
Additionally, when the nanobubbles were equipped with folate residues for active tumor targeting, the signal strength for all three imaging techniques was enhanced, owing to the increased tumor accumulation of the nanobubbles. On the therapeutic side, they were able to completely ablate the MGC-803 xenograft tumor under NIR irradiation. A system with trimodal imaging capabilities undoubtedly has potential for personalized medicine, as it endows doctors with more freedom to incorporate the needs of an individual patient in choosing the appropriate imaging technique.
Like in situ drug depots, theranostics have also proven themselves capable under NIR light, successfully delivering not only combination therapy but also facilitating multimodal imaging. The versatility demonstrated by photo-controllable theranostics is perhaps their greatest asset, as it naturally aligns with the goals of personalized medicine. However, most theranostics discussed here still employed high power density (1 W cm−2) light, which might not be preferrable in a clinical setting (Table 6). Development of photo-controllable theranostics sensitized to light with a lower power density therefore remains of interest.
8. Outlook and conclusion
The progress of in vivo applications of photo-controllable DDS over the past years has been significant, and various promising developments have been made. Notably, DDS have been designed to carry both, multimodal synergistic therapy delivery (chemotherapy, PDT, PTT) and imaging capabilities for facile diagnostics, paving the way to more personalized medicine with high in vivo efficacy. Various DDS have been made compatible with low power density light, crucial for improving the in vivo safety profile. It has also been shown that payload identity is no longer limited to small molecule drugs but could be extended to oligonucleotides and even fully assembled proteins. Additionally, using native cells for targeted drug delivery as demonstrated is a powerful concept.However, photo-controllable DDS still face major hurdles on the path to routine clinical translation. Although great effort has been made to design NIR light controllable DDS, a logical next step would be to advance this trend towards the NIR-II region, which allows for better penetration and has a higher maximum permissible exposure. Another aspect of many DDS discussed in this review is that they must be administered through injections. Therefore, the development of DDS with more convenient routes of administration remains of great interest for further advancement of DDS towards the clinic. Another inconvenient aspect of most circulatory DDS against cancer reported here is their long tumor accumulation time. In most studies the DDS require 24 hours before their maximum tumor accumulation in vivo. With active targeting, DDS can potentially reach a therapeutically relevant concentration more rapidly, owing to their better retention at the diseased site.
Furthermore, a crucial prerequisite for the successful broad clinical translation of photo-controllable nanomedicine is their societal perception, a less commonly discussed topic. While generally held in a favorable view by society at large, not every patient might be comfortable with the idea of taking a “smart nanomedicine”. Indeed, in a survey, only 72% of people said they would be willing to take a drug made of nanomaterials.140 More strikingly, 92% felt that they were not sufficiently informed regarding the risk-benefit aspects of nanotechnology. This knowledge deficit has also been verified in other studies.141 However, only educating the public is likely not sufficient, as greater knowledge about nanotechnology doesn't increase support for it. To facilitate public acceptance of nanomedicine, it will be critical to have an active dialogue between scientists, clinicians, and society in general, in which the perspectives and concerns of each group are respected and addressed.
In the coming years, we anticipate further steps will be taken on the road to clinical translation of photo-controllable DDS with improvements of their versatility and safety profile. Without doubt, in vivo experiments are crucial to elucidate the potential hurdles faced during the translation of DDS and the design of future delivery systems needs to take in vivo performance and efficiency of currently utilized systems into consideration. In this review, we provided an overview of the state-of-the-art from a chemistry perspective with the aim to provide guidance for future design of chemical systems. Furthermore, prolonged in vivo trials will be of great importance to assess the long-term systemic toxicity and efficacy of photo-controllable DDS. Lastly, we urge researchers to think about the potential societal effects of their work and take part in societal dialogue, as the barriers for DDS outside of the lab might not be insignificant to the barriers present within.
Author contributions
R. R. summarized the literature and wrote the manuscript. K. N. wrote the manuscript and supervised the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the funding of Radboud University (start-up package).References
- I. Fratoddi, I. Venditti, C. Cametti and M. V. Russo, J. Mater. Chem. B, 2014, 2, 4204–4220 RSC.
- L. Sercombe, T. Veerati, F. Moheimani, S. Y. Wu, A. K. Sood and S. Hua, Front. Pharmacol., 2015, 6, 286 Search PubMed.
- S. Biswas, P. Kumari, P. M. Lakhani and B. Ghosh, Eur. J. Pharm. Sci., 2016, 83, 184–202 CrossRef CAS PubMed.
- Z. Sun, C. Song, C. Wang, Y. Hu and J. Wu, Mol. Pharm., 2020, 17, 373–391 CAS.
- Y. Dang and J. Guan, Smart Mater. Med., 2020, 1, 10–19 CrossRef PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS PubMed.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, e10143 Search PubMed.
- M. S. Paolini, O. S. Fenton, C. Bhattacharya, J. L. Andresen and R. Langer, Biomed. Microdevices, 2019, 21, 45 CrossRef PubMed.
- S. Wang, R. Liu, Y. Fu and W. J. Kao, Expert Opin. Drug Delivery, 2020, 17, 1289–1304 CrossRef CAS PubMed.
- T. Lammers, F. Kiessling, M. Ashford, W. Hennink, D. Crommelin and G. Storm, Nat. Rev. Mater., 2016, 1, 16069 CrossRef CAS PubMed.
- M. E. R. O'Brien, N. Wigler, M. Inbar, R. Rosso, E. Grischke, A. Santoro, R. Catane, D. G. Kieback, P. Tomczak, S. P. Ackland, F. Orlandi, L. Mellars, L. Alland and C. Tendler, Ann. Oncol., 2004, 15, 440–449 CrossRef PubMed.
- R. M. Rifkin, S. A. Gregory, A. Mohrbacher and M. A. Hussein, Cancer, 2006, 106, 848–858 CrossRef CAS PubMed.
- S. Y. Lee, H. S. Park, K. Y. Lee, H. J. Kim, Y. J. Jeon, T. W. Jang, K. H. Lee, Y. C. Kim, K. S. Kim, I. J. Oh and S. Y. Kim, Clin. Lung Cancer, 2013, 14, 275–282 CrossRef CAS PubMed.
- D. L. Stirland, J. W. Nichols, S. Miura and Y. H. Bae, J. Controlled Release, 2013, 172, 1045–1064 CrossRef CAS PubMed.
- Z. Fang, Y. Shen and D. Gao, New J. Chem., 2021, 45, 4534–4544 RSC.
- L. Xu, Y. Yang, Y. Mao and Z. Li, Adv. Mater. Technol., 2022, 7, 2100055 CrossRef CAS.
- T. L. Rapp and C. A. DeForest, Adv. Healthcare Mater., 2020, 9, 1901553 CrossRef CAS PubMed.
- C. Ash, M. Dubec, K. Donne and T. Bashford, Lasers Med. Sci., 2017, 32, 1909–1918 CrossRef PubMed.
- M. Karimi, P. Sahandi Zangabad, S. Baghaee-Ravari, M. Ghazadeh, H. Mirshekari and M. R. Hamblin, J. Am. Chem. Soc., 2017, 139, 4584–4610 CrossRef CAS PubMed.
- A. Ahmadi, S. Hosseini-Nami, Z. Abed, J. Beik, L. Aranda-Lara, H. Samadian, E. Morales-Avila, M. Jaymand and A. Shakeri-Zadeh, Drug Discovery Today, 2020, 25, 2182–2200 CrossRef CAS PubMed.
- S. M. Mirvakili and R. Langer, Nat. Electron., 2021, 4, 464–477 CrossRef.
- E. Hemmer, A. Benayas, F. Légaré and F. Vetrone, Nanoscale Horiz., 2016, 1, 168–184 RSC.
- X. Zhang, L. An, Q. Tian, J. Lin and S. Yang, J. Mater. Chem. B, 2020, 8, 4738–4747 RSC.
- J. Geng, Y. Zhang, Q. Gao, K. Neumann, H. Dong, H. Porter, M. Potter, H. Ren, D. Argyle and M. Bradley, Nat. Chem., 2021, 13, 805–810 CrossRef CAS PubMed.
- H. Deng, L. Lin, S. Wang, G. Yu, Z. Zhou, Y. Liu, G. Niu, J. Song and X. Chen, Adv. Mater., 2019, 31, 1903443 CrossRef PubMed.
- W. Deng, W. Chen, S. Clement, A. Guller, Z. Zhao, A. Engel and E. M. Goldys, Nat. Commun., 2018, 9, 2713 CrossRef PubMed.
- M. Raoof, S. J. Corr, C. Zhu, B. T. Cisneros, W. D. Kaluarachchi, S. Phounsavath, L. J. Wilson and S. A. Curley, Nanomedicine, 2014, 10, 1121–1130 CrossRef CAS PubMed.
- K. Tamarov, W. Xu, L. Osminkina, S. Zinovyev, P. Soininen, A. Kudryavtsev, M. Gongalsky, A. Gaydarova, A. Närvänen, V. Timoshenko and V.-P. Lehto, J. Controlled Release, 2016, 241, 220–228 CrossRef CAS PubMed.
- X. Zhang, X. Liang, X. Ma, R. Hou, X. Li and F. Wang, Biomater. Sci., 2019, 7, 2873–2888 RSC.
- Y. Zheng, Y. Liang, D. Zhang, Z. Zhou, J. Li, X. Sun and Y.-N. Liu, Chem. Commun., 2018, 54, 13805–13808 RSC.
- Y. Zhang, X. Zhang, W. Chen, Y. He, Y. Liu and H. Ju, J. Controlled Release, 2021, 336, 469–479 CrossRef CAS PubMed.
- Q. Liu, J. Tian, Y. Tian, Q. Sun, D. Sun, D. Liu, F. Wang, H. Xu, G. Ying, J. Wang, A. K. Yetisen and N. Jiang, Acta Biomater., 2021, 127, 287–297 CrossRef CAS PubMed.
- Z. Chu, H. Chen, P. Wang, W. Wang, J. Yang, J. Sun, B. Chen, T. Tian, Z. Zha, H. Wang and H. Qian, ACS Nano, 2022, 16, 4917–4929 CrossRef CAS PubMed.
- J. Yang, S. Zhai, H. Qin, H. Yan, D. Xing and X. Hu, Biomaterials, 2018, 176, 1–12 CrossRef CAS PubMed.
- G. Yang, J. Tian, C. Chen, D. Jiang, Y. Xue, C. Wang, Y. Gao and W. Zhang, Chem. Sci., 2019, 10, 5766–5772 RSC.
- C. Liu, K. K. Ewert, N. Wang, Y. Li, C. R. Safinya and W. Qiao, Biomaterials, 2019, 221, 119412 CrossRef CAS PubMed.
- Z. Xiao, Y. You, Y. Liu, L. He, D. Zhang, Q. Cheng, D. Wang, T. Chen, C. Shi and L. Luo, ACS Appl. Mater. Interfaces, 2021, 13, 35376–35388 CrossRef CAS PubMed.
- S. Uthaman, S. Pillarisetti, A. P. Mathew, Y. Kim, W. K. Bae, K. M. Huh and I.-K. Park, Biomaterials, 2020, 232, 119702 CrossRef CAS PubMed.
- K. Long, Y. Yang, W. Lv, K. Jiang, Y. Li, A. C. Y. Lo, W. C. Lam, C. Zhan and W. Wang, Adv. Sci., 2021, 8, 2101754 CrossRef CAS PubMed.
- W. Lv, K. Long, Y. Yang, S. Chen, C. Zhan and W. Wang, Adv. Healthcare Mater., 2020, 9, 2001118 CrossRef CAS PubMed.
- C. Liu, X. Guo, C. Ruan, H. Hu, B.-P. Jiang, H. Liang and X.-C. Shen, Acta Biomater., 2019, 96, 281–294 CrossRef CAS PubMed.
- G. L. Seah, J. H. Yu, B. I. Koo, D. J. Lee and Y. S. Nam, J. Mater. Chem. B, 2018, 6, 7737–7749 RSC.
- P. Pei, C. Sun, W. Tao, J. Li, X. Yang and J. Wang, Biomaterials, 2019, 188, 74–82 CrossRef CAS PubMed.
- Q. Li, W. Li, H. Di, L. Luo, C. Zhu, J. Yang, X. Yin, H. Yin, J. Gao, Y. Du and J. You, J. Controlled Release, 2018, 277, 114–125 CrossRef CAS PubMed.
- X. Ma, X. Li, J. Shi, M. Yao, X. Zhang, R. Hou, N. Shao, Q. Luo, Y. Gao, S. Du, X. Liang and F. Wang, Adv. Healthcare Mater., 2019, 8, 1900661 CrossRef PubMed.
- S. Palvai, C. T. Moody, S. Pandit and Y. Brudno, Mol. Pharm., 2021, 18, 3920–3925 CrossRef CAS PubMed.
- C. Ruan, C. Liu, H. Hu, X.-L. Guo, B.-P. Jiang, H. Liang and X.-C. Shen, Chem. Sci., 2019, 10, 4699–4706 RSC.
- W. Zhang, T. Ji, Y. Li, Y. Zheng, M. Mehta, C. Zhao, A. Liu and D. S. Kohane, Nat. Commun., 2020, 11, 2323 CrossRef CAS PubMed.
- A. Qu, X. Wu, S. Li, M. Sun, L. Xu, H. Kuang and C. Xu, Adv. Mater., 2020, 32, 2000184 CrossRef CAS PubMed.
- M. Wu, J. Li, X. Lin, Z. Wei, D. Zhang, B. Zhao, X. Liu and J. Liu, Biomater. Sci., 2018, 6, 1457–1468 RSC.
- E. M. Zywot, N. Orlova, S. Ding, R. R. Rampersad, E. M. Rabjohns, V. A. Wickenheisser, Q. Wang, J. G. Welfare, L. Haar, A. M. Eudy, T. K. Tarrant and D. S. Lawrence, Adv. Ther., 2022, 5, 2100159 CrossRef CAS PubMed.
- Y. Huang, Z. Guan, X. Dai, Y. Shen, Q. Wei, L. Ren, J. Jiang, Z. Xiao, Y. Jiang, D. Liu, Z. Huang, X. Xu, Y. Luo and C. Zhao, Nat. Commun., 2021, 12, 4310 CrossRef CAS PubMed.
- T. L. Rapp and C. A. DeForest, Adv. Drug Delivery Rev., 2021, 171, 94–107 CrossRef CAS PubMed.
- W. Zhao, Y. Zhao, Q. Wang, T. Liu, J. Sun and R. Zhang, Small, 2019, 15, 1903060 CrossRef CAS PubMed.
- A. M. Diez-Pascual and A. Rahdar, ChemMedChem, 2022, 17, e202200142 CrossRef CAS PubMed.
- G. Housman, S. Byler, S. Heerboth, K. Lapinska, M. Longacre, N. Snyder and S. Sarkar, Cancers, 2014, 6, 1769–1792 CrossRef PubMed.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS PubMed.
- P. Wu, X. Wang, Z. Wang, W. Ma, J. Guo, J. Chen, Z. Yu, J. Li and D. Zhou, ACS Appl. Mater. Interfaces, 2019, 11, 18691–18700 CrossRef CAS PubMed.
- W. Sun, Y. Wen, R. Thiramanas, M. Chen, J. Han, N. Gong, M. Wagner, S. Jiang, M. S. Meijer, S. Bonnet, H.-J. Butt, V. Mailänder, X.-J. Liang and S. Wu, Adv. Funct. Mater., 2018, 28, 1804227 CrossRef.
- X. Lai, X.-L. Liu, H. Pan, M.-H. Zhu, M. Long, Y. Yuan, Z. Zhang, X. Dong, Q. Lu, P. Sun, J. F. Lovell, H.-Z. Chen and C. Fang, Adv. Mater., 2022, 34, 2106682 CrossRef CAS PubMed.
- A. S. Abu Lila, H. Kiwada and T. Ishida, J. Controlled Release, 2013, 172, 38–47 CrossRef CAS PubMed.
- F. Lopes-Coelho, F. Martins, S. A. Pereira and J. Serpa, Int. J. Mol. Sci., 2021, 22, 3765 CrossRef CAS PubMed.
- G. Szakács, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 5, 219–234 CrossRef PubMed.
- G. K. Balendiran, R. Dabur and D. Fraser, Cell Biochem. Funct., 2004, 22, 343–352 CrossRef CAS PubMed.
- J. Chen, T. Fan, Z. Xie, Q. Zeng, P. Xue, T. Zheng, Y. Chen, X. Luo and H. Zhang, Biomaterials, 2020, 237, 119827 CrossRef CAS PubMed.
- R. Falk-Mahapatra and S. O. Gollnick, Photochem. Photobiol., 2020, 96, 550–559 CrossRef CAS PubMed.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed.
- N. Umezawa, K. Arakane, A. Ryu, S. Mashiko, M. Hirobe and T. Nagano, Arch. Biochem. Biophys., 1997, 342, 275–281 CrossRef CAS PubMed.
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef PubMed.
- K. M. Comerford, T. J. Wallace, J. Karhausen, N. A. Louis, M. C. Montalto and S. P. Colgan, Cancer Res., 2002, 62, 3387–3394 CAS.
- M. Dunne, M. Regenold and C. Allen, Adv. Drug Delivery Rev., 2020, 163–164, 98–124 CrossRef CAS PubMed.
- D. An, J. Fu, B. Zhang, N. Xie, G. Nie, H. Ågren, M. Qiu and H. Zhang, Adv. Funct. Mater., 2021, 31, 2101625 CrossRef CAS.
- K. Ariga, E. Ahn, M. Park and B.-S. Kim, Chem. – Asian J., 2019, 14, 2553–2566 CrossRef CAS PubMed.
- Y. Deng, F. Käfer, T. Chen, Q. Jin, J. Ji and S. Agarwal, Small, 2018, 14, 1802420 CrossRef PubMed.
- X. Sun, L. Li, H. Zhang, M. Dong, J. Wang, P. Jia, T. Bu, X. Wang and L. Wang, Adv. Healthcare Mater., 2021, 10, 2100546 CrossRef CAS PubMed.
- A. N. Bashkatov, E. A. Genina, V. I. Kochubey and V. V. Tuchin, J. Phys. D: Appl. Phys., 2005, 38, 2543–2555 CrossRef CAS.
- Y. Dai, B. Wang, Z. Sun, J. Cheng, H. Zhao, K. Wu, P. Sun, Q. Shen, M. Li and Q. Fan, ACS Appl. Mater. Interfaces, 2019, 11, 39410–39423 CrossRef CAS PubMed.
- Z. Wang, Y. Ju, Z. Ali, H. Yin, F. Sheng, J. Lin, B. Wang and Y. Hou, Nat. Commun., 2019, 10, 4418 CrossRef PubMed.
- J. Wang, W. Mao, L. L. Lock, J. Tang, M. Sui, W. Sun, H. Cui, D. Xu and Y. Shen, ACS Nano, 2015, 9, 7195–7206 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- N. M. Anderson and M. C. Simon, Curr. Biol., 2020, 30, R921–R925 CrossRef CAS PubMed.
- Z. Akram, F. Javed, M. Hosein, M. A. Al-Qahtani, F. Alshehri, A. I. Alzahrani and F. Vohra, Photodermatol., Photoimmunol. Photomed., 2018, 34, 167–174 CrossRef CAS PubMed.
- Y. Li, B. Wang, S. Zheng and Y. He, Photodiagn. Photodyn. Ther., 2019, 25, 17–22 CrossRef PubMed.
- Y.-C. Chang and C.-H. Yu, Photodiagn. Photodyn. Ther., 2014, 11, 127–129 CrossRef PubMed.
- Y. Han, S. Xu, J. Jin, X. Wang, X. Liu, H. Hua, X. Wang and H. Liu, Front. Physiol., 2018, 9, 1911 CrossRef PubMed.
- A. Gulzar, S. Gai, P. Yang, C. Li, M. B. Ansari and J. Lin, J. Mater. Chem. B, 2015, 3, 8599–8622 RSC.
- B. Liu and S. Thayumanavan, Cell Rep. Phys. Sci., 2020, 1, 100271 CrossRef CAS.
- S. Ikehara, R. N. Pahwa, G. Fernandes, C. T. Hansen and R. A. Good, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 886–888 CrossRef CAS PubMed.
- T. Tsuchiyama, Y. Nakamoto, Y. Sakai, Y. Marukawa, M. Kitahara, N. Mukaida and S. Kaneko, J. Immunol., 2007, 178, 574–583 CrossRef CAS PubMed.
- H.-Y. Yeung, P.-C. Lo, D. K. P. Ng and W.-P. Fong, Cell. Mol. Immunol., 2017, 14, 223–234 CrossRef CAS PubMed.
- A. Gandioso, M. Cano, A. Massaguer and V. Marchán, J. Org. Chem., 2016, 81, 11556–11564 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- S. Wen, J. Zhou, K. Zheng, A. Bednarkiewicz, X. Liu and D. Jin, Nat. Commun., 2018, 9, 2415 CrossRef PubMed.
- M. Almáši, A. A. Matiašová, M. Šuleková, E. Beňová, J. Ševc, L. Váhovská, M. Lisnichuk, V. Girman, A. Zeleňáková, A. Hudák and V. Zeleňák, Sci. Rep., 2021, 11, 20191 CrossRef PubMed.
- X. Li, X. Wang, M. Hua, H. Yu, S. Wei, A. Wang and J. Zhou, ACS Biomater. Sci. Eng., 2019, 5, 2399–2408 CrossRef CAS PubMed.
- H. He, J. Zhou, Y. Liu, S. Liu, Z. Xie, M. Yu, Y. Wang and X. Shuai, ACS Appl. Mater. Interfaces, 2018, 10, 7413–7421 CrossRef CAS PubMed.
- S. Li, J. Liu, G. Li, X. Zhang, F. Xu, Z. Fu, L. Teng, Y. Li and F. Sun, Eur. J. Pharm. Biopharm., 2019, 141, 1–11 CrossRef CAS PubMed.
- T. R. Damase, R. Sukhovershin, C. Boada, F. Taraballi, R. I. Pettigrew and J. P. Cooke, Front. Bioeng. Biotechnol., 2021, 9, 628137 CrossRef PubMed.
- M. M. Zhang, R. Bahal, T. P. Rasmussen, J. E. Manautou and X.-B. Zhong, Biochem. Pharmacol., 2021, 189, 114432 CrossRef CAS PubMed.
- A. Yamamoto, H. Ukai, M. Morishita and H. Katsumi, Pharmacol. Ther., 2020, 211, 107537 CrossRef CAS PubMed.
- W. B. Wan and P. P. Seth, J. Med. Chem., 2016, 59, 9645–9667 CrossRef CAS PubMed.
- S.-J. Cao, Z.-Q. Lv, S. Guo, G.-P. Jiang and H.-L. Liu, J. Drug Deliv. Sci. Technol, 2021, 61, 102124 CrossRef CAS.
- H. Sies, Curr. Opin. Toxicol., 2018, 7, 122–126 CrossRef.
- H. Chu, J. Zhao, Y. Mi, Z. Di and L. Li, Nat. Commun., 2019, 10, 2839 CrossRef PubMed.
- S. Huo, N. Gong, Y. Jiang, F. Chen, H. Guo, Y. Gan, Z. Wang, A. Herrmann and X.-J. Liang, Sci. Adv., 2019, 5, eaaw6264 CrossRef CAS PubMed.
- D. G. A. Burton and V. Krizhanovsky, Cell. Mol. Life Sci., 2014, 71, 4373–4386 CrossRef CAS PubMed.
- H. Maeda, K. Tsukigawa and J. Fang, Microcirculation, 2016, 23, 173–182 CrossRef CAS PubMed.
- S. M. Swain, F. S. Whaley and M. S. Ewer, Cancer, 2003, 97, 2869–2879 CrossRef CAS PubMed.
- P. S. Rawat, A. Jaiswal, A. Khurana, J. S. Bhatti and U. Navik, Biomed. Pharmacother., 2021, 139, 111708 CrossRef CAS PubMed.
- A. Singh, M. Talekar, A. Raikar and M. Amiji, J. Controlled Release, 2014, 190, 515–530 CrossRef CAS PubMed.
- L. W. Kleiner, J. C. Wright and Y. Wang, J. Controlled Release, 2014, 181, 1–10 CrossRef CAS PubMed.
- C. Bastiancich, E. Bozzato, I. Henley and B. Newland, J. Controlled Release, 2021, 337, 296–305 CrossRef CAS PubMed.
- X. Wang and D. J. Burgess, Adv. Drug Delivery Rev., 2021, 178, 113912 CrossRef CAS PubMed.
- L. Solorio, D. Sundarapandiyan, A. Olear and A. A. Exner, J. Pharm. Sci., 2015, 104, 3471–3480 CrossRef CAS PubMed.
- L. Solorio and A. A. Exner, J. Pharm. Sci., 2015, 104, 4322–4328 CrossRef CAS PubMed.
- R. R. S. Thakur, H. L. McMillan and D. S. Jones, J. Controlled Release, 2014, 176, 8–23 CrossRef CAS PubMed.
- Y. Yang, K. Long, Y. Wang, L. Li, J. Shi, J. Liu, L. Kong, L. Yu, J. Ding, Z. Huang, W. Wang and C. Zhan, Adv. Healthcare Mater., 2022, 11, 2102362 CrossRef CAS PubMed.
- X. He, Z. Yuan, S. Gaeke, W. W. Y. Kao, S. K. Li, D. Miller, B. Williams and Y. C. Park, ACS Appl. Bio Mater., 2021, 4, 1461–1469 CrossRef CAS PubMed.
- M. Hou, R. Yang, L. Zhang, L. Zhang, G. Liu, Z. Xu, Y. Kang and P. Xue, ACS Biomater. Sci. Eng., 2018, 4, 4266–4277 CrossRef CAS PubMed.
- M. Qiu, D. Wang, W. Liang, L. Liu, Y. Zhang, X. Chen, D. K. Sang, C. Xing, Z. Li, B. Dong, F. Xing, D. Fan, S. Bao, H. Zhang and Y. Cao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 501–506 CrossRef CAS PubMed.
- G. Song, G. Jiang, T. Liu, X. Zhang, Z. Zeng, R. Wang, P. Li and Y. Yang, ACS Biomater. Sci. Eng., 2020, 6, 4116–4125 CrossRef CAS PubMed.
- Y. Wu, H. Wang, F. Gao, Z. Xu, F. Dai and W. Liu, Adv. Funct. Mater., 2018, 28, 1801000 CrossRef.
- Y. Cao, J.-H. Dou, N.-J. Zhao, S. Zhang, Y.-Q. Zheng, J.-P. Zhang, J.-Y. Wang, J. Pei and Y. Wang, Chem. Mater., 2017, 29, 718–725 CrossRef CAS.
- T. Lammers, L. Y. Rizzo, G. Storm and F. Kiessling, Clin. Cancer Res., 2012, 18, 4889–4894 CrossRef CAS PubMed.
- P. S. Steeg, Nat. Rev. Cancer, 2016, 16, 201–218 CrossRef CAS PubMed.
- P. Sharmiladevi, K. Girigoswami, V. Haribabu and A. Girigoswami, Mater. Adv., 2021, 2, 2876–2891 RSC.
- S. Jeyamogan, N. A. Khan and R. Siddiqui, Arch. Med. Res., 2021, 52, 131–142 CrossRef CAS PubMed.
- S. Song, Y. Chong, H. Fu, X. Ning, H. Shen and Z. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 33867–33878 CrossRef CAS PubMed.
- S. Mallidi, G. P. Luke and S. Emelianov, Trends Biotechnol., 2011, 29, 213–221 CrossRef CAS PubMed.
- C. Zhang, D. Li, P. Pei, W. Wang, B. Chen, Z. Chu, Z. Zha, X. Yang, J. Wang and H. Qian, Biomaterials, 2020, 237, 119835 CrossRef CAS PubMed.
- S. S. Gambhir, Nat. Rev. Cancer, 2002, 2, 683–693 CrossRef CAS PubMed.
- S. Goel, C. G. England, F. Chen and W. Cai, Adv. Drug Delivery Rev., 2017, 113, 157–176 CrossRef CAS PubMed.
- X. Dong, W. Yin, X. Zhang, S. Zhu, X. He, J. Yu, J. Xie, Z. Guo, L. Yan, X. Liu, Q. Wang, Z. Gu and Y. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 4271–4284 CrossRef CAS PubMed.
- Y. Zhong, J. Zhang, R. Cheng, C. Deng, F. Meng, F. Xie and Z. Zhong, J. Controlled Release, 2015, 205, 144–154 CrossRef CAS PubMed.
- X. Ding, T. Wang, S. Bai, Y. Wan, S. Zhu, T. Li, N. Peng, T. Qiu and Y. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 27733–27742 CrossRef CAS PubMed.
- M. V. Swain and J. Xue, Int. J. Oral Sci., 2009, 1, 177–188 CrossRef PubMed.
- A. B. E. Attia, G. Balasundaram, M. Moothanchery, U. S. Dinish, R. Bi, V. Ntziachristos and M. Olivo, Photoacoustics, 2019, 16, 100144 CrossRef PubMed.
- L. Zhang, M. Zhang, L. Zhou, Q. Han, X. Chen, S. Li, L. Li, Z. Su and C. Wang, Biomaterials, 2018, 181, 113–125 CrossRef CAS PubMed.
- T. Li, J. Zhou, C. Zhang, X. Zhi, J. Niu, H. Fu, J. Song and D. Cui, NPG Asia Mater., 2018, 10, 1046–1060 CrossRef CAS.
- M. Bottini, N. Rosato, F. Gloria, S. Adanti, N. Corradino, A. Bergamaschi and A. Magrini, Int. J. Nanomed., 2011, 6, 3473–3485 CrossRef PubMed.
- A. R. Binder, M. A. Cacciatore, D. A. Scheufele, B. R. Shaw and E. A. Corley, Public Underst. Sci., 2012, 21, 830–847 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |