
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mucoadhesive chitosan–methylcellulose oral patches for the treatment of local mouth bacterial infections†
Lorenzo
Bonetti
 *a,
Alice
Caprioglio
a,
Nina
Bono
ab,
Gabriele
Candiani
ab and
Lina
Altomare
*ab
*a,
Alice
Caprioglio
a,
Nina
Bono
ab,
Gabriele
Candiani
ab and
Lina
Altomare
*ab
aDepartment of Chemistry, Materials and Chemical Engineering “G. Natta”, Politecnico di Milano, Piazza Leonardo da Vinci 32, 20133, Milan, Italy. E-mail: lorenzo.bonetti@polimi.it
bNational Interuniversity Consortium of Materials Science and Technology (INSTM), Via Giuseppe Giusti 9, 50121 Florence, Italy. E-mail: lina.altomare@polimi.it
First published on 25th January 2023
Abstract
Mucoadhesive buccal patches are dosage forms promising for successful drug delivery. They show the distinctive advantages of long residence time on the oral mucosa and increased in situ drug bioavailability. In this context, electrophoretic deposition (EPD) of chitosan (CS) has been demonstrated as a simple and easily tunable technique to produce mucoadhesive buccal patches. However, CS-based buccal patches may suffer from weak mucoadhesion, which can impair their therapeutic effect. In this work, methylcellulose (MC), a widely investigated biopolymer in the biomedical area, was exploited to increase the mucoadhesive characteristic of pristine CS patches. CS–MC patches were obtained in a one-pot process via EPD, and the possibility of incorporating gentamicin sulfate (GS) as a model of a broad-spectrum antibiotic in the so-obtained patches was investigated. The resulting CS–MC patches showed high stability in a water environment and superior mucoadhesive characteristic (σadh = 0.85 ± 0.26 kPa, Wadh = 1192.28 ± 602.36 Pa mm) when compared with the CS control samples (σadh = 0.42 ± 0.22 kPa, Wadh = 343.13 ± 268.89 Pa mm), due to both the control of the patch porosity and the bioadhesive nature of MC. Furthermore, GS-loaded patches showed no in vitro cytotoxic effects by challenging L929 cells with material extracts and noteworthy antibacterial activity on both Gram-positive and Gram-negative bacterial strains.
1. Introduction
The oral cavity is a complex ecosystem hosting a diverse microflora made up of over 700 recognized bacterial species.1 Under physiological conditions, the oral microflora lives in homeostasis; unfortunately, imbalance or colonization by other microorganisms (viruses, fungi, or bacteria) can lead to infections and diseases of the oral cavity,2 which can affect any structure of the oral mucosa (i.e., masticatory, lining, and specialized mucosa).3 In this regard, smoking, consumption of alcohol, poor oral hygiene, and trauma are the major risk factors leading to primary bacterial infections. Besides, immunocompromised individuals, such as HIV, cancer, and transplant patients, or those treated with corticosteroids are even more susceptible to complications.4Systemic antibiotic therapy is generally prescribed to treat and eradicate oral infections.5 Local administration of antibiotics may represent an advantage in the management of oral infections as compared to the systemic route. Local administration allows for a high antibiotic dose at the treated site but low levels elsewhere, thus resulting in improving patient compliance and drug side effects. Moreover, bypassing the gastrointestinal tract (i.e., avoiding both acid hydrolysis and hepatic first-pass metabolism) leads to increased drug bioavailability.6
The clinical treatment of oral infections usually relies on conventional pharmaceutical dosage forms, such as solutions, gels, suspensions, and mouthwashes.7,8 However, such treatments are often suboptimal since they are easily washed away by the salivary flow and mechanical stresses. This leads to limited exposure times, unpredictable drug distribution, and threatening drug resistance phenomena.9 Overall, the main challenge for an optimal local administration of antibiotics is maintaining a high concentration within a well-defined area for a sustained period.6 In this regard, the design of drug delivery systems with appropriate mucoadhesive activity can ensure prolonged retention in the oral cavity and enhanced drug availability in situ. Various classes of polymers have thus been investigated to meet the requirements for mucoadhesion;10 cellulose derivatives (e.g., sodium carboxymethylcellulose, methylcellulose (MC) and hydroxypropyl methylcellulose), polyacrylic acids, polyvinylpyrrolidone, polyvinyl alcohol, alginates, hyaluronic acid, and chitosan (CS) are among the most widely investigated in this field.11
To date, a wide range of polymeric drug delivery systems for oral applications have been reported in the literature, including but not limited to tablets,12 gels,13,14 patches,15,16 and films.17,18 In this context, oral films and patches may be preferred over other adhesive systems as they are flexible and soft, yet strong enough and with relevant mucoadhesive characteristic to withstand the stresses within the oral cavity.8 Mucoadhesive films/patches are usually designed to be applied twice a day and their in vivo residence time has been reported to vary from less than 1 hour up to 5 hours, depending on the patch type and the position in the oral cavity (e.g., buccal mucosa, gingivae, and tongue).9 Different mucoadhesive films and patches have been reported in the literature to treat local mouth bacterial infections, ranging from fluconazole-loaded mucoadhesive films prepared through the casting method,19 to multilayered electrospun patches loaded with ciprofloxacin hydrochloride.7
Among other possible techniques investigated to obtain mucoadhesive patches, electrophoretic deposition (EPD) is one of the most straightforward and easily tunable techniques. EPD involves processing charged species (e.g., polymers, particles, or molecules), exploiting their motion under the effect of an applied electric field and subsequent coagulation and precipitation on the electrode surface. Notably, EPD allows room temperature (r.t.) processing, useful to deposit sensitive (bio)materials and drugs.20,21 In this regard, EPD has been previously exploited to produce CS patches for buccal drug delivery,22 but the studies related to EPD processing of CS and antibiotics are mainly limited to obtaining coatings for orthopedic application.23–25 Moreover, even if CS has been extensively reported in the literature for obtaining mucoadhesive systems,26 it may suffer from insufficient mucoadhesive behavior. This is particularly true at physiological (i.e., neutral) pH, where CS exhibits limited mucoadhesive strength and poor water solubility, the two major drawbacks of its use.27 Different strategies have been reported in the literature to tackle such limitations, ranging from blending with other polymers26,28 to CS chemical modification/derivatization.27 CS blending with adhesive polymers represents an easy and effective strategy to improve mucoadhesion. CS has been reported to form polymeric matrices with non-ionic polymers (including cellulose derivatives), increasing the blend swelling and adhesion properties.28 In this regard, MC, due to its adhesive nature,29,30 could take up the challenge of increasing the mucoadhesive characteristic of CS. Nevertheless, to the best of our knowledge, MC has never been explored in EPD processing.
In this work, the EPD technique was exploited to obtain CS–MC buccal patches with enhanced adhesion strength compared with pristine CS. Patches were characterized from a physicochemical point of view (FT-IR, TGA, SEM and swelling/degradation) to investigate their composition, structure, and performance in a physiological-like environment. The mechanical and mucoadhesive properties of CS–MC patches were evaluated as well. The possibility of loading gentamicin sulfate (GS) in the blend to obtain CS–MC/GS patches via a one-pot EPD production process was also investigated. Patches were then in vitro tested to assess their possible cytotoxic effects (on L929 cells) and their antibacterial properties (against S. aureus and E. coli).
2. Experimental section
2.1. Chemicals
Unless stated, all reagents were purchased from Merck (Merck Life Science S.r.l., Italy) and used without further purification.2.2. Sample preparation
| Samples | Deposition time (min) | Time up (s) | Time down (s) | V up (V) | V down (V) |
|---|---|---|---|---|---|
| CS | 30 | 15 | 15 | 100 | 0 |
| CS–MC | 5 | 15 | 15 | 50 | 30 |
| CS–MC/GS | 5 | 15 | 15 | 50 | 30 |
2.3. Sample characterization
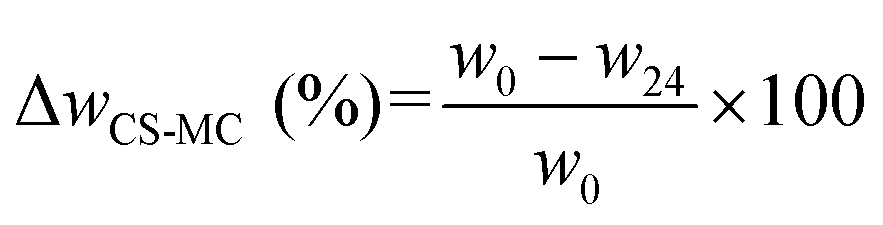 | (1) |
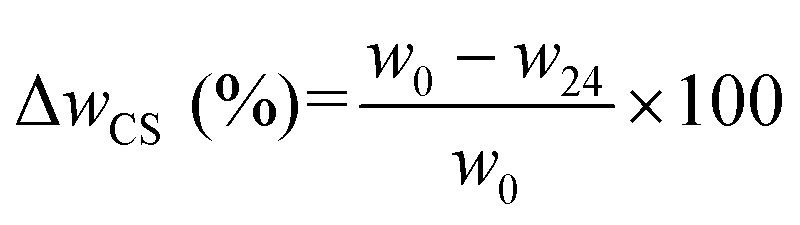 | (2) |
| MC (%) = ΔwCS–MC (%) − ΔwCS (%) | (3) |
For the swelling tests, the samples were fully soaked in artificial saliva (pH = 6.8, ESI 1†). The samples were incubated at 37 °C up to 24 h, and the swelling ratio (SW) was calculated using the following equation (eqn (4)):
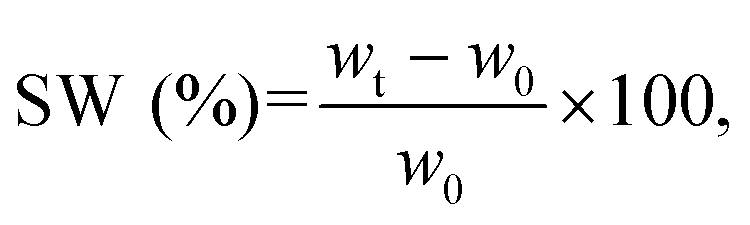 | (4) |
Degradation tests were performed by soaking the samples in artificial saliva at 37 °C up to 24 h. The gel fraction (GF) of the samples was calculated as follows (eqn (5)):
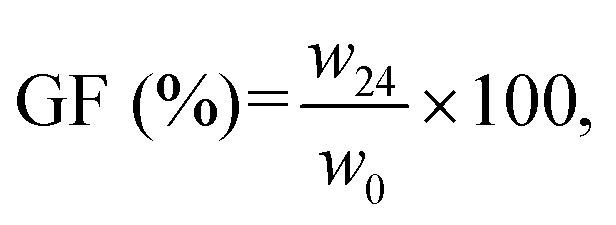 | (5) |
2.3.6.1. Tensile tests. Uniaxial tensile tests (n = 6 per sample type) were performed on 2 × 5 cm2 samples using a dynamic mechanical analyzer (MCR 702, Anton Paar, Austria) at a constant strain rate (
![[small epsi, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a1.gif) ) of 0.2 min−1, for 30 s (preload = 0.01 N). Young's modulus (E), elongation at break (εb), and stress at break (σb) were obtained from the stress/strain curves. The E modulus was determined from the slope of the initial linear portion of the stress/strain curves (ε range of 0–5%). All tests were conducted at r.t.
) of 0.2 min−1, for 30 s (preload = 0.01 N). Young's modulus (E), elongation at break (εb), and stress at break (σb) were obtained from the stress/strain curves. The E modulus was determined from the slope of the initial linear portion of the stress/strain curves (ε range of 0–5%). All tests were conducted at r.t.
2.3.6.2. Ex vivo tack tests. The mucoadhesive properties of the samples were tested in accordance with the ASTM F2258 standard with a modular compact rheometer (MCR 302, Anton Paar). The samples (1 × 1 cm2; n = 5 specimens per sample type) were fixed on the upper disposable plate of the instrument (ø = 25 mm), while the porcine buccal mucosa pieces (1 × 1 cm2) were attached to the lower disposable plate of the device. A compressive preload of 1 N was applied for 10 s to ensure the adhesion between the sample and the mucosa, and the upper plate was then moved upwards at a constant speed of 50 mm min−1.37 All tests were carried out at r.t.
The release pressure (σadh) was calculated as follows (eqn (6)):38
 | (6) |
The mucoadhesion work (Wadh) was calculated according to eqn (7):38
| Wadh = AUC, | (7) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500 in 0.1 M NaHCO3), and incubated at 37 °C for 2 h. The absorbance was measured at λ = 364 nm using a Synergy H1 spectrophotometer (BioTek, Italy), and the GS concentration was obtained from the standard calibration curve (R2 ≥0.99) after blank (fresh PBS mixed with the TNBSA solution) subtraction (ESI 2†).
:500 in 0.1 M NaHCO3), and incubated at 37 °C for 2 h. The absorbance was measured at λ = 364 nm using a Synergy H1 spectrophotometer (BioTek, Italy), and the GS concentration was obtained from the standard calibration curve (R2 ≥0.99) after blank (fresh PBS mixed with the TNBSA solution) subtraction (ESI 2†).
The GS loading efficiency was calculated as follows (eqn (8)):
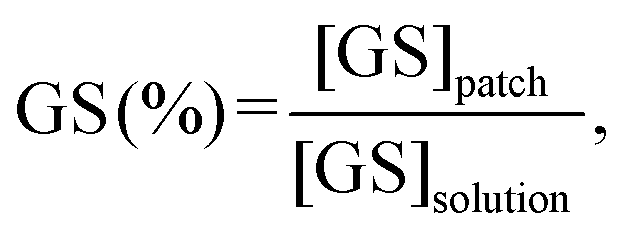 | (8) |
2.3.9.1. Sample preparation. The CS, CS–MC, and CS–MC/GS patches were sanitized via UV irradiation (5 min per side) before the indirect cytotoxicity and indirect antibacterial tests.
For the biological tests (indirect cytotoxicity and antibacterial tests), sample extracts were prepared according to the ISO standard (ISO 10993-12:2012, “Biological evaluation of medical devices – Part 12: Sample preparation and reference materials”). Briefly, material extracts were obtained by placing sterile samples of each experimental group in separated 2 mL Eppendorf tubes, in contact with the extraction vehicle: culture medium for indirect cytotoxicity tests (150 μL + specimen swelling volume); PBS for indirect antibacterial tests (extraction ratio = 3 mg specimen:1 mL PBS). Samples were then incubated at 37 °C for different time periods (3, 6, and 24 h). At each time step, material extracts (eluates) were harvested and stored at −20 °C until use.
2.3.9.2. Indirect cytotoxicity tests. Indirect cytotoxicity tests were performed following the ISO 10993-5:2009 standard. Dulbecco's modified Eagle's medium (DMEM), supplemented with 1 mM sodium pyruvate, 10 mM HEPES buffer, 100 U mL−1 penicillin, 0.1 mg mL−1 streptomycin, 2 mM glutamine, and 10% (v/v) fetal bovine serum (FBS) (hereafter referred to as culture medium), was used as the extraction medium.
L929 (murine fibroblasts from the subcutaneous connective tissue, CCL-1) cell line was purchased from the American Type Culture Collection (ATCC®, Manassas, VA, USA). The L929 cells were seeded in a 96-well culture plate (104 cells per well in 100 μL of the culture medium) and incubated under the standard culture conditions for 24 h. Afterward, the medium was replaced with 100 μL per well of eluates (n = 3 wells per material sample per time point), and the plate was incubated for further 24 h under the standard culture conditions. Cells grown in the culture medium were used as the control (CTRL, n = 3).
Cell viability was assessed using the resazurin assay (Sigma-Aldrich). Fluorescence (λex = 540 nm; λem = 595 nm) was measured using a Synergy H1 spectrophotometer. For each well, cell viability was calculated as follows (eqn (9)):
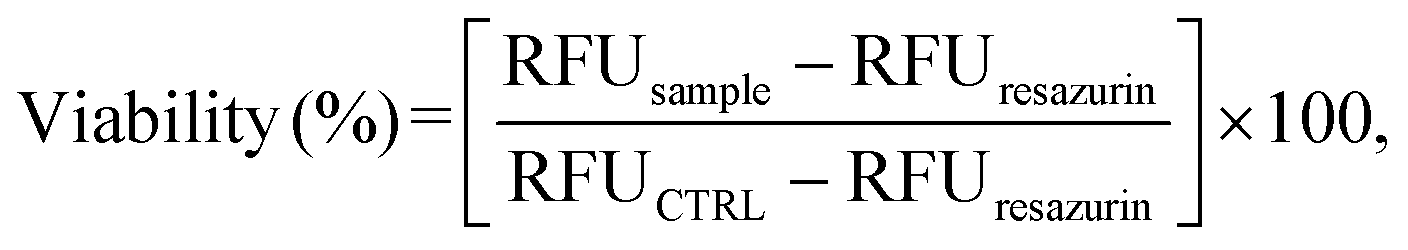 | (9) |
2.3.9.3. Indirect antibacterial tests. Indirect antibacterial tests were performed according to the literature.31Escherichia coli JM109 (E. coli, Gram-negative bacteria, Leibniz Institute DSMZ, German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) and Staphylococcus aureus (S. aureus, Gram-positive bacteria, ATCC 9341, ATCC) were pre-cultured ON in 5 mL of Luria–Bertani (LB) broth and nutrient broth (NB), respectively, at 37 °C under shaking at 130 rpm, until an optical density (OD600nm) of about 1 was reached, corresponding to ≈109 bacteria per mL. The bacterial suspensions were then diluted to a concentration of ∼106 bacteria per mL. Afterward, the bacterial suspensions (50 μL per well) were transferred into 96-well plates at a density of 1.5 × 105 bacteria per cm2 and inoculated with 50 μL of the eluate per well (n = 5 wells per material sample per time point). The plates were then incubated at 37 °C for 24 h. The bacteria inoculated in 100 μL per well of LB were used as a positive control for bacterial growth (CTRL+), while the bacteria inoculated in 50 μL per well of the GS solutions (range: 0.03–256 μg mL−1) were used as internal references. In particular, for the antibiotic, the minimal inhibitory concentration (MIC) was the lowest concentration of GS that prevents the visible growth of bacteria within a 24 h-incubation period.39
The antibacterial efficacy of each eluate was evaluated using the turbidity method (i.e., OD600nm measurements). Briefly, 24 h post-inoculum, the OD600 (nm) of each well (n = 5 per eluate) was read utilizing a GENios Plus reader (Tecan Group Ltd., Switzerland). The antibacterial efficiency was calculated according to eqn (10):
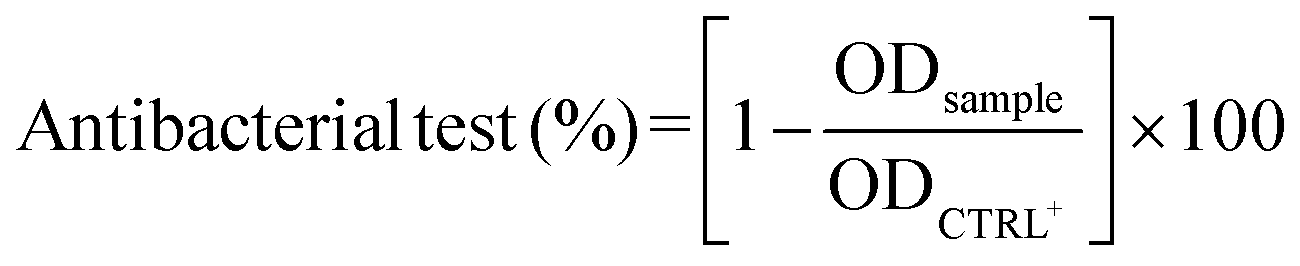 | (10) |
2.4. Statistical analysis
Unless stated, the tests were run in triplicate (n = 3). Data are expressed as mean ± standard deviation (SD). One-way ANOVA with Tukey's post hoc tests were used to compare the data, considering a significance level of α = 0.05.3. Results and discussion
In this work, CS and CS–MC patches were obtained via EPD by selecting the optimal processing parameters: the electrolyte composition, the applied voltage, the deposition time, and the duty cycle. The optimal set of parameters (Table 1) was identified based on previous works from our research group dealing with the production of self-standing scaffolds via EPD, starting from CS32 and CS–gelatin blends.31 The final purpose of this work was to obtain patches that were thick enough to be peeled off from the cathode surface and with the desired porosity. Interestingly, we report here for the first time the co-deposition of a CS–MC blend via EPD, and we further demonstrate the possibility of a one-pot deposition of the GS-loaded CS–MC patches.3.1. Structural analysis: morphology
The morphology of the lyophilized CS and CS–MC samples was examined through SEM. All specimens appeared homogeneous, with random porosity created by the evolution of the H2 bubbles.31,40,41 The difference in the surface morphology between the CS and the CS–MC samples was apparent and can be seen in Fig. 1. The porosity of the patches qualitatively increased in the CS–MC samples compared with the CS ones due to both the EPD processing conditions (i.e., 50–30 V vs. 100–0 V square waveform) and the addition of MC. In this regard, it has been widely reported in the literature that controlling the EPD parameters can contribute to obtaining scaffolds with different porosities.21,42 Moreover, it has also been reported how the addition of MC can lead to an increase in the porosity of MC-containing blends.43,44 Similar observations have also been reported when other cellulose derivatives (e.g., hydroxypropylmethylcellulose)45 were added to CS. Overall, the increase in porosity, coupled with the noteworthy adhesive properties of MC,29 can endow the CS–MC patches with a significant advantage for buccal applications, thanks to the possibility of increasing both the patch contact time and in situ drug bioavailability.46–49 | ||
| Fig. 1 SEM micrographs of the (a and c) CS and (b and d) CS–MC samples. (a and b) Scale bar = 200 μm; (c and d) scale bar = 20 μm. | ||
3.2. Structural analysis: FT-IR
The spectra of the samples (ESI 3†) were divided into three regions of interest, i.e., 850–1235 cm−1 (Fig. 2a), 1235–1850 cm−1 (Fig. 2b), and 1850–4000 cm−1 (Fig. 2c), to highlight the representative functional groups of the polymers. The peaks of all spectra were assigned to the appropriate vibrational motions, characteristic of the functional groups of CS and MC, in agreement with the literature. For the CS–MC samples, CS presence was confirmed by the peak of amides at 1569 cm−1.50–55 The presence of MC in the samples was confirmed by the appearance of two peaks representing the C–O–C stretching of the glucose ring (1117 cm−1) and the asymmetric C–H bending of CH3 of MC (1458 cm−1).51,56–59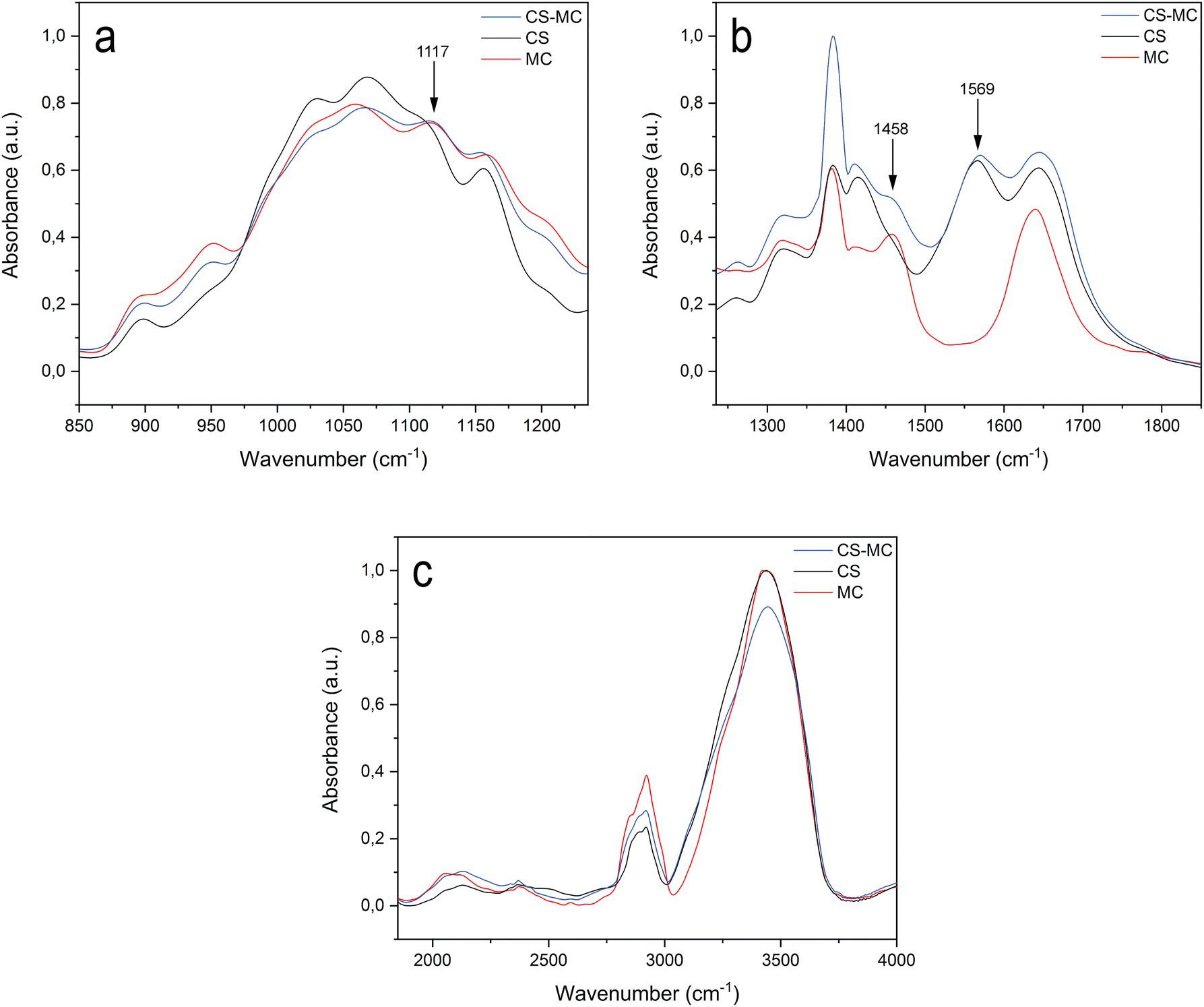 | ||
| Fig. 2 FT-IR spectra of the CS, MC, and CS–MC samples. The spectra were divided into three regions of interest. (a) 850–1235 cm−1, (b) 1235–1850 cm−1, and (c) 1850–4000 cm−1. | ||
Beyond these distinctive peaks, the spectrum of the CS–MC sample was superimposable with both the spectra of CS and MC, presenting small or no shifts in the position of their characteristic bands. Such observations suggest that a low interaction occurred between CS and MC.54 Furthermore, the CS–MC spectrum did not show any additional absorption peaks compared to pristine CS and MC, suggesting that no covalent bonds occurred between the polymers, but rather weak interactions (e.g., hydrogen bond and hydrophobic interactions) took place.54,55,60
The co-deposition via EPD of CS with other polymers has been reported in the literature. For instance, the CS–gelatin blend has been deposited through the formation of polyelectrolyte complexes in solution due to an electrostatic interaction between the –NH3+ moieties of chitosan and the –COO− groups of gelatin.21 Similar to MC, uncharged polyvinyl alcohol (PVA)61 has been co-deposited with CS, possibly exploiting the formation of weak interactions among the blended polymers. In light of the FT-IR analysis, the deposition mechanism of the CS–MC blend can also be explained by the establishment of weak interactions between the two polymers.54 CS, dragging MC toward the cathode, resulted in physically trapping MC in the insoluble deposit formed on the electrode surface due to a local increase in the pH.
The CS–MC/GS samples were also analyzed via FT-IR spectroscopy (data not shown). As expected, the CS–MC/GS spectrum revealed no differences compared to the CS–MC samples, due to both the low GS:CS–MC weight ratio and the overlapping of the N–H and O–H bands of GS and CS.23
3.3. Sample composition: TGA and the CS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :MC weight ratio
:MC weight ratio
TGA was carried out to preliminarily investigate the composition of the samples and assess their thermal stability. The TGA curves of all the sample types showed a two-stage weight loss (Fig. 3). The initial weight reduction occurred due to water evaporation, whereas the second reduction was due to the degradation of the polymer backbone.
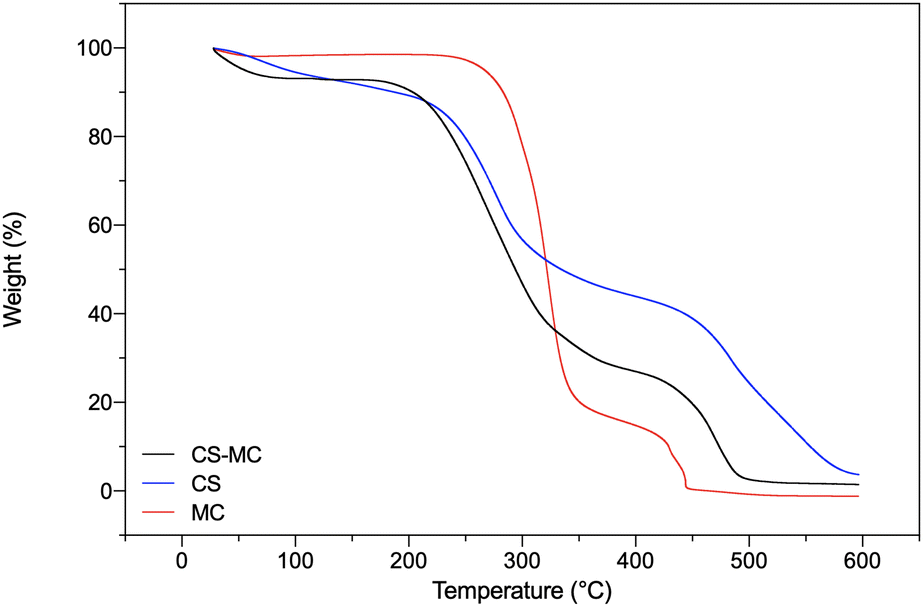 | ||
| Fig. 3 TGA curves of the CS, MC, and CS–MC samples. | ||
The CS samples showed a first weight loss stage of ∼17% at 113 °C, attributable to the water loss from the polar moieties of CS molecules.62–64 A second thermal event, starting at 195 °C, was related to the depolymerization of CS chains.62 The MC samples instead showed a first weight loss of ∼7% at 100 °C, due to the evaporation of unbound water, followed by a second thermal event at about 300 °C related to MC degradation, involving the two parallel processes of dehydration and demethoxylation.65,66 The CS–MC samples showed a thermal behavior between the two pristine polymers, with faster polymeric chain decomposition up to 400 °C compared to pristine CS, indicating the presence of MC in the electrodeposited patches.
Furthermore, the residues of the CS, MC, and CS–MC samples at 400 °C were 43.91%, 14.75%, and 27.00%, respectively, suggesting that the presence of MC in the CS–MC samples resulted in lower thermal stability at elevated temperatures (Table 2) in agreement with the findings of Khan et al.55
| Sample type | T 50 (°C) | Residue, 400 °C (%) | Residue, 600 °C (%) |
|---|---|---|---|
| CS | 272.91 | 43.91 | 3.71 |
| CS–MC | 273.50 | 27.00 | 1.45 |
| MC | 317.74 | 14.75 | 0.00 |
The exact composition of the CS–MC patches (i.e., the wt% of CS and MC in the patches) was evaluated by selective MC dissolution. In fact, MC undergoes fast dissolution in water at low temperatures (i.e., at T ≪ transition temperature (Tt) of MC).34 In this regard, 4 °C is usually the temperature selected to achieve complete hydration of the MC powder in the water solution.34,67
Gravimetric analysis revealed a predominant presence of CS in the patches, with a mean CS fraction of 66.8%. The chemical composition was further confirmed using a ninhydrin assay carried out on both CS and CS–MC specimens (ESI 4†).
3.4. Swelling and degradation behaviors
Fig. 4 shows the swelling rate (SW) values over time of the CS and CS–MC samples soaked in artificial saliva (pH = 6.8) at 37 °C.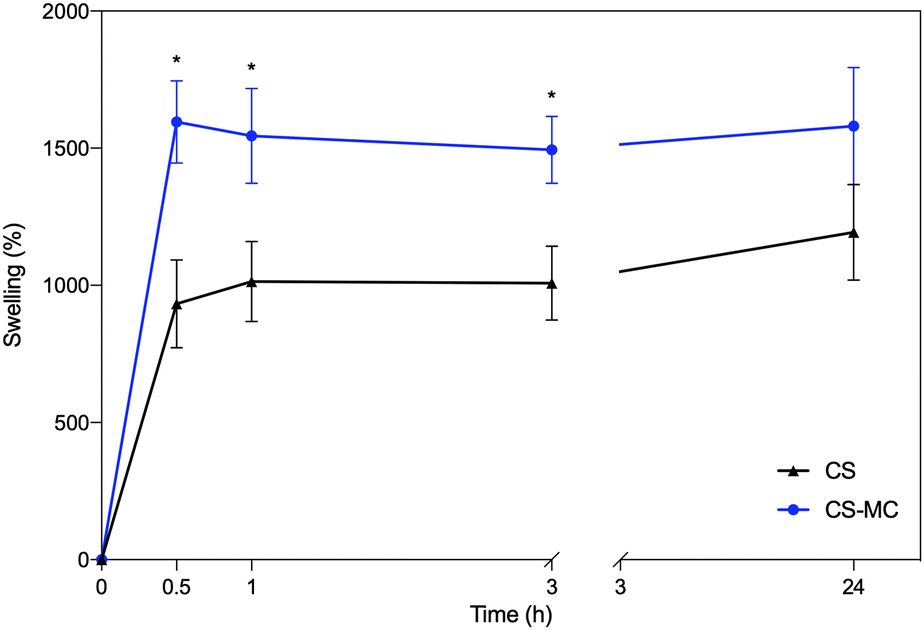 | ||
| Fig. 4 The swelling rate over time for the CS and CS–MC samples in artificial saliva (T = 37 °C) (*p < 0.05). | ||
From these plots, it is possible to notice that the CS–MC samples showed higher SW values compared to CS. This behavior can be attributed to the hydrophilic nature of MC at the test temperature (37 °C; i.e., T < Tt).34 In this regard, it has been widely reported that different factors (i.e., physical and chemical interactions) can contribute to mucoadhesion.68 Hydrophilic polymers, undergoing significant swelling, can contribute to the partial dehydration of the mucosal surface at the contact area, eventually resulting in strong mucoadhesiveness.68 This behavior has been reported when hydrophilic polymers were added to CS.69
GF values of 92.87 ± 4.98 and 91.76 ± 0.63 were obtained after a 24 h soaking period in artificial saliva for the CS and CS–MC samples, respectively. The GF values obtained are compatible with a minor dissolution of CS and MC within the timeframe of the test due to water penetration into the polymeric matrix and the consequent destruction of weak bonds (e.g., hydrogen and van der Waals bonds). No significant differences (p > 0.05) in the GF values between the CS and the CS–MC samples were found, indicating that the addition of the MC did not affect the water stability of the blend, highlighting their potential for application as oral patches.
3.5. Mechanical performance
Uniaxial tensile tests were performed on the swollen patches of CS and CS–MC soaked in artificial saliva (30 min, T = 37 °C) before testing. A swelling study at short time points in artificial saliva was performed to ascertain whether the patches were at the swelling equilibrium (data not shown). Elongation at break εb (%), stress at break σb (MPa), and Young's modulus (E) obtained from the uniaxial tensile tests are presented in Table 3.| Sample type | ε b (%) | σ b (MPa) | E (MPa) |
|---|---|---|---|
| CS | 49.38 ± 6.13 | 0.11 ± 0.04* | 0.16 ± 0.05# |
| CS–MC | 35.77 ± 7.06 | 0.01 ± 0.01* | 0.04 ± 0.01# |
CS samples obtained here exhibited mechanical performances in line with or lower than the porous CS samples reported in the literature.22,70 However, these differences in the mechanical properties can be ascribed to the differences in the CS source, CS solutions, and the selected EPD parameters, which determine the structure and porosity of the patches.
The CS–MC samples showed a significant reduction in their mechanical properties compared to the control samples (i.e., CS), as σb and E were nearly one order of magnitude lower than the CS values.
This behavior is consistent with the work of Kanimozhi et al.71 who studied the effect of MC incorporation in CS–PVA porous scaffolds, and found that the mechanical properties (i.e., E, tensile strength, and εb) of the wet samples decreased by increasing the MC concentration. This means that the significant decrease in the mechanical properties reported here may be related to the low mechanical behavior of MC in a water environment.34
Since the delivery platforms for the oral cavity are not exposed to significant mechanical loads, the obtained CS–MC patches were considered suitable for this application. In addition, the CS–MC samples were stable and did not break during the test, making them suitable for handling by the patient.72,73
Adequate mucoadhesion is essential to guarantee a prolonged residence time of the patches in the target site and, consequently, a prolonged in situ drug release.74 Therefore, the obtained patches were further tested for mucoadhesive properties.
Fig. 5 shows the release pressure (σadh) and the mucoadhesion work (Wadh) of both CS and CS–MC samples. The CS–MC samples exhibited significantly higher σadh (0.85 ± 0.26 kPa) and Wadh (1192.28 ± 602.36 Pa mm) compared to the CS control samples (σadh = 0.42 ± 0.22 kPa and Wadh = 343.13 ± 268.89 Pa mm) (p < 0.05).
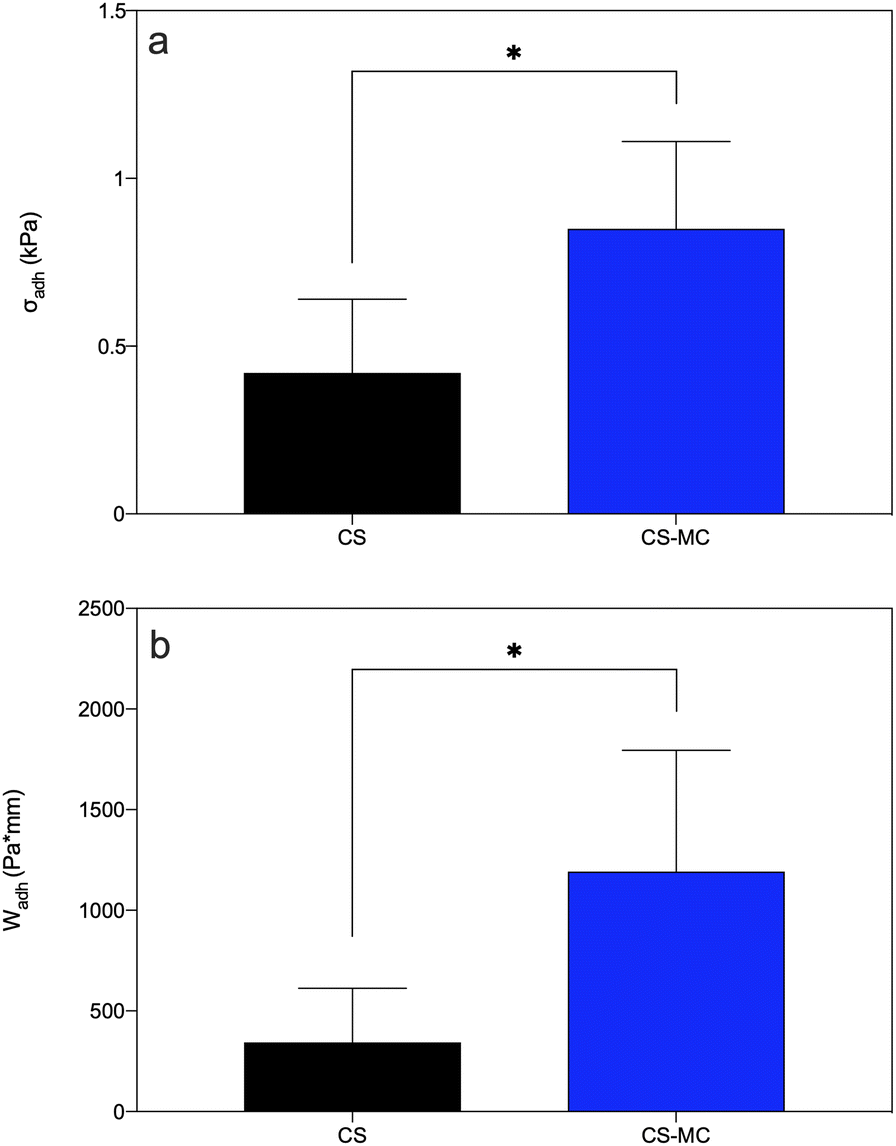 | ||
| Fig. 5 (a) Release pressure (σadh) and (b) mucoadhesion work (Wadh) for the CS and CS–MC samples (*p < 0.05). | ||
Mucoadhesion is a complex phenomenon that relies on different mechanisms (i.e., physical and chemical interactions) taking place at the material–mucus interface.75 The interaction between CS and mucin has been reported to be primarily due to electrostatic interactions and supported by other forces, such as hydrogen bonding and hydrophobic interactions.75 In particular, electrostatic interactions between positively charged amino groups of CS and negatively charged sialic acid residues of mucin play a crucial role in adhesion.75,76 Moreover, the formation of hydrogen bonds between the amine and hydroxyl groups of CS and the mucus layer present on the mucosa,77,78 together with the hydrophobic interactions between CS and mucin glycoproteins,75 strengthen the adhesion of the patch.
The addition of MC increased the mucoadhesion of the CS–MC samples ex vivo, primarily because of the bioadhesive nature of MC.75,79,80 Hydrophobic interactions and the hydrogen and van der Waals bonds established between non-ionic MC and porcine buccal mucosa,75 along with the abovementioned CS–mucin bonds, led to superior mucoadhesive properties of the CS–MC samples compared with the CS ones. Comparing the σadh showed by the CS–MC samples with the data found in the literature, MC performance was similar to that of other adhesive cellulose derivatives. In this regard, Tejada et al.28 developed and characterized mucoadhesive films based on CS and hydroxypropyl methylcellulose (HPMC). The observed σadh were comparable to those of the CS–MC samples (i.e., 0.81 ± 0.12 kPa vs. 0.85 ± 0.26 kPa for CS–HPMC and CS–MC, respectively). Moreover, focusing on the mucoadhesion work (Wadh), our CS–MC samples exhibited values comparable to the CS films reported by Kharenko et al.38 (i.e., 2086 ± 243 Pa mm vs. 1192.28 ± 602.36 Pa mm for the CS and CS–MC samples, respectively).
To the best of our knowledge, we are the first to demonstrate the potential of MC as a mucoadhesion-enhancer agent for patches obtained via EPD. Overall, the produced MC–CS patches, thanks to their noteworthy adhesive characteristic, lend themselves well as candidates for the delivery of drugs to the oral cavity.
3.6. Evaluation of GS loading and release for the CS–MC patches
GS was selected as a model drug in light of its wide-spectrum antibiotic activity and extensive use in medical applications via both parenteral and topical administration.81 In EPD processing, GS has been investigated in coatings for orthopedic implants, primarily for the treatment/prevention of periprosthetic infections.23,82,83 Furthermore, GS has also been reported for the treatment of infections affecting mucosal tissues (e.g., oral mucosa84 and nasal mucosa85). Thus, the idea of this work was to combine this information to obtain, via a one-pot process, GS-loaded patches for the treatment of infections affecting oral mucosal tissues.GS loading in the CS–MC/GS patches obtained via EPD was assessed by a TNBSA assay, and our results showed a GS concentration of 62.1 ± 4.5 mg g−1 (wGS/wCS–MC) in the patches. Considering that 200 mg of GS was added to the EPD solution (V = 100 mL, [GS] = 2 mg mL−1, [CS–MC] = 1% (w/v)), it can be concluded that the drug loading efficiency of the EPD process was ∼30%. However, it is not trivial to compare this value with the literature, mainly because in most studies dealing with the EPD of GS there is no quantification of antibiotics loaded in the samples.82,86 However, it was possible to calculate the amount of GS loaded per cm2 of the sample and compare it with the work of Pishbin et al.,23 who quantified the GS amount in their samples after EPD. The GS loading was 145 and 50 μg cm−2 for the work of Pishbin et al. and the present study, respectively. The lower GS loading could be ascribed to the different EPD solutions used (CS/bioactive glass/GS vs. CS–MC/GS), the EPD parameters, and the thickness of the deposits.
For the CS–MC/GS samples, GS deposition was attributed to the electrophoretic mobility of the antibiotic. In fact, the pKa values of the amino groups of GS have been reported to be in the range of 5.5–9, causing GS to be cationic under the working pH conditions (i.e., pH = 3.4) and leading to mobility toward the negatively charged cathode.23,87 Moreover, the stability of GS has been reported in the 2–10 pH range,88 further supporting its applicability to the EPD process.
GS release from the CS–MC/GS samples was then assessed by the TNBSA assay. As shown in Fig. 6, the drug release kinetics can be described by a burst release. In particular, about 65% of GS was released within 10 min, while a complete (∼100%) release was achieved 30 min post-soaking. This behavior is compatible with the rapid swelling of the CS–MC samples, which reached the swelling equilibrium 30 min after immersion (Fig. 4). It is worth noting that the burst release can be regarded as an optimal delivery mechanism in many circumstances, such as for delivery systems with a limited residence time (minutes to hours).89 In our study, the burst release of GS from the patches allowed an early-stage, high antibiotic bioavailability, which is promising for the rapid eradication of the infection and the prevention of antimicrobial resistance.
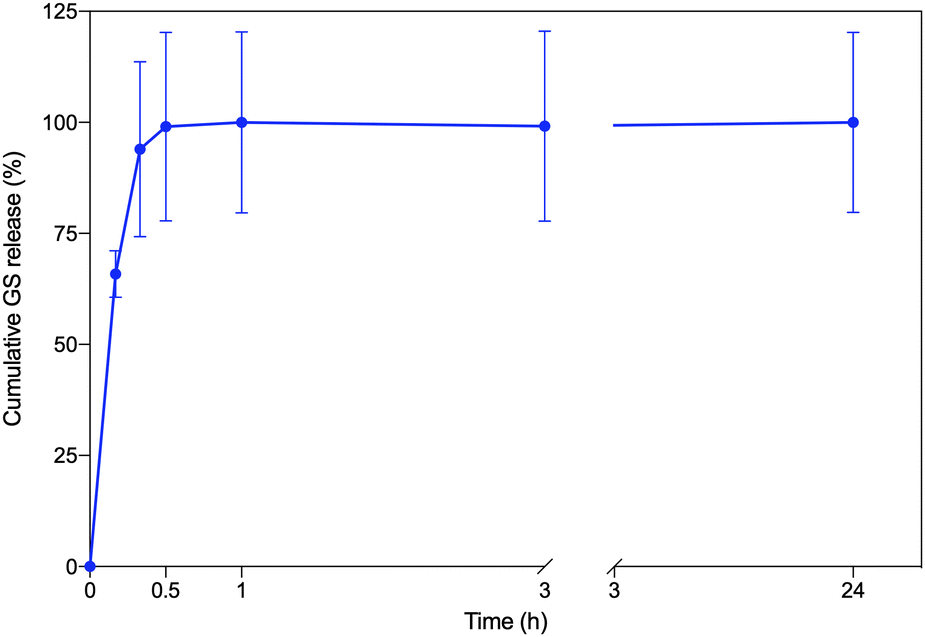 | ||
| Fig. 6 The in vitro GS cumulative release (%) profile for the CS–MC/GS samples. | ||
3.7. In vitro biological performance
Indirect cytotoxicity tests were carried out on L929 cells to evaluate, in a short period of 24 h (that is, the time patches would be in contact with the buccal mucosa), any possible cytotoxic effects of the leached substance(s), e.g., GS. It is worth noting that such tests avoid taking into account some other issues related to the features of the materials themselves, for example, the inherent adhesive properties that can vary among the different materials. Indeed, in our case, direct cytotoxicity tests, in which cells are seeded onto biomaterial specimens, might provide unreliable data as both the CS and CS–MC patches were not stable over time due to the swelling, such that cell adhesion might be affected. This, in turn, would lead to underestimation of the cell viability.Fig. 7 shows that the viability of cells incubated with the material extracts was always >70%, ruling out any possible cytotoxic effects of the substance(s) leached from the patch or from the released GS, following the ISO standard 10993-5:2009 (“Biological evaluation of medical devices—Part 5: Tests for in vitro cytotoxicity”). Such results are in line with the published literature about CS-based patches90 and coatings91 obtained via EPD. As expected, the addition of MC (considered “Generally recognized as safe (GRAS)” by the U.S. Food and Drug Administration92 and extensively reported in diverse biomedical applications30) resulted in a biomaterial as safe as its pristine counterpart.
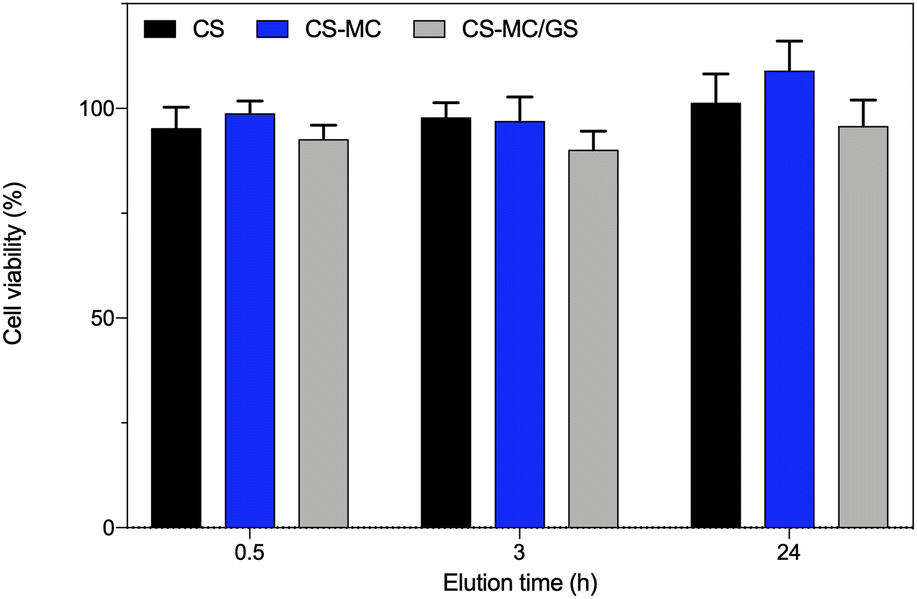 | ||
| Fig. 7 In vitro indirect cytotoxicity tests. The CS, CS–MC, and CS–MC/GS samples were incubated in complete DMEM for different time points (3, 6, and 24 h). Then, the eluates (material extracts) were tested on the L929 cells (*p < 0.05). | ||
In this study, we also evaluated the antibacterial activity of the CS–MC/GS patches. It is well known that GS is an aminoglycoside antibiotic exhibiting wide-spectrum bactericidal activity, especially against aerobic bacteria and most microorganisms with facultative metabolism.93 The mode of action of GS relies on its ability to electrostatically bind with negatively charged phospholipid head groups and, once it has penetrated inside the cell, to interfere with protein synthesis.
In this work, we decided to use GS as the loading drug, which, in comparison with other aminoglycosides, shows lower bacterial resistance.94 Accordingly, in this study, we tested the antibacterial activity of the CS–MC/GS patches against both Gram-negative (E. coli) and Gram-positive (S. aureus) bacterial strains.
As shown in Fig. 8, the eluates obtained from the CS and CS–MC samples did show low-to-negligible antibacterial activity against both bacterial strains. This is not surprising as similar dressings made of pure CS only did not show antibacterial effects.90,95,96 Again, the addition of MC did not have any effect on the microbiological performances of the electrodeposited patches, as MC does not show inherent antibacterial properties.97 Interestingly, when the CS–MC patches were loaded with GS, the resulting materials showed a remarkable antibacterial activity against E. coli, as ∼100% antibacterial activity was found for every condition (Fig. 8a). Similar results were found with Gram-positive S. aureus (Fig. 8b), although the antibacterial activity (%) was lower. This is not surprising as GS is generally much more effective against Gram-negative E. coli (MIC = 2 μg mL−1)98 as compared to their Gram-positive counterparts (MIC = 16 μg mL−1).99
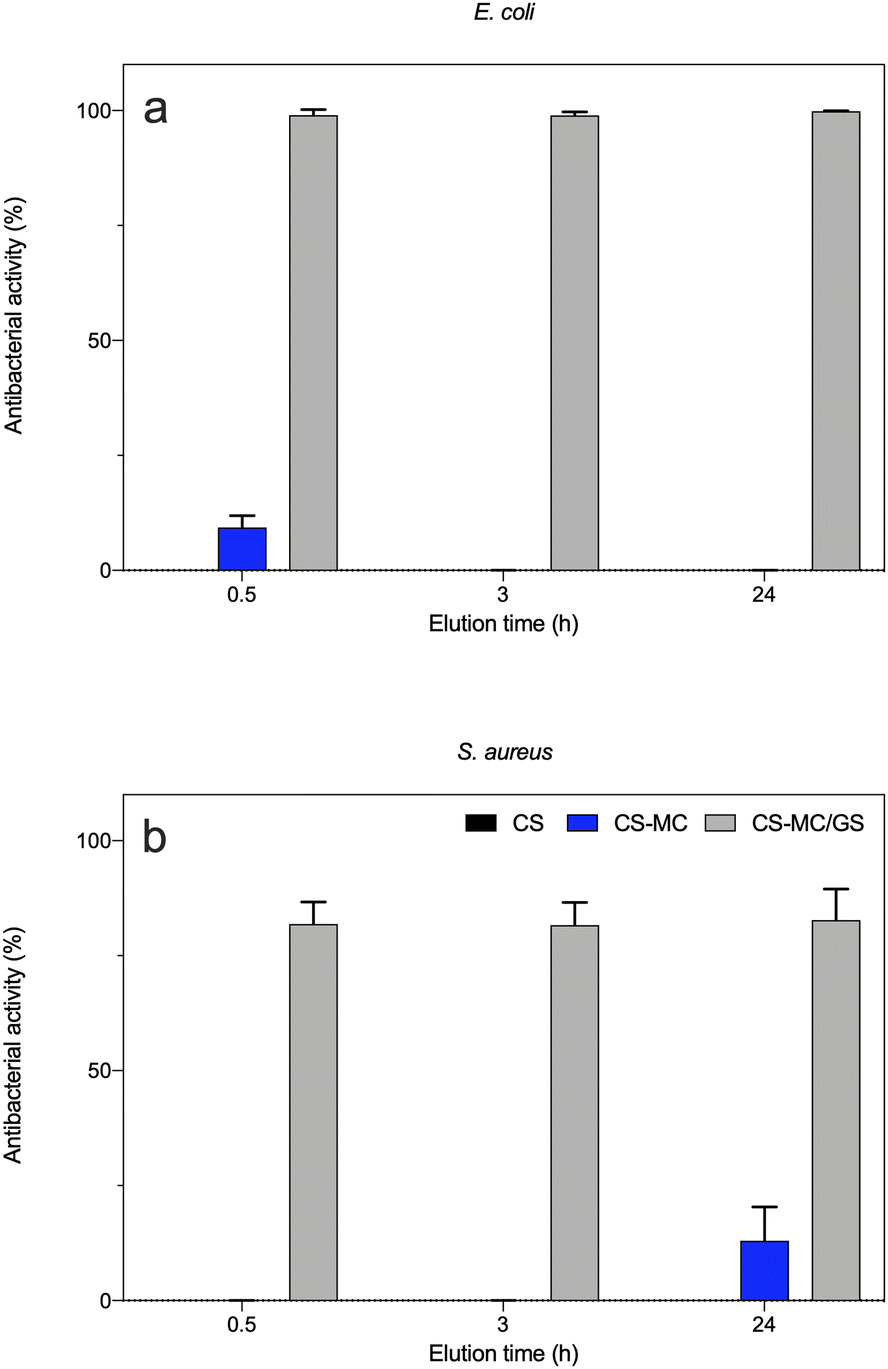 | ||
| Fig. 8 The antibacterial activity of the eluates (material extracts) against the (a) Gram-negative (E. coli) and (b) Gram-positive (S. aureus) bacterial strains. The CS, CS–MC, and CS–MC/GS samples were incubated with PBS, and the material extracts (eluates) were collected at 0.5, 3, and 24 h after incubation (*p < 0.05). | ||
More interestingly, our results confirmed a burst release of the antibiotic already after 0.5 h, which means that the CS–MC/GS patches are beneficial for elevating the bioavailability of the antibiotic in situ in the early stage of patch adhesion to the buccal mucosa. This, in turn, may prevent the onset of the bacterial infection in the site of interest as well as biofilm formation, while avoiding bacterial adhesion and subsequent colonization.
4. Conclusions
In this study, we disclosed the possibility of obtaining novel mucoadhesive patches with broad antibacterial activity via a one-pot EPD process. The non-trivial co-deposition of positively charged CS and neutral MC was achieved, thanks to the establishment of weak interactions between the two polymers, and then exploiting CS migration towards the cathode region as a driving force. Moreover, GS was added to the electrolyte, and a one-pot EPD process of CS–MC/GS patches was achieved. The presence of MC endowed the CS–MC samples with superior mucoadhesive behavior, while GS ensured broad antibacterial activity on the selected bacterial strains (E. coli and S. aureus). Overall, these results disclose the CS–MC/GS patches as potential mucoadhesive delivery systems to treat bacterial infections affecting the oral cavity.Author contributions
Conceptualization, methodology, formal analysis, investigation, writing – original draft, writing – review and editing, and visualization, L.B.; methodology and investigation, A.C.; methodology, investigation, writing – original draft, and writing – review and editing, N.B.; conceptualization and writing – review and editing, G.C.; conceptualization, resources, writing – review and editing, supervision, L.A.; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was carried out within the Agritech National Research Center and received funding from the European Union Next-GenerationEU (PIANO NAZIONALE DI RIPRESA E RESILIENZA (PNRR) – MISSIONE 4 COMPONENTE 2, INVESTIMENTO 1.4 – D.D. 1032 17/06/2022, CN00000022). This manuscript reflects only the authors' views and opinions, neither the European Union nor the European Commission can be considered responsible for them.References
- J. A. Aas, B. J. Paster, L. N. Stokes, I. Olsen and F. E. Dewhirst, J. Clin. Microbiol., 2005, 43, 5721–5732 CrossRef PubMed.
- S. Minhas, A. Sajjad, M. Kashif, F. Taj, H. Alwadaani and Z. Khurshid, Open Access Maced. J. Med. Sci., 2019, 7, 3341–3347 CrossRef PubMed.
- F. Rivera-Hidalgo and T. W. Stanford, Periodontology 2000, 1999, 21, 106–124 CrossRef CAS PubMed.
- P. Jain and I. Jain, J. Clin. Diagn. Res., 2014, 8, ZE18-21 Search PubMed.
- M. E. Levi and V. D. Eusterman, Otolaryngol. Clin. North Am., 2011, 44, 57–78 CrossRef PubMed.
- A. Mombelli and L. P. Samaranayake, Int. Dent. J., 2004, 54, 3–14 CrossRef PubMed.
- J. Teno, M. Pardo-Figuerez, K. J. Figueroa-Lopez, C. Prieto and J. M. Lagaron, J. Funct. Biomater., 2022, 13, 170 CrossRef CAS PubMed.
- M. Gihhotra, M. Ikrama, S. Srivastavaa and N. Gilhotra, J. Biomed. Res., 2014, 28, 81–97 Search PubMed.
- H. E. Colley, Z. Said, M. E. Santocildes-Romero, S. R. Baker, K. D'Apice, J. Hansen, L. S. Madsen, M. H. Thornhill, P. V. Hatton and C. Murdoch, Biomaterials, 2018, 178, 134–146 CrossRef CAS PubMed.
- R. M. Obaidat, Sci. Pharm., 2011, 79, 197–212 CrossRef CAS PubMed.
- G. P. Andrews, T. P. Laverty and D. S. Jones, Eur. J. Pharm. Biopharm., 2009, 71, 505–518 CrossRef CAS PubMed.
- I. A. Sogias, A. C. Williams and V. V. Khutoryanskiy, Int. J. Pharm., 2012, 436, 602–610 CrossRef CAS PubMed.
- A. C. Marques, A. I. Rocha, P. Leal, M. Estanqueiro and J. M. S. Lobo, Int. J. Pharm., 2017, 533, 455–462 CrossRef CAS PubMed.
- C. Pagano, S. Giovagnoli, L. Perioli, M. C. Tiralti and M. Ricci, Eur. J. Pharm. Sci., 2020, 142, 105125 CrossRef CAS PubMed.
- L. Perioli, V. Ambrogi, F. Angelici, M. Ricci, S. Giovagnoli, M. Capuccella and C. Rossi, J. Controlled Release, 2004, 99, 73–82 CrossRef CAS PubMed.
- P. Sanghai, T. Nandgude and S. Poddar, J. Drug Delivery, 2016, 2016, 1–9 CrossRef PubMed.
- M. Ansari, B. Sadarani and A. Majumdar, J. Drug Delivery Sci. Technol., 2018, 44, 278–288 CrossRef CAS.
- T. H. Kim, J. S. Ahn, H. K. Choi, Y. J. Choi and C. S. Cho, Arch. Pharmacal Res., 2007, 30, 381–386 CrossRef CAS PubMed.
- S. Yehia, O. El-Gazayerly and E. Basalious, Curr. Drug Delivery, 2009, 6, 17–27 CrossRef CAS PubMed.
- M. A. U. Rehman, M. A. Munawar, D. W. Schubert and A. R. Boccaccini, Surf. Coat. Technol., 2019, 358, 976–986 CrossRef CAS.
- E. Avcu, F. E. Baştan, H. Z. Abdullah, M. A. U. Rehman, Y. Y. Avcu and A. R. Boccaccini, Prog. Mater. Sci., 2019, 103, 69–108 CrossRef CAS.
- A. Ghalayani Esfahani, L. Altomare, E. M. Varoni, S. Bertoldi, S. Farè and L. De Nardo, J. Mater. Sci. Mater. Med., 2019, 30, 40 CrossRef PubMed.
- F. Pishbin, V. Mouriño, S. Flor, S. Kreppel, V. Salih, M. P. Ryan and A. R. Boccaccini, ACS Appl. Mater. Interfaces, 2014, 6, 8796–8806 CrossRef CAS PubMed.
- K. D. Patel, A. El-Fiqi, H.-Y. Lee, R. K. Singh, D.-A. Kim, H.-H. Lee and H.-W. Kim, J. Mater. Chem., 2012, 22, 24945 RSC.
- J. Song, Q. Chen, Y. Zhang, M. Diba, E. Kolwijck, J. Shao, J. A. Jansen, F. Yang, A. R. Boccaccini and S. C. G. Leeuwenburgh, ACS Appl. Mater. Interfaces, 2016, 8, 13785–13792 CrossRef CAS PubMed.
- B. Fonseca-Santos and M. Chorilli, Mater. Sci. Eng., C, 2018, 86, 129–143 CrossRef CAS PubMed.
- T. M. Ways, W. Lau and V. Khutoryanskiy, Polymers, 2018, 10, 267 CrossRef PubMed.
- G. Tejada, M. G. Barrera, G. N. Piccirilli, M. Sortino, A. Frattini, C. J. Salomón, M. C. Lamas and D. Leonardi, AAPS PharmSciTech, 2017, 18, 936–946 CrossRef CAS PubMed.
- P. Nasatto, F. Pignon, J. Silveira, M. Duarte, M. Noseda and M. Rinaudo, Polymers, 2015, 7, 777–803 CrossRef CAS.
- L. Bonetti, L. De Nardo and S. Farè, Tissue Eng., Part B, 2021, 27, 486–513 CrossRef CAS PubMed.
- L. Bonetti, L. Altomare, N. Bono, E. Panno, C. E. Campiglio, L. Draghi, G. Candiani, S. Farè, A. R. Boccaccini and L. De Nardo, J. Mater. Sci. Mater. Med., 2020, 31, 43 CrossRef CAS PubMed.
- D. J. Lomboni, A. Steeves, S. Schock, L. Bonetti, L. De Nardo and F. Variola, Soft Matter, 2021, 17, 5284–5302 RSC.
- L. Bonetti, L. De Nardo and S. Farè, Gels, 2021, 7, 141 CrossRef CAS PubMed.
- L. Bonetti, L. De Nardo, F. Variola and S. Fare’, Soft Matter, 2020, 16, 5577–5587 RSC.
- S. Pistone, F. M. Goycoolea, A. Young, G. Smistad and M. Hiorth, Eur. J. Pharm. Sci., 2017, 96, 381–389 CrossRef CAS PubMed.
- J. Gal, Talanta, 2001, 53, 1103–1115 CrossRef CAS PubMed.
- H. Yuk, C. E. Varela, C. S. Nabzdyk, X. Mao, R. F. Padera, E. T. Roche and X. Zhao, Nature, 2019, 575, 169–174 CrossRef CAS PubMed.
- E. A. Kharenko, N. I. Larionova and N. B. Demina, Pharm. Chem. J., 2008, 42, 392–399 CrossRef CAS.
- EN ISO 20776-1:2020 Susceptibility testing of infectious agents and evaluation of performance of antimicrobial susceptibility test devices – Part 1, Broth micro-dilution reference method for testing the in vitro activity of antimicrobial agents against rapidly growing aerobic bacteria involved in infectious diseases.
- I. Zhitomirsky and A. Hashambhoy, J. Mater. Process. Technol., 2007, 191, 68–72 CrossRef CAS.
- A. R. Boccaccini, S. Keim, R. Ma, Y. Li and I. Zhitomirsky, J. R. Soc., Interface, 2010, 7, S581–613 CrossRef CAS PubMed.
- L. Altomare, L. Draghi, R. Chiesa and L. De Nardo, Mater. Lett., 2012, 78, 18–21 CrossRef CAS.
- K. Schütz, A.-M. Placht, B. Paul, S. Brüggemeier, M. Gelinsky and A. Lode, J. Tissue Eng. Regener. Med., 2017, 11, 1574–1587 CrossRef PubMed.
- M. Nadour, F. Boukraa, A. Ouradi and A. Benaboura, Mater. Res., 2017, 20, 339–348 CrossRef CAS.
- J. Rotta, E. Minatti and P. L. M. Barreto, Cienc. Tecnol. Aliment., 2011, 31, 450–455 CrossRef.
- S. Roy, K. Pal, A. Anis, K. Pramanik and B. Prabhakar, Des. Monomers Polym., 2009, 12, 483–495 CrossRef CAS.
- N. A. Peppas and J. J. Sahlin, Biomaterials, 1996, 17, 1553–1561 CrossRef CAS PubMed.
- J. D. Smart, Crit. Rev. Ther. Drug Carrier Syst., 2004, 21, 319–344 CrossRef CAS PubMed.
- A. Rohani Shirvan, A. Bashari and N. Hemmatinejad, Eur. Polym. J., 2019, 119, 541–550 CrossRef CAS.
- F. Gebhardt, S. Seuss, M. C. Turhan, H. Hornberger, S. Virtanen and A. R. Boccaccini, Mater. Lett., 2012, 66, 302–304 CrossRef CAS.
- A. Sionkowska, Biomaterials, 2004, 25, 795–801 CrossRef CAS PubMed.
- T. Wu, S. Zivanovic, F. A. Draughon, W. S. Conway and C. E. Sams, J. Agric. Food Chem., 2005, 53, 3888–3894 CrossRef CAS PubMed.
- A. Pinotti, M. A. García, M. N. Martino and N. E. Zaritzky, Food Hydrocolloids, 2007, 21, 66–72 CrossRef CAS.
- Y. Tang, X. Wang, Y. Li, M. Lei, Y. Du, J. F. Kennedy and C. J. Knill, Carbohydr. Polym., 2010, 82, 833–841 CrossRef CAS.
- R. A. Khan, S. Salmieri, D. Dussault, N. Tufenkji, J. Uribe-Calderon, M. R. Kamal, A. Safrany and M. Lacroix, J. Polym. Environ., 2012, 20, 43–52 CrossRef CAS.
- S. Ahmed and S. Ikram, Achiev. Life Sci., 2016, 10, 27–37 Search PubMed.
- S. Mura, F. Corrias, G. Stara, M. Piccinini, N. Secchi, D. Marongiu, P. Innocenzi, J. Irudayaraj and G. F. Greppi, J. Food Sci., 2011, 76, N54–N60 CrossRef CAS PubMed.
- R. L. Oliveira, J. G. Vieira, H. S. Barud, R. M. N. Assunção, G. R. Filho, S. J. L. Ribeiro and Y. Messadeqq, J. Braz. Chem. Soc., 2015, 26, 1861–1870 CAS.
- R. A. Khan, S. Salmieri, D. Dussault, J. Uribe-Calderon, M. R. Kamal, A. Safrany and M. Lacroix, J. Agric. Food Chem., 2010, 58, 7878–7885 CrossRef CAS PubMed.
- T. Wang, L. Chen, T. Shen and D. Wu, Int. J. Biol. Macromol., 2016, 93, 775–782 CrossRef CAS PubMed.
- E. Vafa, R. Bazargan-Lari and M. E. Bahrololoom, Prog. Org. Coat., 2021, 151, 106059 CrossRef CAS.
- M. Bajpai, S. K. Bajpai and P. Jyotishi, Int. J. Biol. Macromol., 2016, 84, 1–9 CrossRef CAS PubMed.
- N. L. Calvo, L. A. Svetaz, V. A. Alvarez, A. D. Quiroga, M. C. Lamas and D. Leonardi, Int. J. Pharm., 2019, 556, 181–191 CrossRef CAS PubMed.
- H. Nagahama, H. Maeda, T. Kashiki, R. Jayakumar, T. Furuike and H. Tamura, Carbohydr. Polym., 2009, 76, 255–260 CrossRef CAS.
- J. Yin, K. Luo, X. Chen and V. V. Khutoryanskiy, Carbohydr. Polym., 2006, 63, 238–244 CrossRef CAS.
- V. V. Khutoryanskiy, M. G. Cascone, L. Lazzeri, Z. S. Nurkeeva, G. A. Mun and R. A. Mangazbaeva, Polym. Int., 2003, 52, 62–67 CrossRef CAS.
- L. Altomare, A. Cochis, A. Carletta, L. Rimondini and S. Farè, J. Mater. Sci. Mater. Med., 2016, 27, 95 CrossRef PubMed.
- N. A. Peppas, J. B. Thomas and J. McGinty, J. Biomater. Sci., Polym. Ed., 2009, 20, 1–20 CrossRef CAS PubMed.
- M. B. Stie, J. R. Gätke, F. Wan, I. S. Chronakis, J. Jacobsen and H. M. Nielsen, Carbohydr. Polym., 2020, 242, 116428 CrossRef CAS PubMed.
- L. Altomare, E. Guglielmo, E. M. Varoni, S. Bertoldi, A. Cochis, L. Rimondini and L. De Nardo, Biomatter, 2014, 4, e29506 CrossRef PubMed.
- K. Kanimozhi, S. Khaleel Basha and V. Sugantha Kumari, Mater. Sci. Eng., C, 2016, 61, 484–491 CrossRef CAS PubMed.
- D. Steiner, J. F. Emmendörffer and H. Bunjes, Pharmaceutics, 2021, 13, 2162 CrossRef CAS PubMed.
- M. Preis, C. Woertz, K. Schneider, J. Kukawka, J. Broscheit, N. Roewer and J. Breitkreutz, Eur. J. Pharm. Biopharm., 2014, 86, 552–561 CrossRef CAS PubMed.
- D. M. Shinkar, A. S. Dhake and C. M. Setty, PDA J. Pharm. Sci. Technol., 2012, 66, 466–500 CrossRef PubMed.
- S. Mansuri, P. Kesharwani, K. Jain, R. K. Tekade and N. K. Jain, React. Funct. Polym., 2016, 100, 151–172 CrossRef CAS.
- S. S. Timur, S. Yüksel, G. Akca and S. Şenel, Int. J. Pharm., 2019, 559, 102–112 CrossRef CAS PubMed.
- N. Iglesias, E. Galbis, M. Díaz-Blanco, R. Lucas, E. Benito and M.-V. De-Paz, Int. J. Mol. Sci., 2019, 20, 398 CrossRef PubMed.
- J. Smart, Adv. Drug Delivery Rev., 2005, 57, 1556–1568 CrossRef CAS PubMed.
- Y. Sudhakar, K. Kuotsu and A. K. Bandyopadhyay, J. Controlled Release, 2006, 114, 15–40 CrossRef CAS PubMed.
- D. Pal and A. K. Nayak, AAPS PharmSciTech, 2011, 12, 1431–1441 CrossRef CAS PubMed.
- C. Chen, Y. Chen, P. Wu and B. Chen, J. Formosan Med. Assoc., 2014, 113, 72–82 CrossRef CAS PubMed.
- L. Altomare, L. Visai, N. Bloise, C. R. Arciola, L. Ulivi, G. Candiani, A. Cigada, R. Chiesa and L. De Nardo, Int. J. Artif. Organs, 2012, 35, 876–883 CrossRef CAS PubMed.
- S. Seuss and A. R. Boccaccini, Biomacromolecules, 2013, 14, 3355–3369 CrossRef CAS PubMed.
- K. Osipowicz, P. Wychowanski, P. Nieckula, S. Shamsa, K. Wertheim-Tysarowska, K. Wozniak and C. Kowalewski, Adv. Dermatol. Allergol., 2021, 38, 979–984 CrossRef PubMed.
- C. Hasçiçek, N. Gönül and N. Erk, Farmaco, 2003, 58, 11–16 CrossRef PubMed.
- R. Rahighi, M. Panahi, O. Akhavan and M. Mansoorianfar, Mater. Chem. Phys., 2021, 272, 125018 CrossRef CAS.
- P. Baudoux, N. Bles, S. Lemaire, M.-P. Mingeot-Leclercq, P. M. Tulkens and F. Van Bambeke, J. Antimicrob. Chemother., 2006, 59, 246–253 CrossRef PubMed.
- T. W. Schafer, A. Pascale, G. Shimonaski and P. E. Came, Appl. Microbiol., 1972, 23, 565–570 CrossRef CAS PubMed.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef CAS PubMed.
- A. Ghalayani Esfahani, B. Lazazzera, L. Draghi, S. Farè, R. Chiesa, L. De Nardo and F. Billi, J. Appl. Microbiol., 2019, 126, 87–101 CrossRef CAS PubMed.
- M. A. Akhtar, K. Ilyas, I. Dlouhý, F. Siska and A. R. Boccaccini, Int. J. Mol. Sci., 2020, 21, 2637 CrossRef CAS PubMed.
- M. L. Coughlin, L. Liberman, S. P. Ertem, J. Edmund, F. S. Bates and T. P. Lodge, Prog. Polym. Sci., 2021, 112, 101324 CrossRef CAS.
- B. J. Chaves and P. Tadi, Gentamicin, in StatPearls [Internet]. StatPearls Publishing, Treasure Island (FL), 2022 Search PubMed.
- E. Kovács, T. Savopol, M.-M. Iordache, L. Săplăcan, I. Sobaru, C. Istrate, M.-P. Mingeot-Leclercq and M.-G. Moisescu, Bioelectrochemistry, 2012, 87, 230–235 CrossRef PubMed.
- M. Li, Q. Chen, M. Ma, X. Liu, K. Dong, Y. Zhang and T. Lu, Mater. Lett., 2019, 239, 29–32 CrossRef CAS.
- T. Aydemir, L. Liverani, J. I. Pastore, S. M. Ceré, W. H. Goldmann, A. R. Boccaccini and J. Ballarre, Mater. Sci. Eng., C, 2020, 115, 111062 CrossRef CAS PubMed.
- K. Kolarova, D. Samec, O. Kvitek, A. Reznickova, S. Rimpelova and V. Svorcik, Jpn. J. Appl. Phys., 2017, 56, 06GG09 CrossRef.
- T. Tada, K. Uechi, I. Nakasone, K. Shimada, M. Nakamatsu, T. Kirikae and J. Fujita, Int. J. Infect. Dis., 2017, 63, 21–22 CrossRef CAS PubMed.
- E. Akturk, H. Oliveira, S. B. Santos, S. Costa, S. Kuyumcu, L. D. R. Melo and J. Azeredo, Antibiotics, 2019, 8, 103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01540d |
| This journal is © The Royal Society of Chemistry 2023 |