
Engineered bacteria for augmented in situ tumor vaccination
Xinyuan
Shen†
a,
Chaojie
Zhu†
 ab,
Xutao
Liu†
c,
Hanqi
Zheng
a,
Qing
Wu
ad,
Jijin
Xie
e,
Hao
Huang
f,
Ziyan
Liao
a,
Jiaqi
Shi
ad,
Kewang
Nan
g,
Junxia
Wang
a,
Xuming
Mao
e,
Zhen
Gu
*adhij and
Hongjun
Li
*abd
ab,
Xutao
Liu†
c,
Hanqi
Zheng
a,
Qing
Wu
ad,
Jijin
Xie
e,
Hao
Huang
f,
Ziyan
Liao
a,
Jiaqi
Shi
ad,
Kewang
Nan
g,
Junxia
Wang
a,
Xuming
Mao
e,
Zhen
Gu
*adhij and
Hongjun
Li
*abd
aKey Laboratory of Advanced Drug Delivery Systems of Zhejiang Province, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, China. E-mail: guzhen@zju.edu.cn; hongjun@zju.edu.cn
bDepartment of Hepatobiliary and Pancreatic Surgery the Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310009, China
cDepartment of Bioengineering, University of California, Los Angeles, California 90095, USA
dLiangzhu Laboratory, Zhejiang University Medical Center, Hangzhou 311121, China
eInstitute of Pharmaceutical Biotechnology, School of Medicine, Zhejiang University, Hangzhou 310058, China
fCollege of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China
gDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
hDepartment of General Surgery, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou 310016, China
iJinhua Institute of Zhejiang University, Jinhua 321299, China
jMOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
First published on 5th January 2023
Abstract
In situ tumor vaccination has aroused tremendous interest with its capability for eliciting strong and systemic antitumor immune responses. Unlike traditional cancer vaccines, in situ tumor vaccination avoids the laborious process of tumor antigen identification and can modulate tumor immunosuppressive microenvironment at the same time. In recent years, bacteria have been used as both efficient tumor-targeted delivery vehicles and potent adjuvants. Regarding the rapid development in this area, in this review, we summarize recent advances in the application of bacteria for in situ cancer vaccination. We illustrate the mechanisms of bacteria as both efficient tumor immunogenic cell death inducers and tumor-targeted delivery platforms. Then we comprehensively review the engineering strategies for designing bacteria-based in situ vaccination, including chemical modification, nanotechnology, and genetic engineering. The current dilemma and future directions are discussed at the end of this review.
1. Introduction
Cancer haunts public health with its high mortality rate.1 Though exhibiting certain therapeutic benefits, traditional chemotherapy and radiotherapy are rarely capable of curing such diseases and often correlate to severe side effects.2,3 As an alternative approach, cancer immunotherapy builds on priming endogenous immune systems to improve the recognition and elimination of tumors.4,5 Current cancer immunotherapy mainly includes four categories: cytokine-based therapies, monoclonal antibody-based therapies, tumor vaccines, and cell therapies.6 Among them, tumor vaccines have received wide attention, due to previous success in using vaccines to treat various infectious diseases.7 The formulation of cancer vaccines consists of tumor antigens and adjuvants. However, the laborious procedure of tumor antigen identification, tumor heterogeneity, and tumor immunosuppressive microenvironment significantly impact the therapeutic efficacy of cancer vaccines.8In recent years, the concept of tumor in situ vaccination (ISV) has been put forward.9 ISV strategies seek to induce immunogenic cell death (ICD) of tumor cells and generate immune responses against tumor antigens. During ICD, tumor debris and diverse danger-associated molecular patterns (DAMPs) are produced, which are capable of facilitating dendritic cell (DC) uptake and tumor antigen presentation and maturation, respectively.10 Compared to the traditional tumor vaccines, the immune responses elicited by in situ vaccination (ISV) could be more comprehensive and effective.11 In addition, the arduous tumor antigen identification process is also avoided at the same time. And during ICD, the released DAMPs can regulate the tumor immune suppressive microenvironment, which further facilitates the tumoricidal effects brought about by the activation of the ISV-elicited DCs and priming of tumor-specific cytotoxic T lymphocytes.12
During the development of in situ vaccination-oriented strategies, diverse delivery platforms have been used, including bacteria, nanoparticles, and cell components.13–15 Among them, bacteria-based platforms have demonstrated unique properties as a living therapeutic. In 1891, Coley utilized attenuated Streptococcus pyogenes for the treatment of malignant sarcoma and bacteria-based therapy demonstrated certain therapeutic benefits.16 In 1898, the first bacteria-based formulation of the Bacillus Calmette–Guerin (BCG) vaccine was approved by the FDA for bladder cancer therapy.17 In the last decade, the tumor-targeting capability of bacteria has been unraveled and is employed for anticancer drug delivery.18 In addition, bacteria contains diverse pathogen-associated molecular patterns (PAMPs), which are immunomodulatory and are capable of serving as vaccine adjuvants for boosting immune responses.19 Summarizing the above, the tumor-targeting potential and immunomodulatory capability of bacteria render the possibility of their integration into in situ vaccination formulation to augment cancer therapeutic efficacy.20 In this review, we first introduce the mechanisms of in situ vaccination. Then we discuss how bacteria can engage in the current process of ISV. Recent advances in developing bacteria as ISV components are surveyed, including chemical engineering of whole cells, tailoring bacteria into the nanoscale, and genetic engineering of bacteria to carry out therapeutic functions in situ (Fig. 1). Current limitations and future avenues are discussed at the end of the review.
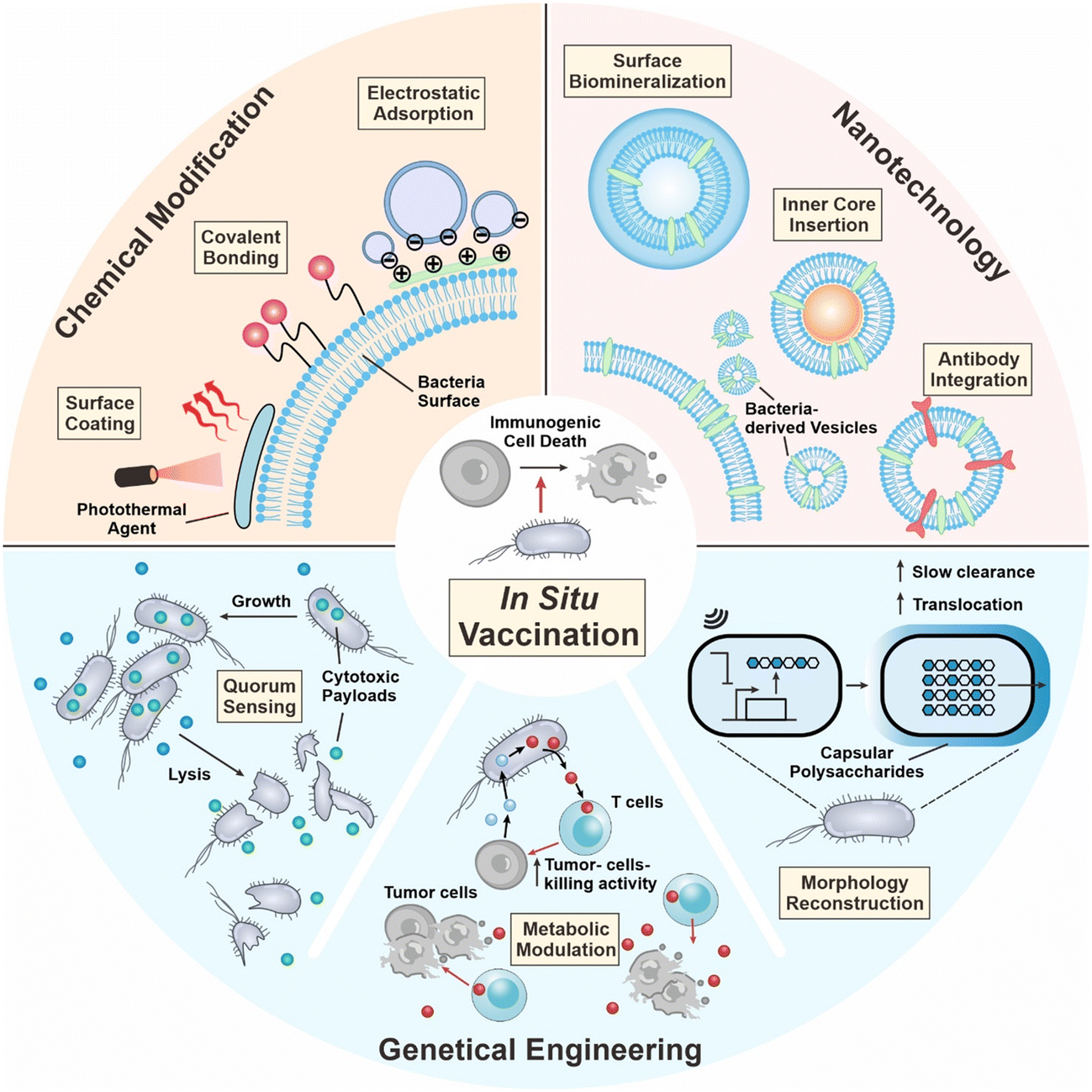 | ||
| Fig. 1 Engineering strategies of bacteria for in situ tumor vaccination. Firstly, the surface of bacteria can be chemically modified with diverse functional moieties through covalent linkage, electrostatic adsorption, and surface coating. Secondly, bacteria-derived membrane vesicles can be functionalized via surface biomineralization, inner core insertion, and antibody integration. Thirdly, bacteria can be genetically engineered to perform various functions, including quorum sensing lysis, metabolic modulatory capabilities, and morphology reconstruction. | ||
2. Mechanism: roles of bacteria in in situ tumor vaccination
The tumor microenvironment (TME) of solid tumors is characterized by the dense protection of the extracellular matrix, abnormal angiogenesis, and disordered metabolism.21,22 How to deliver drugs into the TME and maintain their efficacy have been heated questions for decades. Bacteria demonstrated strong capabilities for TME-targeted drug delivery. Their autonomous nature endows them with mobility and adaptivity to target and carry out certain functions within the TME. As for in situ vaccination, engineered bacteria-based strategies display superiority over other delivery strategies because of their tumor selectivity and cell death induction characteristics.2.1 Tumor targeting
Tropism, mobility, and anaerobic metabolism endow bacteria with tumor-targeting properties. In the periphery, bacteria can be rapidly cleared by lymphocytes and neutrophils, but the TME features a lack of immune clearance due to the immunosuppression caused by tumor cells and immunosuppressive cells.23 Compared with normal tissues, bacteria prefer to breed and replicate in tumor tissue. Bacterial structures like flagella and pili enable engineered bacteria to acquire proactive targeting capabilities towards the tumor, which is a unique feature of living bacteria-based strategies.24 With the help of these bacterial flagellar motors, engineered bacteria could penetrate the extracellular matrix of solid tumors, and execute their therapeutic functions within the TME. Moreover, because of the disordered vasculatures and high tissue density, the TME usually lacks oxygen, which hinders the infiltration and proliferation of many immune cells. Anaerobes like Salmonella, Bacillus subtilis, and Escherichia coli (E. coli) favor the hypoxic environment in solid tumors, while some of them can survive in oxygenated conditions, permitting their spread into the tumor periphery or distant tumors.25,26 Besides as tumor-targeting vectors, engineered bacteria have also been explored as tumor biosensors. Through genetic reconstruction, bacteria can be equipped with biosensors that only permit their proliferation in TME-specific environmental conditions. Acidity, hypoxia, and TME-related metabolite biosensors are possibly being integrated into the fundamental gene circuits in the bacterial genome, thereby developing platforms that target tumor-specific delivery.272.2 Immunogenic cell death induction
The crucial step in realizing ISV is to induce immunogenic cell death (ICD) of cancer cells, which could be caused by bacteria through either indirect immunogenicity enhancement or direct cytotoxicity generation. Unlike conventional vaccine preparation, which requires careful identification and isolation of tumor antigens, ICD can provide more comprehensive tumor-associated antigens (TAAs) directly from the dying tumor cells. During ICD, the circulating TAAs are exposed on the dying cancer cells, while DAMPs such as ATP and calreticulin (CRT) are released, increasing the immunogenicity of cell death. Thus, the attraction and activation of DCs are promoted and followed by the priming of T cells. However, cancer cell debris alone always fails to initiate sufficient immune response and escape from immune surveillance. Living bacteria could serve as an adjuvant to trigger immune responses in vivo. Pathological components of bacteria such as flagellin B (FlaB) and lipopolysaccharide (LPS) could activate the recognition of PAMPs in solid tumors, which subsequently induces the recruitment of T cells, NK cells, and macrophages into the TME and potentiates them for exerting immune surveillance.28 Local inflammation could be induced as innate immune pathways such as the toll-like receptor (TLR) and the stimulator of interferon genes (STING) are activated. Levels of pro-inflammatory cytokines like interleukins, IFN-γ, and TNF-α increase. And lymphocyte infiltration is promoted, converting a ‘cold tumor’ into a ‘hot tumor’.29,30 A typical example of approved bacteria-based ISVs is the BCG vaccine, which activates the TLR2 and TLR4 pathways, improving the efficacy of bacteria-induced ICD.31 Moreover, exotoxins secreted by the bacteria colonized in the TME can damage the membrane structures of cancer cells or disrupt normal cellular metabolism when internalized. In this way, the bacteria could kill cancer cells directly through the activation of TNF-related apoptosis.323. Engineering strategies
Despite the innate targeting property, the biosafety and body circularity of bacteria are still of great concern. Different engineering approaches can enhance the safety and efficacy of bacterial therapeutics, as well as moderate the immunogenicity, and improve intratumor survival. Nowadays, various engineering strategies are being used, such as chemical modification of the bacterial surface, harnessing nanotechnology to tailor bacteria into lifeless vesicles, or genetic modifications that endow bacteria with desired functions.3.1 Chemical modification
Due to the immunosuppressive and hypoxic tumor microenvironment and the motility of bacteria, therapeutic bacterial agents exhibit outstanding tumor targeting capability, rendering them as capable of serving as delivery vehicles for cancer in situ vaccination. The loading methods can be broadly divided into four categories, electrostatic adsorption, covalent binding, antibody–antigen specific interaction, membrane coating, and metabolic labeling.20 In this part, we will briefly introduce the engineering approaches and their mechanisms of conjugating drugs with bacteria and emphasize how bacteria can be leveraged as adjuvants for augmented cancer in situ vaccination.Photothermal therapy has been clinically verified to possess the ability to elicit anti-cancer immune responses.33In situ photothermal immunotherapy offers a feasible method for inducing ICD in solid tumors.34 In addition, it can be combined with other adjuvants for augmented cancer in situ vaccination. For example, in 2020, Yi et al. unexpectedly discovered that tumor thrombosis occurred and was darkened upon i.v. injection of ΔppGpp S. typhimurium, which could be leveraged for further photothermal therapy (Fig. 2A).35 The study first verified the tumor-homing capability of ΔppGpp S. typhimurium (Fig. 2B). Bacterial concentrations in all organs decreased except for tumor tissue. And upon intravenously injecting over 5 × 106 CFU, the darkness of the tumor can be easily distinguished (Fig. 2C). In the CT26 tumor model, the bacteria with the laser treatment group displayed increased DC maturation ratios in tumor-draining lymph nodes (Fig. 2D). As a result, the bacteria + laser + anti-CTLA4 treatment almost completely eradicated both the primary and the secondary CT26 tumors. Taking a further step, the bacterial surface can be modified with photothermal agents for augmented photothermal immunotherapy, for example, polydopamine (pDA). Chen et al. generated pDA-coated VNP20009 through surface dopamine oxidation and self-polymerization.36 With the advantage of the inherent tumor homing capability of VNP20009, the photothermal agents can be efficiently delivered to tumor site. With a single injection and laser irradiation, B16F10 melanoma tumors were completely eradicated by inducing tumor cell apoptosis and necrosis (Fig. 2E). Apart from surface chemical modification, bacteria can also be genetically engineered to produce photothermal agents. Meanwhile, chemical modifications of the bacterial surface well collaborate with other engineering approaches, for example, genetic engineering. Wang et al. transformed tyrosinase-encoded plasmids into E. coli with surface modification of dopamine and anti-PD1 antibodies (BacMel) (Fig. 2G).37 Intratumoral injection of BacMel exhibited uniform distribution inside 4T1 tumors. Such homogeneous distribution made the localization of photothermal agents (melanin) spatiotemporally controllable, resulting in a moderate and uniform heating treatment of tumors (Fig. 2F). As a result, this engineering strategy significantly suppressed the growth of 4T1 orthotopic murine tumors (Fig. 2H).
 | ||
| Fig. 2 Leveraging bacteria for augmented in situ photothermal immunotherapy. (A) VNP20009 induced thrombosis in tumors and can be served for photothermal immunotherapy. (B) VNP20009 colonization in mouse organs after i.v. injection. (C) Photographs of mice before and after i.v. injection of different doses of VNP20009. (D) The maturation of DC cells in tumor-draining lymph nodes after different treatments. (a) Surgery; (b) PLGA-PEG-ICG + surgery; (c) PLGA-PEG-ICG + laser; (d) bacteria + surgery; (e) bacteria + laser. Reproduced from ref. 35. Copyright 2020 American Association for the Advancement of Science. (E) Hypoxia-targeting bacteria deliver polydopamine for cancer photoimmunotherapy. Reproduced from ref. 36. Copyright 2018 American Chemical Society. (F) Spatiotemporal controlled distribution of bacteria, photothermal agents, and immune checkpoint inhibitors can augment cancer in situ vaccination via photothermal immunotherapy. (G) Surface engineering strategy and TEM images of engineered bacteria. Scale bar = 1 μm. (H) 4T1 orthotopic tumor growth and survival curves after receiving different treatments. Reproduced from ref. 37 Copyright 2022 John Wiley & Sons, Inc. | ||
Besides photothermal therapy, bacteria can also engage in current radiotherapy to elicit therapeutic efficacy. According to a clinic report, patients with melanoma receiving local irradiation combined with CTLA-4 antibodies (ipilimumab) exhibited dramatic abscopal responses.38 Recently, researchers have poured their efforts into chemical engineering living bacteria to synergize with current irradiation therapy (IRT). For example, Pei et al. loaded radioiodine (125I or 131I) labeled VNP20009 via the iodogen oxidation method (I-VNP) (Fig. 3A).39 Intratumoral injection of I-VNP realized sustained retention of irradiation-sensitizing agents for over 168 hours, possibly due to the tropism of bacteria and the engulfing of I-VNP by tumor-resident macrophages. However, in the control group, radioiodine-loaded bovine serum albumin (BSA) exhibited rapid clearance in vivo. After five days of post-irradiation treatments, the spleens of the I-VNP group were found to contain the highest content of 2′,3′-cGAMP, which correlated with the signaling potency of the cGAS-STING pathway and was beneficial for eliciting the subsequent adaptive anticancer immune responses. In addition, they verified that abscopal tumor cells upregulated expression of PD-L1 after IRT on primary tumors, which was potentially induced by extracellular vesicles released by the primary tumor cells. Accordingly, they further combined ICB therapy and realized 60% survival for up to 60 days in the CT26 tumor model, with none of the subjects in the control group remaining to live on day 30. Recently, Wang et al. taking a different perspective, leveraged bacteria as a tumor antigen delivery vehicle for eliciting augmented systematic anticancer immune responses (Fig. 3B).40 The study loaded positively charged nanoparticles (polyamidoamine dendrimer) on VNP20009 through electrostatic adsorption. Electrostatic adsorption could capture tumor antigens in situ through ionic forces. VNP20009 are highly mobile through converting chemical energy into mechanical energy via flagella. After applying local IRT, tumor cells underwent immunogenic cell death and released tumor antigens along with various DAMPs. Such antigen-capturing bacteria can transport the tumor antigens derived from the inner necrotic core to tumor marginal tissues, where a large number of normal DCs are localized. This strategy enhanced the crosstalk between tumor antigens induced by IRT and peripheral DC cells and achieved a prolonged survival time of up to 265 days in the CT26 tumor mouse model.
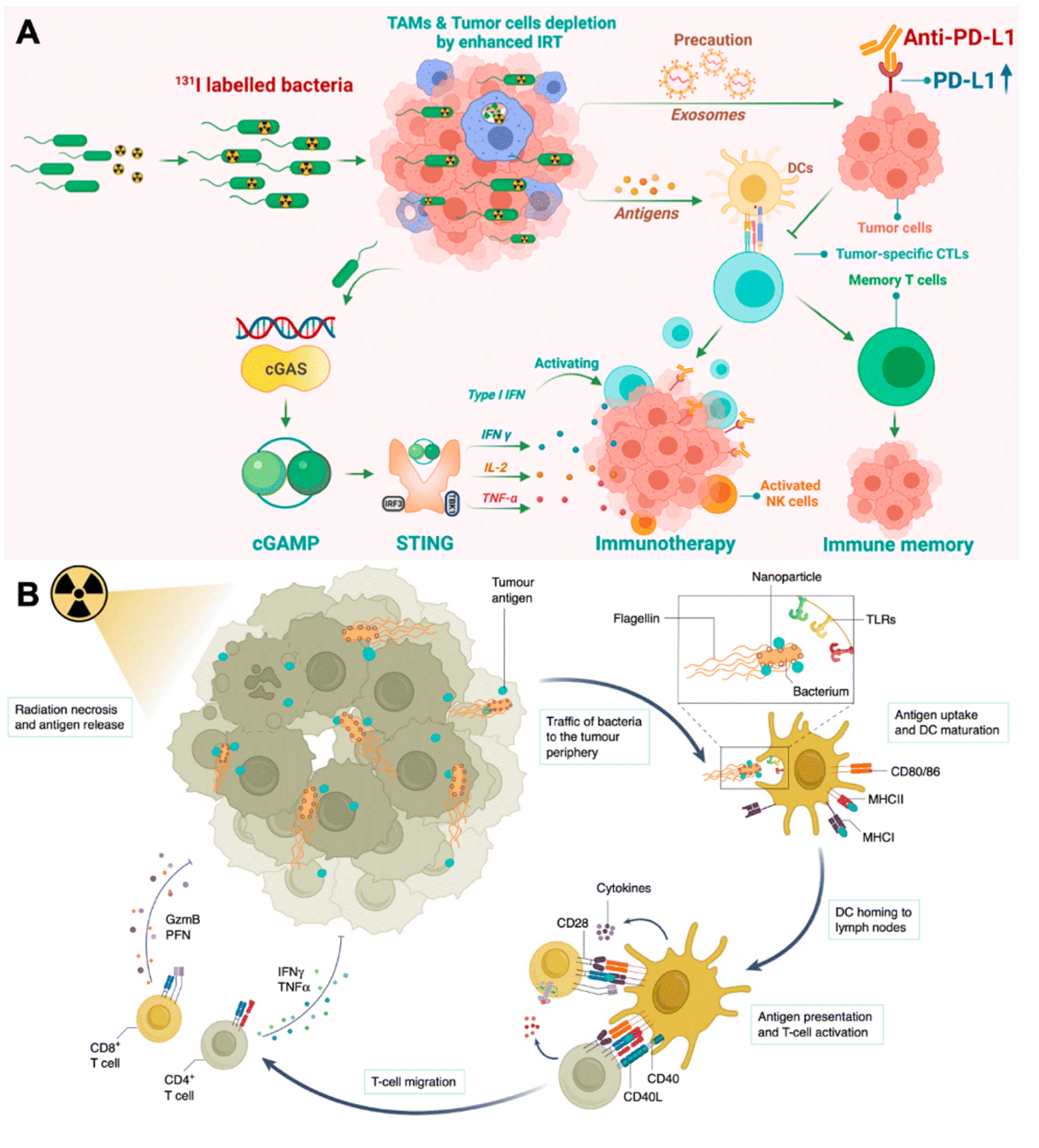 | ||
| Fig. 3 Engineering bacteria for augmenting IRT-induced in situ tumor vaccination. (A) Therapeutic mechanisms of I-VNP for potentiating IRT-induced adaptive anticancer immune response. Reproduced from ref. 39. Copyright 2022 American Chemical Society. (B) Antigen-capturing bacteria transport IRT-induced tumor antigens from inner core in the tumor periphery for augmented in situ cancer vaccination. Reproduced from ref. 41. Copyright 2022 Nature Publishing group. | ||
Overall, bacteria can be chemically engineered or genetically engineered to overcome the current limits of photothermal and irradiation therapy. Bacteria can serve as delivery vehicles to transport acquired sensitive agents to the targeted lesions for promoting therapeutic efficacy. Importantly, the intratumoral motility of bacteria has been leveraged to enhance the crosstalk between ICD-released tumor antigens to DCs. This strategy has provided more options in using bacteria to serve as novel adjuvants for cancer in situ vaccination.
3.2 Nanotechnology
Bacteria display various pathogen-associated molecular patterns, which are immunostimulatory and can be leveraged to reverse tumor local immune suppression.42 The recent advancements in nanotechnology have facilitated vaccine development in many aspects.43,44 Bacteria can be tailored to the nanoscale and termed bacteria-derived nanovesicles. Such nanovesicles preserve the immune stimulatory properties of whole bacteria. However, they are deficient in proliferation, and hence are less pathogenic. Based on the sourcing and the manufacturing process, bacteria-derived nanovesicles can be divided into five categories, including membrane vesicles (G+ bacteria release), outer membrane vesicles (G− bacteria produce), double-layered membrane vesicles (via chemical/physical breakdown), protoplast-derived nanovesicles (derived from bacteria inner membranes), and minicells (originate from abnormal bacteria division).45 Despite their different biogenesis routes, they all contain membrane proteins, lipoproteins, peptidoglycans, lipopolysaccharides, and nucleic acid, with different composition ratios.46 Therefore, in this section, we will not strictly distinguish the nanovesicle type, but emphasize the nanotechnology-based engineering strategies for strengthening these bacteria-derived nanovesicles for cancer immunotherapy.In 2017, Kim et al. explored the therapeutic capability of bacteria-derived outer membrane vesicles (OMVs).47 In this study, they genetically inactivated lipopolysaccharide expression in E. coli (E. coli msbB−/−, ΔmsbB) for safety assurance. The E. coli outer membrane nanovesicles derived from the wild-type and ΔmsbB mutant strains exhibited similar diameters of 38.6 ± 3.6 nm and 38.7 ± 4.2 nm, respectively (Fig. 4A & B). Systematic administration of OMVs resulted in strong tumor accumulation, possibly due to the enhanced permeability and retention (EPR) effect of the nanoparticle (Fig. 4C). Different i.v. injection dosages of OMV induced variable tumor inhibition, the highest of which (5 μg) almost completely regressed CT26 murine colon adenocarcinoma (Fig. 4D). The therapeutic potential of OMVs was later discovered to be interferon-γ (IFN-γ) dependent. However, IFN-γ can induce local immune suppression at the tumor site, which may impact the final therapeutic potency of OMVs. Li et al. genetically engineered ollie. Coli to produce OMVs that are surface modified with mouse PD-1, denoted as OMV-PD1.48 Such modified OMV-PD1 not only preserved immunostimulatory functions as bare OMVs but also could bind to the PD-L1 on tumor cells for internalization and reduction, and subsequently avoided T cell exhaustion induced by the PD1/PD-L1 immune inhibitory axis (Fig. 4E).
 | ||
| Fig. 4 Leveraging OMV for cancer immunomodulation. (A) TEM images of OMVs derived from wild-type and ΔmsbB mutant strain of E. coli. (B) Dynamic light scattering of measuring the size of OMVs. (C) Biodistribution of OMV via systematic injection. (D) Tumor inhibition capability of OMV. Reproduced from ref. 47. Copyright 2017 Nature Publishing Group. (E) Engineering method and therapeutic mechanisms of E. coli OMV. Reproduced from ref. 48. Copyright 2020 American Chemical Society. | ||
Bacteria-derived membrane nanovesicles can also be combined with other therapies for synergistic therapeutic effects, such as photothermal therapy. Hwang et al. designed E. coli mimetic AuNRs, which were made of Escherichia coli membrane proteins and adhesion protein-encapsulated gold nanorods.49 Gold nanorods could convert near-infrared light irradiation into heat for tumor local photothermal therapy. In the study, they discovered that the E. coli adhesion protein (FimH) increased the number of DCs and induced their maturation in the tumor-draining lymph node. As a result, when combined with direct tumoricidal efficacy of hyperthermia induced by NIR irradiation, such combinatory therapy successfully inhibited CT26 tumors and could also protect the body from tumor relapse. Intervening in the highly immunosuppressive tumor microenvironment often requires comprehensive perspectives for the approaches. And it is inadequate to simply focus on tumor local cytokine profiles. Tumor-resident immune cells, serving as both the producers and receivers of tumor-derived cytokines, also play a key role during tumor progression.
Tumor-associated macrophages are typical targets for immunomodulation, which usually exhibit the M2 phenotype in the tumor microenvironment for promoting tumor progression. Qing et al. fabricated a calcium phosphate (CaP) biomineralized, indocyanine green-loaded OMV (OMV@CaP-ICG) (Fig. 5A).50 The highly pH-responsive CaP shell could effectively neutralize tumor acidity, which led to strong tumor-associated macrophage modulation from the M2 to the M1 phenotype. In vivo experiment discovered that the OMV@CaP-ICG plus laser treatment elicited CRT expression, ATP secretion, and HMGB1 release of 4T1 tumor cells (Fig. 5B). Upon systematic injection, OMV@CaP-ICG accumulated in the tumor due to the responsiveness of the CaP shell and EPR effect. When exposed to 808 nm irradiation, local hyperthermia-induced immunogenic tumor cell death, together with the OMV adjuvant-induced tumor cell debris uptake and maturation of DCs. As a result, the OMV@CaP-ICG plus laser treatment completely inhibited 4T1 tumor growth in mice (Fig. 5C). However, when applying photothermal therapy to induce tumor cell ICD, the exposure of phosphatidylserine (PS) on the outer tumor cell membrane leaflet exposes the ‘eat me’ signals to the surrounding macrophages. And subsequently, these macrophages release the immunosuppressive cytokines, transforming growth factor-β (TGF-β) and interleukin-10 (IL-10) in particular, which suppress tumor local inflammation and inhibit the maturation of DCs. To elicit a stronger tumor immune response, Li et al. innovated an apoptotic cell blockade strategy. In the study, they fabricated annexin A5 (ANX5)-loaded, hyaluronate, and OMV-coated hollow mesoporous organosilica nanocapsules (HMSeN–ANX5@HOMV).51 ANX5 can bind to surface phosphatidylserine (PS) of apoptotic tumor cells, which leads to secondary necrosis and are more suitable for yielding a variety of tumor cell antigens and DAMPs (Fig. 5D). Mechanistically, once laser irradiation was applied, systematic injected HMSeN–ANX5@HOMV induced secondary necrosis in tumor cells. And the debris of the primary tumor could act as an in situ tumor antigen depot and activate systematic anticancer immune responses together with the adjuvant DAMPs and OMVs. As a result, the strategy achieved 50% tumor eradication in mice with orthotopic breast tumors.
 | ||
| Fig. 5 Combinatory OMV-based cancer immunotherapy. (A) Therapeutic strategy of OMV@CaP-ICG. (B) CRT exposure, ATP release, and HMGB1 release of 4T1 tumor cells under different treatment conditions. (C) Survival curves of 4T1 tumor-bearing mouse receiving different treatments. Reproduced from ref. 50. Copyright 2022 John Wiley & Sons, Inc. (D) Mechanisms of HMSeN–ANX5@HOMV-based therapy. Reproduced from ref. 51. Copyright 2020 Nature Publishing Group. | ||
The abovementioned strategies all targeted tumor local immune modulation. And the tumor antigens that dead tumor cells release are unattached to the adjuvant OMVs. Empirical experience indicates that vaccines are more potent when the adjuvant and antigens are integrated into a tangible formulation, although the mechanisms remain to be explored. And the tumor antigen-capturing strategy sheds new light on OMV-based in situ tumor vaccination. For example, Patel et al. designed bacterial membrane-coated PC7A/CpG polyplex core nanoparticles with a surface display of imide groups denoted as bacterial membrane-coated nanoparticles (BNPs).52 After tumors are subjected to radiation therapy, cancer cells release neoantigens. Intratumoral administration of BNPs could react with the thiol group on the neoantigens to form the BNP–neoantigen complex. The bacterial membrane coating stimulated TLR2 on the DC membrane and facilitated highly efficient DC uptake. The CpG inside could stimulate the maturation of DC cells via engaging with TLR9 (Fig. 6A). Similarly, Li et al. established 1-methyl-tryptophan-loaded OMVs joined with maleimide groups (1-MT@OMV-Mal).53 After applying photothermal therapy at the primary tumor, intratumoral administration of 1-MT@OMV-Mal could capture tumor antigens via the formation of stable thioether bonds. In addition, 1-MT could inhibit tumor immune suppression through suppressing indoleamine 2,3-dioxygenase-mediated T cell regulation post photothermal therapy (Fig. 6B). As a result, 54% of mice in the 1-MT@OMV-Mal group survived for more than 42 days post drug injection, while mice in the other groups all died before 35 days.
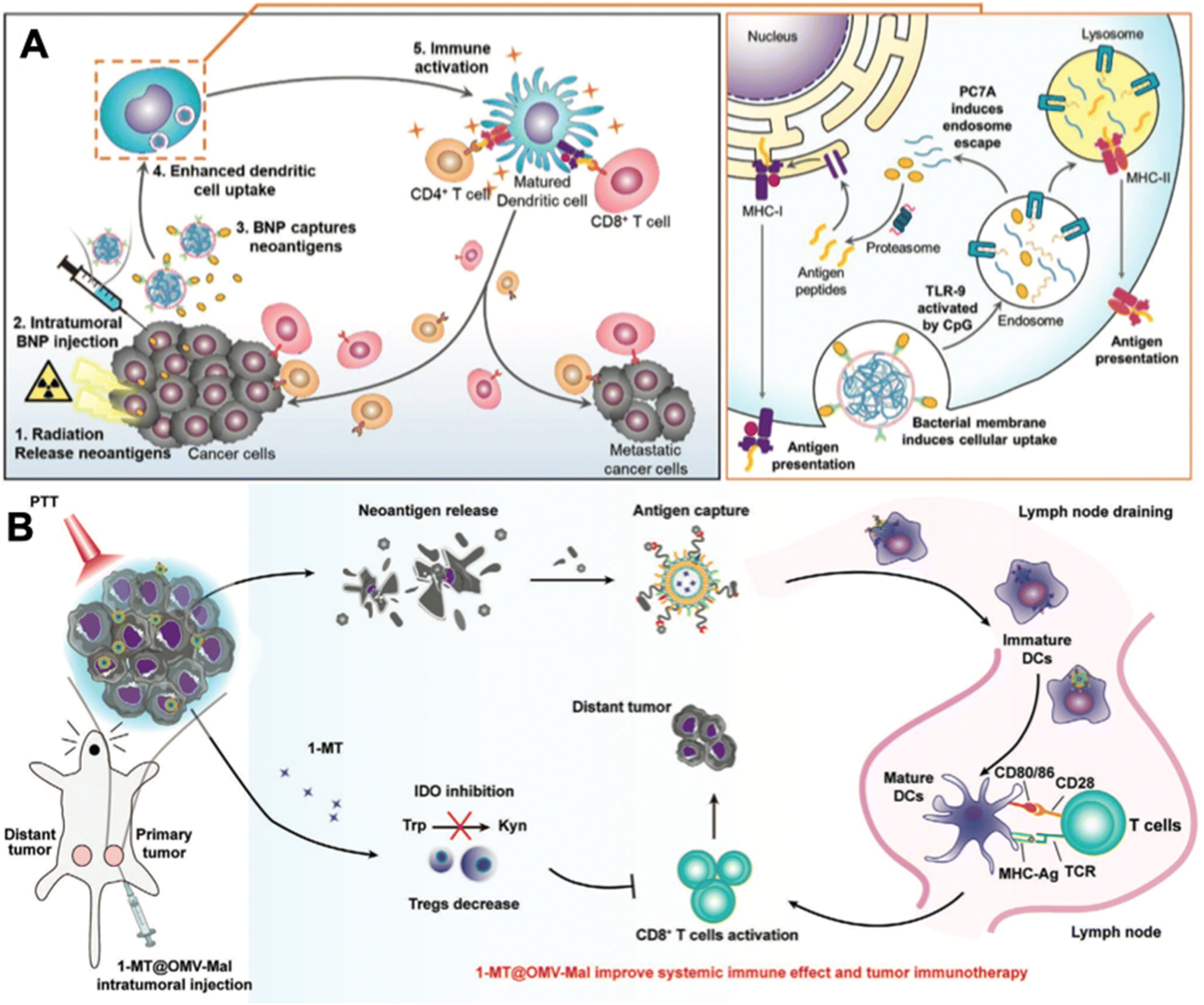 | ||
| Fig. 6 In situ tumor antigen-capturing OMV-based cancer immunotherapy. (A) Therapeutic mechanisms of BNPs. BNPs can capture tumor antigens in situ. Bacteria membrane coating facilitate the VNP-neoantigens uptake by DC cells. CpG can stimulate the maturation of DC cells. Reproduced from ref. 52. Copyright 2019 John Wiley & Sons, Inc. (B) 1-MT@OMV-Mal for cancer immunotherapy. Surface maleimide groups can bind to tumor antigens by forming thioether bonds. OMVs can facilitate the maturation of DC cells. The loaded 1-MT can inhibit IDO-mediated metabolism from tryptophan (Trp) to kynurenine (Kyn), which decreases the number of Tregs for augmenting CD8+ T cell activity. Reproduced from ref. 53. Copyright 2022 John Wiley & Sons, Inc. | ||
In conclusion, OMV-based in situ vaccination exhibits therapeutic potential when converged with advancing nanotechnology. Firstly, bare OMVs can suppress tumor progression through the IFN-γ mediated way. Secondly, OMVs can be further genetically engineered, or surface modified with other functional moieties, for example, PD-1, and CaP nanoshell. These groups can neutralize the tumor immunosuppressive microenvironment, which can cooperate with the CD8+ T cells elicited by OMV-based in situ tumor vaccination for better therapeutic performance. Thirdly, OMVs can serve as membrane coating materials to promote the uptake of nanoparticles by DCs. The OMV membrane can also directly engage with the TLR2 of DCs to elicit their maturation. Fourthly, OMV-based vaccines can be combined with current therapy including chemotherapy and photothermal therapy for better therapeutic performance. Apart from in situ vaccination, researchers recently innovated a hybrid membrane strategy for cancer vaccines. Chen et al. designed hybrid nanoparticles with tumor cell membranes and E. coli cytoplasm membranes.54 Tumor cell membranes provided tumor antigens, and E. coli cytoplasm membranes acted as adjuvants. This method avoided the need for identifying and isolating tumor neoantigens, exhibiting great promise in serving as next-generation patient-specific cancer vaccines with high feasibility and therapeutic efficacy. Besides, OMVs can be genetically engineered using protein plug-and-display systems.55 Such systems allow OMVs specifically binding with tagged neoantigens via an isopeptide connection between the tag and the catcher, forming viable off-the-shelf tumor vaccines.
3.3 Genetic engineering
Bacteria, with their tendency to colonize the TME and programmable genetic circuits, are versatile platforms that can be genetically engineered to possess various functions. Bacteria have available metabolic pathways that can be utilized for artificial modification.56,57 Notably, many of these pathways naturally interfere with tumor metabolism processes such as the tricarboxylate cycle, lactic acid metabolism, and transfer of electrons. Combining the technologies of synthetic biology, we can reconstruct the bacteria to realize diverse functions like in situ syntheses of antitumoral molecules, responsive secretion, and metabolic regulation in the tumor microenvironment.The most basic idea to achieve antitumoral function on engineered bacteria is releasing genetically encoded cargos into the TME, which may serve as antigens or adjuvants for in situ vaccination. In 2016, Omar Din et al. engineered Salmonella with a synchronized lysis circuit (SLC) to achieve the controlled release of payloads and to limit their population.58 The Salmonella were programmed to attain synchronized cycle lysis via the bacteriophage lysis gene (φX174 E), which is controlled by the autoinducer acyl-homoserine-lactone (AHL) that co-expressed with a cytotoxic agent (Fig. 7A). Once the bacteria reached the quorum threshold according to critical AHL concentration, AHLs enable the transcription of the φX174 E gene. Cytotoxic payloads like hemolysin, pro-apoptotic peptides, and chemokines were incorporated into the engineered Salmonella strains for antitumoral function. The SLC circuit enabled the bacteria to release its cargo in a pulsatile delivery manner in vivo (Fig. 7B). The engineered Salmonella could be orally administered alone or as co-therapy with chemotherapeutic agents, exhibiting strong efficacy in a mouse model. In another study, the SLC circuit was deployed on a non-pathogenic E. coli strain for in situ synthesis and release of CD47 nanobodies at the tumor site (Fig. 7C).59 The bacterial lysate alone is an adjuvant that encouraged macrophage phagocytosis and TLR agonism. When used in combination with CD47 nanobody blockade, the total increase in phagocytosis was 90% higher, suggesting a synergetic effect of the SLC–CD47 nanobody encoding ollie. Coli strain (Fig. 7D). Consequently, the therapy induced robust adaptive antitumor immune responses, and therefore inhibited tumor growth in local and distant tumors upon intratumoral (i.t.) injection (Fig. 7E). In 2020, E. coli Nissle 1917 (EcN), a safe probiotic was engineered to express PD-L1 and CTLA4 nanobodies in the same manner (Fig. 7F).60 The SLC circuit was transcriptionally stabilized by replacing the previous two-plasmid system with a stretch of DNA that integrated into the genome of EcN-lux (Fig. 7G). The copy number of different types of plasmids encoding quorum-sensing gene were simulated and examined, and the plasmid resulting in the optimal therapeutic release mechanism was selected. Two strains of EcN encoding PD-L1 nanobodies and CTLA4 nanobodies were administered upon a single injection when the tumor reached 150 mm3, and the therapeutic efficacy of the engineered bacteria was significantly stronger compared to that of merely PD-L1 mAb and CTLA4 mAb co-therapy (Fig. 7H).
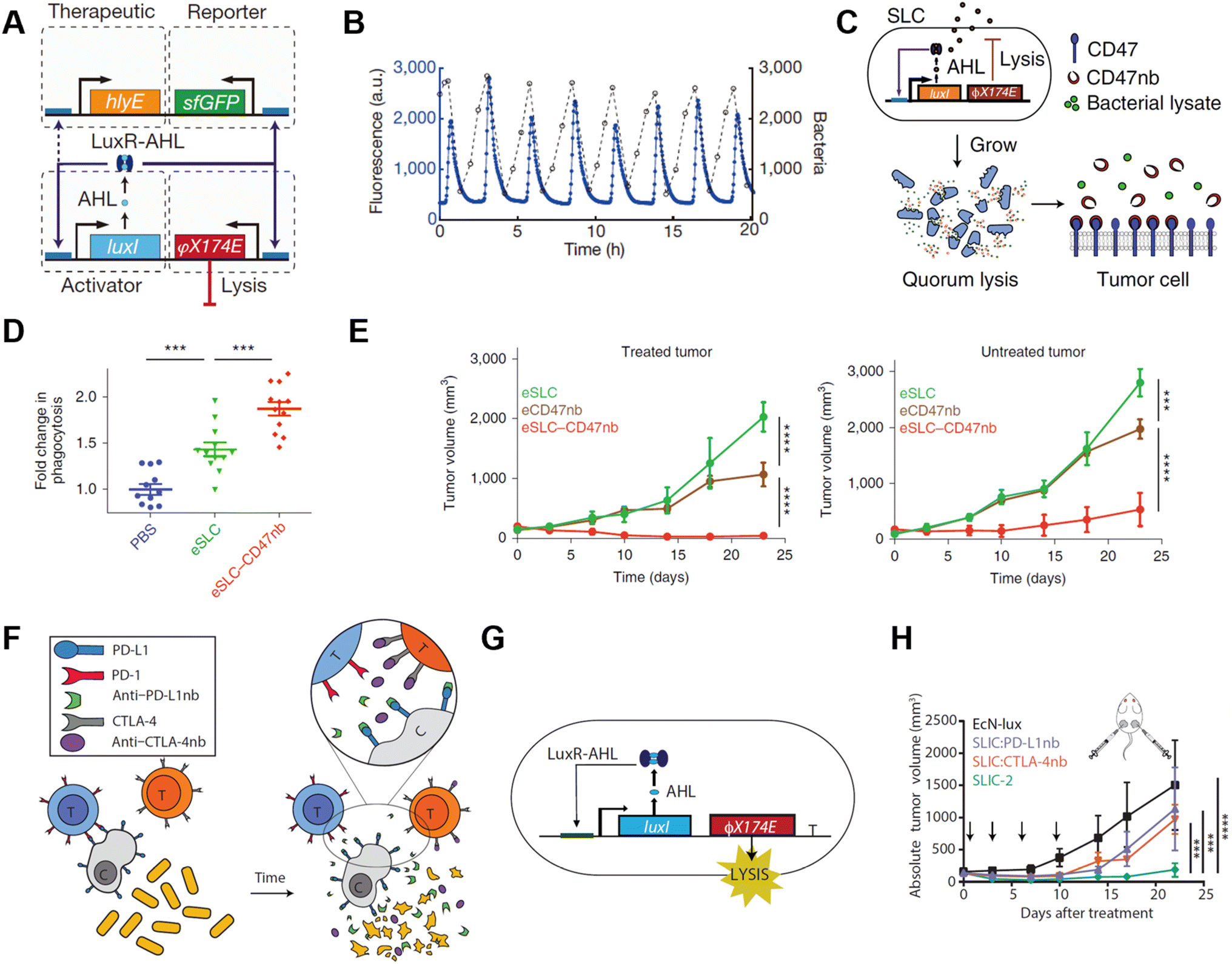 | ||
| Fig. 7 Quorum sensing bacteria for in situ vaccination. (A) The circuit consists of a hlyE, sfGFP-based reporter module, and a φX174 E-based quorum lysis model, and they are driven by the luxI promoter. (B) A typical population trajectory is tracked by fluorescence via a microfluidic experiment. Reproduced from ref. 58. Copyright 2016 Nature Publishing Group. (C) Synchronized lysis circuit was constructed to achieve the constitutive release of CD47 nanobodies. (D) DiI-labeled A20 cells were used to assess in vitro phagocytosis by BDMDs, cancer cells are pretreated with PBS or lysate of engineered bacteria. (E) Tumor growth curves on treated and untreated A20 tumors administered unilateral i.t. injections of engineered bacteria. Reproduced from ref. 59. Copyright 2019 Nature Publishing Group. (F) Schematic illustration of the intratumoral release of PD-L1 and CTLA-4 nanobodies released from the engineered EcN. (G) The quorum lysis module is integrated in the genome of EcN. (H) Tumor growth curves on A20 tumor-bearing BALB/c mice, mice received i.t. injections of EcN-lux (control), CTLA-4nb expressing strain, PD-L1nb expressing strain, or a combination of the latter two. Reproduced from ref. 60. Copyright 2020 American Chemical Society. | ||
Genetically modified bacteria also demonstrated strong potency in metabolic modulation of the TME, either enhancing antitumoral immune response or removing tumor cell metabolites. Studies have revealed that EcN is a non-pathogenic strain that colonizes a wide range of tumor types, which makes it an ideal platform for sustained bacteria-based modulation within the TME.61–63 Leventhal et al. reconstructed EcN to stimulate the STING pathway for potentiating phagocytic and antigen-presenting cell-related immune responses. The engineered EcN strain, termed SYNB1891, could express cyclic di-AMP (CDA)-producing enzymes when the tetracycline-inducible promoter (Ptet) is activated, elevating the CDA level within the TME to initiate macrophage phagocytosis (Fig. 8A).64 Thymidine and diaminopimelic acid-related genes were depleted from the EcN genome for safety and regulatory purposes. IFN and other T cell-associated cytokines were found spiked on B16·F10 tumor-bearing mice after being treated with a single i.t. injection of 109 colony-forming units (CFUs) of SYNB1891. Consequently, SYNB1891 treatment slowed tumor growth and eliminated tumors in some mice (Fig. 8B). Another attempt to modulate tumor metabolism with genetically modified EcN focused on producing L-arginine in the TME to regulate T cell metabolism (Fig. 8C).65 Canale et al. integrated ArgAfbr gene into EcN to continuously express N-acetyl glutamate synthase without being suppressed by the high arginine concentration.66 The high L-arginine level enhanced the survival and antitumor activity of CD4+/CD8+ T cells, resulting in a nearly two-fold higher T cell population in MC38 tumor models (Fig. 8D). When the L-arg bacteria therapy synergized with PD-L1 blockade, it exhibited tumor-curing efficacy upon i.t. injection. The co-therapy can be administered systemically, showing its broader application prospects as complementary strategies for adoptive T cell therapies.
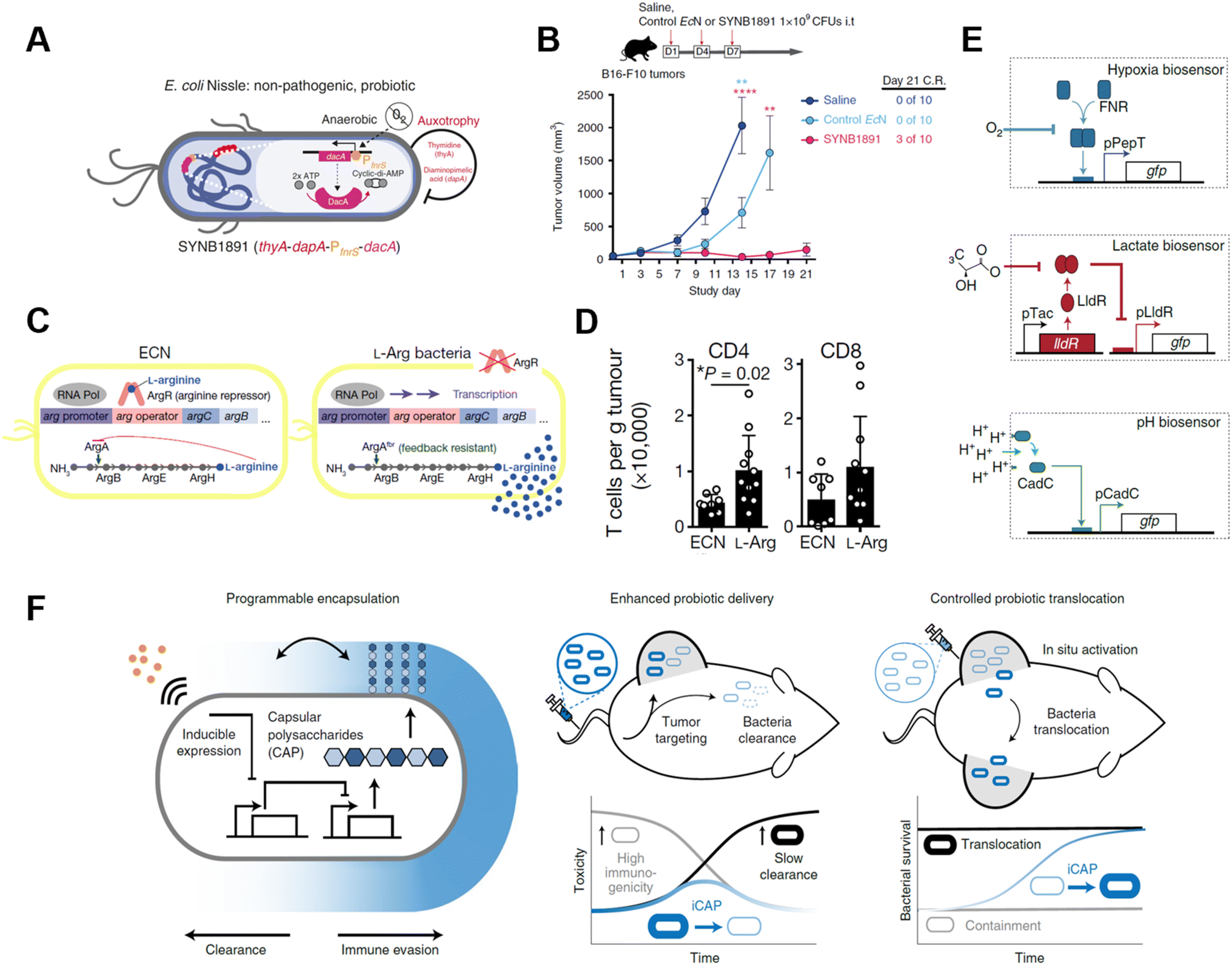 | ||
| Fig. 8 Genetically engineered bacteria for tumor metabolism modulation and tumor tropism enhancement. (A) Schematic illustration of the engineered bacteria strain, the gene encoding CDA-producing enzyme under the control of Ptet was encoded in the genome of EcN. (B) Tumor growth curves of B16·F10 tumor-bearing mice treated with i.t. injection of saline, control EcN or engineered EcN. Reproduced from ref. 64. Copyright 2020 Nature Publishing Group. (C) Schematic illustration of l-Arg producing bacteria, showing the differences between modified genome and normal EcN genome. (D) CD4/CD8+ T cell infiltration on the MC38 tumor was measured via flow cytometry. Reproduced from ref. 66. Copyright 2021 Nature Publishing Group. (E) Biosensor of hypoxia, lactate consists of modified native bacteria promoters and a GFP-expressing unit. Reproduced from ref. 27. Copyright 2022 Nature Publishing Group. (F) Schematic illustration of engineered bacteria with a CAP expressing unit. Reproduced from ref. 67. Copyright 2022 Nature Publishing Group. | ||
As engineered bacteria for cancer immunotherapy are under active research, scientists have been improving the targetability and responsiveness of strains, hoping to build versatile and tumor-penetrating platforms. Hypoxia and acidity are the two most irrepressible traits of the TME. In a recent study, bacteria were equipped with biosensors to preferentially harbor tumors. Hypoxia-sensing promoters, pH-sensitive promoters, and lactate promoters were constructed to control the essential genes for bacterial amino acid biosynthesis (Fig. 8E).27 An AND gate logic circuit was designed between each of two biosensors to permit bacterial growth only when two environmental conditions simultaneously occured. The multiplexed biosensor circuits augmented the specific colonization within the TME when orally administered to mice. Also, the immunogenicity of bacteria induces an innate immune response in vivo, which limits the maximum tolerated dose of bacteria. To tackle this problem, Harimoto et al. designed a programmable encapsulation system that allows the bacteria to escape from immune attack temporarily, which resulted in a 10-fold increase in the maximum tolerated dose of the bacteria (Fig. 8F).67 Capsular polysaccharides (CAPs) expressed on bacterial surfaces promote their survival and colonization by protecting bacteria from immune factors in the human body. When the CAP expression system was constructed to regulate the bacterial surface, bacterial immunogenicity and survival ability were controlled and their in situ trafficking capabilities were augmented. The system was regulated with an external inducer, which ensured safety and enhanced efficacy. When treated with CAP-modified EcN in vivo, translocation of bacteria to distal tumors was observed and survival of the bacteria was extended as well.
Using genetically engineered bacteria as drug delivery vehicles is still in its infancy, and most current studies are focused on the expression of therapeutic proteins or delivery of nucleic acid-based drugs, while delivery of small molecule therapeutic agents is rare. Given the abundance of microbially encoded small molecule natural product biosynthetic gene clusters, it is hoped that the direct delivery of small molecule compounds with engineered bacterial strains will eventually be possible, which should expand the function of genetically engineered bacteria for ISVs. Meanwhile, genetic modifications are expected to endow bacteria with stronger capabilities to target tumor tissue, reduce toxicity and exhibit drug-like properties.
4. Conclusion and perspectives
With their unique properties, bacteria-based platforms have demonstrated strong potential for cancer treatment through various engineering strategies. Firstly, living bacteria can be chemically engineered with diverse functional moieties. For example, liposomes, oncolytic viruses, and dopamine can be embedded on their surfaces.68–70 With the intrinsic tumor tropism of bacteria, these therapeutic agents can be carried into tumor tissues, and further elicit robust tumor ICD. Moreover, direct chemical editing of the bacterial surface endows bacteria with more therapeutic functions as an emerging strategy.71 Secondly, the bacteria-derived components can also be used as a nanoscale delivery platform. For example, OMVs preserved the bacterial characteristics like PAMPs on their surface, while reducing the possibility of infection. Thirdly, genetic engineering provides bacteria with more capabilities. For example, through genetic circuit programming, bacteria are equipped with new functions like quorum-lysis, drug production, and enhanced tropism into the TME. With the help of these strategies, bacteria-based platforms are endowed with extraordinary antitumor capacities. They can be employed as adjuvants or antigenic carriers for ISVs to reconstruct the TME and provide a relatively cell-friendly environment for CD8+ T cells to eliminate tumor cells.72In this review, we comprehensively summarized bacteria being leveraged as therapeutic in situ tumor vaccination for cancer treatments. Compared to other intratumoral immunotherapies including TLR agonist, glucan, cytokine, or nanoparticle-based formulations, bacteria as ‘living therapeutic platforms’ can proliferate in the hypoxic tumor regions and produce targeted-gene encoded therapeutic agents within tumor sites, which could improve the local concentration of drugs continuously for augmented therapeutic efficacy.73–75 In addition, bacteria comprise diverse PAMP molecules like polysaccharides and nucleic acids, which can activate a broad class of innate-sensing signaling pathways to sensitize the tumor microenvironment. However, direct intravenous injection of bacteria may induce sepsis and can cause death. The dosage and administration route should be taken into careful consideration. Besides, other living microorganisms like yeast can also exert immunomodulation functions within the tumor microenvironment with the help of the danger signals provided by β-glucan and chitin. In 2022, Xu et al. leveraged yeast cell wall-derived nanoparticles (YCW NPs) for cancer immunotherapy. YCW NPs can effectively remodel both the environment of the tumor microenvironment and tumor-draining lymph nodes for improved therapeutic efficacy against melanoma. This strategy inspires further exploration of existing microorganism-derived cellular components as effective immunomodulation agents for cancer immunotherapy.76
Although bacteria-based platforms have roused great interest among researchers, their clinical applications still face controversial arguments.20 The safety concerns for delivering living bacteria into human bodies is one of the greatest uncertainties for bacteria-based strategies. Chemically or genetically engineered live microorganisms can multiply within the internal environment and interact with other microbes in the human body or gain unexpected mutations, which may disrupt the human microbiomes.86 Besides, bacterial fractions are usually antigenic substances, which may trigger an uncertain degree of immune response. There might be excessive immune responses like bacteremia and cytokine storm that threaten patients’ lives and continuous infection for the immunocompromised cancer patients. To tackle these problems, we need to choose bacterial strains for engineering wisely, for example, some probiotics present in the human body including E. coli, L. lactis and some yeasts, are more suitable for bacteria-based strategies.87,88 Designing the bacteria to survive in specific tissues or organs through genetic modifications, or using nanotechnology to engineer inanimate bacterial components, could minimize these problems. Genetic modification can also endow bacterial therapeutics with limited lifespan and responsiveness towards tumor-associated molecules or exogenous signals, and further increase their safety. In addition, for the manufacturing process of engineered bacteria, quality control and production efficiency still need optimization to generate more stable and reliable products. In the past two decades, a few engineered bacteria have entered clinical trials for human (Table 1). Most of the strategies include attenuated bacterial strains with various genetic modifications, and the bacterial strains are mostly anaerobes. Though small in number, these trials demonstrated that engineered bacteria could be tolerated in the human body and stressed the importance of robust colonization of the bacteria. Clinical trials of bacteria-derived vesicles or chemically engineered bacteria for cancer treatment are still lacking. However, bacteria-derived vesicles have been extensively tested on humans for infectious diseases such as respiratory diseases and meningococcal diseases, and their safety and immunogenicity have been proven. Thus, their future applications for tumor vaccination are promising. On the other hand, very few chemically engineered bacteria have been administered on humans. The effect of chemical modification on the innate immune system needs further investigation.
| Bacteria strain | Condition | Phase | Treatment | Functionalization | Status & Ref. |
|---|---|---|---|---|---|
| S. Typhimurium VNP20009 | Melanoma or RCC | Phase I | i.v. infusion (1 dose) of the engineered strain | Attenuated by genetic deletion of the purI and msbB genes | Published77 |
| S. Typhimurium VNP2009 | Head and neck SCC or oesophageal adenocarcinoma | Phase I | i.t. injection (multiple cycle) with 5-FC administration | Genetically modified to express cytosine deaminase to converse 5-FC to 5-FU | Published78 |
| S. Typhimurium TAPET-CD | Unspecified solid tumors | Phase I | i.v. infusion with dose escalation | — | Completed79 |
| S. Typhimurium SalpIL2 | Liver metastases of solid tumors | Phase I | Oral administration with dose escalation | Genetically modified to express IL-2 | Completed80 |
| Listeria CRS-207 | Metastatic pancreatic cancer | Phase II | i.v. infusion combined with cyclophosphamide | Genetically modified to express mesothelin | Published81 |
| Streptococcus pyogenes, Serratia marcescens | NY-ESO-1 Expressing Cancer | Phase I | i.m. injection with dose escalation | — | Published82 |
| C. novyi-NT | Malignant solid tumors | Phase I | i.t. injection with dose escalation | — | Published83 |
| C. novyi-NT | Treatment-refractory Advanced solid tumors | Phase Ib | i.t. injection with anti-PD1 immunotherapy | — | Recruiting84 |
| B. longum APS001F | Advanced/metastatic solid tumors | Phase I/II | i.v. infusion with maltose and 5-FC | Genetically modified to express cytosine deaminase | Recruiting85 |
Nowadays, advances in chemical engineering, nanotechnology, and synthetic biology make it possible to obtain bacteria with the desired functions to fight more types of cancers. Some of these strategies have taken a further step to clinical evaluation.89 The adjuvanticity and tumor-targeting capability of bacteria make them attractive platforms for in situ tumor vaccination, overcoming the heterogenicity of different tumors. Moreover, the development of these technologies has facilitated the production of bacterial therapeutics. Chemistry approaches like click-chemistry and bioorthogonal chemistry provide neat paths to modify selected strains in an efficient and reliable manner.90 The developments in nanotechnology and microbiology provide more methods to assist the production and quality control of bacteria-derived vesicles, while other technologies could endow bacterial therapeutics with brand new properties.91,92 For instance, the application of biomineralization on biomaterials has provided an innovative route to endow bacteria with magnetotactic characteristics. As we are gaining a deeper understanding of the structure and genome of bacteria, future bacteria-based engineering strategies are oriented to safety, precision, and effectiveness. More complex and precise bacteria-based strategies will undoubtedly be developed for next-generation in situ tumor vaccination.
Conflicts of interest
Z.G. is the co-founder of Zenomics Inc., Zencapsule Inc., Lizen Inc., Wskin Inc., and ZCapsule Inc. and the other authors declare no conflict of interest.Acknowledgements
The authors would like to acknowledge the support from the National Key R&D Program of China (2021YFA0909900), the National Natural Science Foundation of China (52173142), and the grants from the Startup Package of Zhejiang University.References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2020, 70, 7–30 CrossRef PubMed.
- V. Schirrmacher, Int. J. Oncol., 2019, 54, 407–419 Search PubMed.
- L. Barazzuol, R. P. Coppes and P. Luijk, Mol. Oncol., 2020, 14, 1538–1554 CrossRef PubMed.
- D. N. Khalil, E. L. Smith, R. J. Brentjens and J. D. Wolchok, Nat. Rev. Clin. Oncol., 2016, 13, 273–290 CrossRef PubMed.
- A. D. Waldman, J. M. Fritz and M. J. Lenardo, Nat. Rev. Immunol., 2020, 20, 651–668 CrossRef PubMed.
- A. D. Fesnak, C. H. June and B. L. Levine, Nat. Rev. Cancer, 2016, 16, 566–581 CrossRef.
- S. A. Rosenberg, J. C. Yang and N. P. Restifo, Nat. Med., 2004, 10, 909–915 CrossRef PubMed.
- Z. Hu, P. A. Ott and C. J. Wu, Nat. Rev. Immunol., 2018, 18, 168–182 CrossRef.
- A. Marabelle, L. Tselikas, T. de Baere and R. Houot, Ann. Oncol., 2017, 28, xii33–xii43 CrossRef.
- A. D. Garg, L. Galluzzi, L. Apetoh, T. Baert, R. B. Birge, J. M. B.-S. Pedro, K. Breckpot, D. Brough, R. Chaurio, M. Cirone, A. Coosemans, P. G. Coulie, D. D. Ruysscher, L. Dini, P. de Witte, A. M. Dudek-Peric, A. Faggioni, J. Fucikova, U. S. Gaipl, J. Golab, M.-L. Gougeon, M. R. Hamblin, A. Hemminki, M. Herrmann, J. W. Hodge, O. Kepp, G. Kroemer, D. V. Krysko, W. G. Land, F. Madeo, A. A. Manfredi, S. R. Mattarollo, C. Maueroder, N. Merendino, G. Multhoff, T. Pabst, J.-E. Ricci, C. Riganti, E. Romano, N. Rufo, M. J. Smyth, J. Sonnemann, R. Spisek, J. Stagg, E. Vacchelli, P. Vandenabeele, L. Vandenberk, B. J. V. den Eynde, S. V. Gool, F. Velotti, L. Zitvogel and P. Agostinis, Front. Immunol., 2015, 6, 588 Search PubMed.
- L. Hammerich, T. U. Marron, R. Upadhyay, J. Svensson-Arvelund, M. Dhainaut, S. Hussein, Y. Zhan, D. Ostrowski, M. Yellin, H. Marsh, A. M. Salazar, A. H. Rahman, B. D. Brown, M. Merad and J. D. Brody, Nat. Med., 2019, 25, 814–824 CrossRef PubMed.
- L. Hammerich, A. Binder and J. D. Brody, Mol. Oncol., 2015, 9, 1966–1981 CrossRef.
- Y. Wang, J. Chen, R. Duan, R. Gu, W. Wang, J. Wu, H. Lian, Y. Hu and A. Yuan, Adv. Mater., 2022, 34, 2109726 CrossRef.
- Y. Wang, N. Gong, C. Ma, Y. Zhang, H. Tan, G. Qing, J. Zhang, Y. Wang, J. Wang, S. Chen, X. Li, Q. Ni, Y. Yuan, Y. Gan, J. Chen, F. Li, J. Zhang, C. Ou, Y. Zhao, X. Liu and X.-J. Liang, Nat. Commun., 2021, 12, 4964 CrossRef.
- C. Zhu, Z. Ji, J. Ma, Z. Ding, J. Shen and Q. Wang, Pharmaceutics, 2021, 13, 940 CrossRef PubMed.
- K. H. Gupta, C. Nowicki, E. F. Giurini, A. L. Marzo and A. Zloza, Vaccines, 2021, 9, 1497 CrossRef PubMed.
- S. Guallar-Garrido and E. Julián, ImmunoTargets Ther., 2020, 9, 1–11 CrossRef PubMed.
- N. S. Forbes, Nat. Rev. Cancer, 2010, 10, 785–794 CrossRef PubMed.
- B. Pulendran, P. S. Arunachalam and D. T. O'Hagan, Nat. Rev. Drug Discovery, 2021, 20, 454–475 CrossRef.
- X. Huang, J. Pan, F. Xu, B. Shao, Y. Wang, X. Guo and S. Zhou, Adv. Sci., 2021, 8, 2003572 CrossRef.
- C. Roma-Rodrigues, R. Mendes, P. Baptista and A. Fernandes, Int. J. Mol. Sci., 2019, 20, 840 CrossRef PubMed.
- W. He, Q. Wang, X. Tian and G. Pan, Exploration, 2022, 2, 20210093 CrossRef.
- K. Nakamura and M. J. Smyth, Cell. Mol. Immunol., 2020, 17, 1–12 CrossRef.
- V. Lagage and S. Uphoff, Nat. Rev. Microbiol., 2018, 16, 584–584 CrossRef.
- M. Zhao, J. Geller, H. Ma, M. Yang, S. Penman and R. M. Hoffman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10170–10174 CrossRef CAS.
- B. F.-L. Sieow, K. S. Wun, W. P. Yong, I. Y. Hwang and M. W. Chang, Trends Cancer, 2021, 7, 447–464 CrossRef CAS.
- T. Chien, T. Harimoto, B. Kepecs, K. Gray, C. Coker, N. Hou, K. Pu, T. Azad, A. Nolasco, M. Pavlicova and T. Danino, Nat. Biomed. Eng., 2022, 6, 94–104 CrossRef CAS PubMed.
- I. A. Hajam, P. A. Dar, I. Shahnawaz, J. C. Jaume and J. H. Lee, Exp. Mol. Med., 2017, 49, e373–e373 CrossRef CAS.
- H. Zhao, L. Wu, G. Yan, Y. Chen, M. Zhou, Y. Wu and Y. Li, Signal Transduction Targeted Ther., 2021, 6, 263 CrossRef CAS.
- A. Iwasaki and R. Medzhitov, Nat. Immunol., 2015, 16, 343–353 CrossRef PubMed.
- C. Covián, A. Fernández-Fierro, A. Retamal-Díaz, F. E. Díaz, A. E. Vasquez, M. K. Lay, C. A. Riedel, P. A. González, S. M. Bueno and A. M. Kalergis, Front. Immunol., 2019, 10, 2806 CrossRef.
- S. Zhou, C. Gravekamp, D. Bermudes and K. Liu, Nat. Rev. Cancer, 2018, 18, 727–743 CrossRef.
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef PubMed.
- W. Li, J. Yang, L. Luo, M. Jiang, B. Qin, H. Yin, C. Zhu, X. Yuan, J. Zhang, Z. Luo, Y. Du, Q. Li, Y. Lou, Y. Qiu and J. You, Nat. Commun., 2019, 10, 3349 CrossRef.
- X. Yi, H. Zhou, Y. Chao, S. Xiong, J. Zhong, Z. Chai, K. Yang and Z. Liu, Sci. Adv., 2022, 6(33), eaba3546 CrossRef.
- W. Chen, Y. Wang, M. Qin, X. Zhang, Z. Zhang, X. Sun and Z. Gu, ACS Nano, 2018, 12, 11 Search PubMed.
- L. Wang, Z. Cao, M. Zhang, S. Lin and J. Liu, Adv. Mater., 2022, 34, 2106669 CrossRef PubMed.
- E. B. Golden, S. Demaria, P. B. Schiff, A. Chachoua and S. C. Formenti, Cancer Immunol. Res., 2013, 1, 365–372 CrossRef PubMed.
- P. Pei, Y. Zhang, Y. Jiang, W. Shen, H. Chen, S. Yang, Y. Zhang, X. Yi and K. Yang, ACS Nano, 2022, 16, 11325–11337 CrossRef PubMed.
- W. Wang, H. Xu, Q. Ye, F. Tao, I. Wheeldon, A. Yuan, Y. Hu and J. Wu, Nat. Biomed. Eng., 2022, 6, 44–53 CrossRef PubMed.
- A. Redenti, J. Hahn and T. Danino, Nat. Biomed. Eng., 2022, 6, 3–5 CrossRef PubMed.
- F. Guo, J. K. Das, K. S. Kobayashi, Q.-M. Qin, T. A. Ficht, R. C. Alaniz, J. Song and P. D. Figueiredo, J. Immunother. Cancer, 2022, 10, e003760 CrossRef PubMed.
- Y. Guo, Z. Wang, X. Shi and M. Shen, Exploration, 2022, 2, 20210171 CrossRef.
- W. Ziqi, C. Kai, U. Costabel and Z. Xiaoju, Exploration, 2022, 00, 20210082 Search PubMed.
- Q. Long, P. Zheng, X. Zheng, W. Li, L. Hua, Z. Yang, W. Huang and Y. Ma, Adv. Drug Delivery Rev., 2022, 186, 114321 CrossRef CAS PubMed.
- M. Kaparakis-Liaskos and R. L. Ferrero, Nat. Rev. Immunol., 2015, 15, 375–387 CrossRef CAS PubMed.
- O. Y. Kim, H. T. Park, N. T. H. Dinh, S. J. Choi, J. Lee, J. H. Kim, S.-W. Lee and Y. S. Gho, Nat. Commun., 2017, 8, 626 CrossRef PubMed.
- Y. Li, R. Zhao, K. Cheng, K. Zhang, Y. Wang, Y. Zhang, Y. Li, G. Liu, J. Xu, J. Xu, G. J. Anderson, J. Shi, L. Ren, X. Zhao and G. Nie, ACS Nano, 2020, 14, 16698–16711 CrossRef PubMed.
- J. Hwang, E.-K. An, S.-J. Kim, W. Zhang and J.-O. Jin, ACS Nano, 2022, 16, 8472–8483 CrossRef.
- S. Qing, C. Lyu, L. Zhu, C. Pan, S. Wang, F. Li, J. Wang, H. Yue, X. Gao, R. Jia, W. Wei and G. Ma, Adv. Mater., 2020, 32, 2002085 CrossRef.
- L. Li, J. Zou, Y. Dai, W. Fan, G. Niu, Z. Yang and X. Chen, Nat. Biomed. Eng., 2020, 4, 1102–1116 CrossRef PubMed.
- R. B. Patel, M. Ye, P. M. Carlson, A. Jaquish, L. Zangl, B. Ma, Y. Wang, I. Arthur, R. Xie, R. J. Brown, X. Wang, R. Sriramaneni, K. Kim, S. Gong and Z. S. Morris, Adv. Mater., 2019, 31, 1902626 CrossRef PubMed.
- Y. Li, K. Zhang, Y. Wu, Y. Yue, K. Cheng, Q. Feng, X. Ma, J. Liang, N. Ma, G. Liu, G. Nie, L. Ren and X. Zhao, Small, 2022, 18, 2107461 CrossRef.
- L. Chen, H. Qin, R. Zhao, X. Zhao, L. Lin, Y. Chen, Y. Lin, Y. Li, Y. Qin, Y. Li, S. Liu, K. Cheng, H. Chen, J. Shi, G. J. Anderson, Y. Wu, Y. Zhao and G. Nie, Sci. Transl. Med., 2021, 13, eabc2816 CrossRef.
- K. Cheng, R. Zhao, Y. Li, Y. Qi, Y. Wang, Y. Zhang, H. Qin, Y. Qin, L. Chen, C. Li, J. Liang, Y. Li, J. Xu, X. Han, G. J. Anderson, J. Shi, L. Ren, X. Zhao and G. Nie, Nat. Commun., 2021, 12, 2041 CrossRef PubMed.
- X. Liu, M. Wu, M. Wang, Y. Duan, C. Phan, G. Qi, G. Tang and B. Liu, Mater. Horiz., 2021, 8, 1454–1460 RSC.
- Q. Chen, J. Wang, X. Wang, J. Fan, X. Liu, B. Li, Z. Han, S. Cheng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 21562–21570 CrossRef.
- M. O. Din, T. Danino, A. Prindle, M. Skalak, J. Selimkhanov, K. Allen, E. Julio, E. Atolia, L. S. Tsimring, S. N. Bhatia and J. Hasty, Nature, 2016, 536, 81–85 CrossRef PubMed.
- S. Chowdhury, S. Castro, C. Coker, T. E. Hinchliffe, N. Arpaia and T. Danino, Nat. Med., 2019, 25, 1057–1063 CrossRef PubMed.
- C. R. Gurbatri, I. Lia, R. Vincent, C. Coker, S. Castro, P. M. Treuting, T. E. Hinchliffe, N. Arpaia and T. Danino, Sci. Transl. Med., 2020, 12, eaax0876 CrossRef PubMed.
- H. Loessner, S. Leschner, A. Endmann, K. Westphal, K. Wolf, K. Kochruebe, T. Miloud, J. Altenbuchner and S. Weiss, Microbes Infect., 2009, 11, 1097–1105 CrossRef.
- J. Stritzker, S. Weibel, P. J. Hill, T. A. Oelschlaeger, W. Goebel and A. A. Szalay, Int. J. Med. Microbiol., 2007, 297, 151–162 CrossRef.
- Y. Zhang, Y. Zhang, L. Xia, X. Zhang, X. Ding, F. Yan and F. Wu, Appl. Environ. Microbiol., 2012, 78, 7603–7610 CrossRef.
- D. S. Leventhal, A. Sokolovska, N. Li, C. Plescia, S. A. Kolodziej, C. W. Gallant, R. Christmas, J.-R. Gao, M. J. James, A. Abin-Fuentes, M. Momin, C. Bergeron, A. Fisher, P. F. Miller, K. A. West and J. M. Lora, Nat. Commun., 2020, 11, 2739 CrossRef PubMed.
- R. Geiger, J. C. Rieckmann, T. Wolf, C. Basso, Y. Feng, T. Fuhrer, M. Kogadeeva, P. Picotti, F. Meissner, M. Mann, N. Zamboni, F. Sallusto and A. Lanzavecchia, Cell, 2016, 167, 829–842 CrossRef.
- F. P. Canale, C. Basso, G. Antonini, M. Perotti, N. Li, A. Sokolovska, J. Neumann, M. J. James, S. Geiger, W. Jin, J.-P. Theurillat, K. A. West, D. S. Leventhal, J. M. Lora, F. Sallusto and R. Geiger, Nature, 2021, 598, 662–666 CrossRef PubMed.
- T. Harimoto, J. Hahn, Y.-Y. Chen, J. Im, J. Zhang, N. Hou, F. Li, C. Coker, K. Gray, N. Harr, S. Chowdhury, K. Pu, C. Nimura, N. Arpaia, K. W. Leong and T. Danino, Nat. Biotechnol., 2022, 40, 1259–1269 CrossRef.
- D. Akin, J. Sturgis, K. Ragheb, D. Sherman, K. Burkholder, J. P. Robinson, A. K. Bhunia, S. Mohammed and R. Bashir, Nat. Nanotechnol., 2007, 2, 441–449 CrossRef PubMed.
- B. Wei, J. Pan, R. Yuan, B. Shao, Y. Wang, X. Guo and S. Zhou, Nano Lett., 2021, 21, 4231–4240 CrossRef.
- M. Sun, S. Yang, H. Huang, P. Gao, S. Pan, Z. Cheng, Z. He, Z. Wang, J. Sun and F. Liu, Nano Lett., 2022, 22, 5055–5064 CrossRef.
- H. Jia, Y. Zhu, Y. Liu, Y. Guo, S. M. Sayed, X. Zhu, X. Cheng and F. Wu, Exploration, 2022, 20220010 CrossRef.
- Q. Chen, Q. Hu, E. Dukhovlinova, G. Chen, S. Ahn, C. Wang, E. A. Ogunnaike, F. S. Ligler, G. Dotti and Z. Gu, Adv. Mater., 2019, 31, 1900192 CrossRef.
- D. Liu, B. Deng, Z. Liu, B. Ma, X. Leng, D. Kong, T. Ji and L. Liu, Nano Lett., 2021, 21, 3965–3973 CrossRef PubMed.
- Y. Liu, M. Li, H. Zhu, Z. Jing, X. Yin, K. Wang, Z. Hong and W. Zhao, Chin. Chem. Lett., 2021, 32, 1963–1966 CrossRef.
- T. Ji, Y. Li, X. Deng, A. Y. Rwei, A. Offen, S. Hall, W. Zhang, C. Zhao, M. Mehta and D. S. Kohane, Nat. Biomed. Eng., 2021, 5, 1099–1109 CrossRef.
- J. Xu, Q. Ma, Y. Zhang, Z. Fei, Y. Sun, Q. Fan, B. Liu, J. Bai, Y. Yu, J. Chu, J. Chen and C. Wang, Nat. Commun., 2022, 13, 110 CrossRef PubMed.
- J. F. Toso, V. J. Gill, P. Hwu, F. M. Marincola, N. P. Restifo, D. J. Schwartzentruber, R. M. Sherry, S. L. Topalian, J. C. Yang, F. Stock, L. J. Freezer, K. E. Morton, C. Seipp, L. Haworth, S. Mavroukakis, D. White, S. MacDonald, J. Mao, M. Sznol and S. A. Rosenberg, J. Clin. Oncol., 2002, 20, 142–152 CrossRef.
- J. Nemunaitis, C. Cunningham, N. Senzer, J. Kuhn, J. Cramm, C. Litz, R. Cavagnolo, A. Cahill, C. Clairmont and M. Sznol, Cancer Gene Ther., 2003, 10, 737–744 CrossRef PubMed.
- US National Library of Medicine, ClinicalTrials.gov, https://www.clinicaltrials.gov/ct2/show/NCT00006254.
- US National Library of Medicine, ClinicalTrials.gov, https://www.clinicaltrials.gov/ct2/show/NCT01099631.
- D. T. Le, A. Wang-Gillam, V. Picozzi, T. F. Greten, T. Crocenzi, G. Springett, M. Morse, H. Zeh, D. Cohen, R. L. Fine, B. Onners, J. N. Uram, D. A. Laheru, E. R. Lutz, S. Solt, A. L. Murphy, J. Skoble, E. Lemmens, J. Grous, T. Dubensky, D. G. Brockstedt and E. M. Jaffee, J. Clin. Oncol., 2015, 33, 1325–1333 CrossRef.
- J. Karbach, A. Neumann, K. Brand, C. Wahle, E. Siegel, M. Maeurer, E. Ritter, T. Tsuji, S. Gnjatic, L. J. Old, G. Ritter and E. Jäger, Clin. Cancer Res., 2012, 18, 5449–5459 CrossRef PubMed.
- F. Janku, H. H. Zhang, A. Pezeshki, S. Goel, R. Murthy, A. Wang-Gillam, D. R. Shepard, T. Helgason, T. Masters, D. S. Hong, S. A. Piha-Paul, D. D. Karp, M. Klang, S. Y. Huang, D. Sakamuri, A. Raina, J. Torrisi, S. B. Solomon, A. Weissfeld, E. Trevino, G. DeCrescenzo, A. Collins, M. Miller, J. L. Salstrom, R. L. Korn, L. Zhang, S. Saha, A. A. Leontovich, D. Tung, B. Kreider, M. Varterasian, K. Khazaie and M. M. Gounder, Clin. Cancer Res., 2021, 27, 96–106 CrossRef PubMed.
- US National Library of Medicine, ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03435952.
- US National Library of Medicine, ClinicalTrials.gov, https://www.clinicaltrials.gov/ct2/show/NCT01562626.
- U. Wegmann, A. L. Carvalho, M. Stocks and S. R. Carding, Sci. Rep., 2017, 7, 2294 CrossRef.
- Y. Zhou and Y. Han, Eng. Microbiol., 2022, 2, 100034 CrossRef CAS.
- N. Qiao, G. Du, X. Zhong and X. Sun, Exploration, 2021, 1, 20210026 CrossRef.
- Y. Li, Y. Wang, X. Li and C. Liu, Natl. Sci. Rev., 2021, 8, nwab023 CrossRef PubMed.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef.
- M. G. Sartorio, E. J. Pardue, M. F. Feldman and M. F. Haurat, Annu. Rev. Microbiol., 2021, 75, 609–630 CrossRef PubMed.
- L. van der Pol, M. Stork and P. van der Ley, Biotechnol. J., 2015, 10, 1689–1706 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |