
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Injectable local drug delivery systems for glioblastoma: a systematic review and meta-analysis of progress to date†
Yu
Wang
a,
Chiara
Bastiancich
bc and
Ben
Newland
 *a
*a
aSchool of Pharmacy and Pharmaceutical Sciences, Cardiff University, King Edward VII Avenue, Cardiff, CF10 3NB, UK. E-mail: newlandb@cardiff.ac.uk
bAix-Marseille Univ, CNRS, INP, Inst Neurophysiopathol, 13344 Marseille, France
cDepartment of Drug Science and Technology, University of Turin, 10125 Turin, Italy
First published on 6th January 2023
Abstract
Glioblastoma (GBM) is an aggressive malignant cancer associated with bleak prognosis and high mortality. The current standard of care for GBM is maximum surgical resection plus radiotherapy and temozolomide (TMZ) chemotherapy. The blood brain barrier (BBB) remains the main obstacle for chemotherapy and severely limits the choice of therapeutic agents. Local treatment allows drugs to circumvent the BBB and reduces systemic side effects. Despite much research effort, to date, no drug delivery system (DDS) designed to be directly injected into brain tumors has been clinically approved, and a systematic overview of the progress in this field, or lack thereof, is missing. In this review, a systematic search of pre-clinical literature was conducted which resulted in 36 original articles on injectable DDS for local treatment of GBM which met the inclusion criteria. A wide range of injectable DDS have been developed and tested pre-clinically which include nanoparticles, liposomes, microspheres, hydrogels and others. meta-Analyses of the included studies showed that, overall, local administration of injectable DDS was beneficial to increase the animal's survival time. Finally, this review summarized the therapeutic effect after local treatment and discussed the shortcomings of the experimental setting in in vivo studies.
1. Introduction
Glioblastoma (GBM) is a primary malignant brain tumor of the central nervous system, associated with poor clinical outcome and high mortality.1 Surgery is the mainstay of GBM management as it allows to reduce the tumor mass, relieve symptoms and obtain samples to confirm the diagnosis. Moreover, maximum safe resection also impacts the therapeutic outcome, leading to an increase of the patients overall survival.2 However, because of the strong infiltrating nature of GBM cells into the healthy surrounding tissue, debulking of the tumor mass is insufficient to prevent GBM recurrence.3 Moreover, 20–30% of GBM patients are not eligible for resection, either due to the extent of tumor spread, tumor location, impaired clinical condition of the patient or other reasons.4,5 When surgery is not recommended, a stereotactic biopsy should be performed for tissue diagnosis and for clinical decision making.6In 2005, a series of clinical trials were published showing the improvement of median survival (MS) after oral administration of Temozolomide (TMZ) combined with radiotherapy (Stupp protocol), which shifted the paradigm of GBM treatment to a combined-modality regimen.7–9 Since then, the standard of care therapy for newly diagnosed GBM patients is maximum surgical resection followed by the Stupp protocol. At present, GBM is still incurable. The overall MS is only 14.6 to 16.6 months after diagnosis, and only around 5% to 10% of GBM patients survive over five years.9–11 Meanwhile, patients suffer from a decline in their quality of life and loss of cognitive function.12
The efficacy of radiotherapy is hampered by GBM cells showing radioresistance, even at high radiation doses,13 which in part is driven by tumor hypoxia.14 On the other hand, several innate or acquired chemoresistance mechanisms limit the efficacy of chemotherapy. For example, GBM cells can overexpress O6-methylguanine-DNA methyltransferase (MGMT), leading to resistance to alkylating agents such as TMZ.15 MGMT, a DNA repair enzyme, can remove alkyl groups from the O6 position of guanine, which is a crucial target site for alkylating agents. Hence, the gene mutation is reverted, leading to the avoidance of cell death. Due to the limitations of current treatment options, most patients still suffer from recurrence, which often appear within two years.16 Even though no drugs have received regulatory approval for the treatment of newly diagnosed GBM patients since 2005, a non-pharmacological therapeutic approach based on tumor treating fields (TTF) was approved by the Food and Drug Administration in 2015.17 The TTF device is applied to the scalp and delivers low-intensity, alternating electric fields delivered via transducer arrays. When added to TMZ chemotherapy, this approach can slightly improve progression-free survival.18 However, its use in clinical practice is still limited.
Standard of care treatments for recurrent GBM patients are not well defined and may vary depending on the country and the hospital rules. They are established based on previous response to treatment, clinical status of the patient, MGMT promoter methylation and disease progression. Around a quarter of patients are eligible for the second surgery who are diagnosed with symptomatic and circumscribed recurrence more than a half year after the initial surgery.6 Re-irradiation could be performed with an altered fractionated dose depending on the tumor size, while the efficacy remains controversial.19 Systemic chemotherapy (TMZ rechallenge, nitrosoureas, bevacizumab) is also an option for recurrent GBM and patients could be included into clinical trials if they meet the inclusion criteria.6 In addition, although bevacizumab received accelerated approval in the USA, it is not approved for the treatment of recurrent GBM in European countries, due to no sufficient data showing the improvement of overall survival time after the combined treatment between bevacizumab and lomustine compared with lomustine alone.20
GBM is an unmet medical need, and finding effective, safe and long-lasting therapeutic approaches is urgently required. Local drug delivery directly to the tumor or in the resection cavity has been proposed as a means of bypassing the blood–brain barrier (BBB). This strategy allows a higher drug concentration at the tumor site and reduces systemic side effects that often-limited systemic chemotherapy. Up to now, only one drug delivery system (DDS) – Gliadel®, delivering the chemotherapeutic drug carmustine from biodegradable wafers – has been approved for the local treatment of GBM. Gliadel® wafers contain 7.7 mg of carmustine and 192.3 mg of 1,3-bis-(p-carboxyphenoxy)propane (pCPP) and sebacic acid (SA) copolymer (in a ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80) and can be directly positioned into the resection cavity during surgery.21 A systematic review of clinical trials utilizing Gliadel® in combination with radiotherapy and TMZ showed that the MS was extended to 18.2 months compared to radiotherapy plus TMZ (14.6 months) from previous clinical trials.9,22 Whilst Gliadel® wafers show a modest improvement in therapeutic outcomes, its clinical use is still limited. Its rigid structure can lead to mechanical mismatch between the stiff wafer and soft brain tissue causing side effects.23 Moreover, the amount of drug that can be administered depends on the size of the cavity (8 wafers need to be placed to ensure therapeutic dose of carmustine) and the drug release profile is sub-optimal.24–26 Whilst an array of local DDS is being developed to overcome these drawbacks,27 there is still no clinically approved DDS designed for stereotactic injection directly into non-resectionable tumors.
:80) and can be directly positioned into the resection cavity during surgery.21 A systematic review of clinical trials utilizing Gliadel® in combination with radiotherapy and TMZ showed that the MS was extended to 18.2 months compared to radiotherapy plus TMZ (14.6 months) from previous clinical trials.9,22 Whilst Gliadel® wafers show a modest improvement in therapeutic outcomes, its clinical use is still limited. Its rigid structure can lead to mechanical mismatch between the stiff wafer and soft brain tissue causing side effects.23 Moreover, the amount of drug that can be administered depends on the size of the cavity (8 wafers need to be placed to ensure therapeutic dose of carmustine) and the drug release profile is sub-optimal.24–26 Whilst an array of local DDS is being developed to overcome these drawbacks,27 there is still no clinically approved DDS designed for stereotactic injection directly into non-resectionable tumors.
Bypassing the BBB via direct intratumor administration would not only facilitate a high local concentration of a drug to be achieved at the target site, but also allow a wider range of therapeutics to be investigated. These factors allow a reduction in dose-limiting systemic toxicity whilst opening possibilities for re-purposing existing anti-cancer therapeutics, previously unsuitable due to BBB impermeability. Several methods such as convection enhanced delivery (CED) or local injection of hydrogels or colloid systems are widely investigated in pre-clinical studies. However, to date, there is no systematic overview of the current state of these delivery systems nor a meta-analysis of their efficacy at improving the MS in GBM animal models.
The aim of this review was to undertake a systematic review of the pre-clinical literature to tabulate injectable DDS that have been developed for GBM therapies. Furthermore, we aimed to analyze whether injectable DDS directly administered intratumorally in animals bearing GBM tumors showed therapeutic efficacy compared to negative controls (no treatment or vehicle only controls). We also analyzed the role that the delivery system plays in comparison to direct injection of a free drug. Finally, we sought to compare local drug administration with systemic administration.
2. Methods
2.1 Search strategy
Three databases (PubMed, Web of Science and Scopus) were used in this review to systemically search articles which were relevant to the topic. The search for this review was completed on 9th June 2022.The three search strategies were as follows:
Pubmed: ((glioblastoma[Title/Abstract] OR “brain tumo*” [Title/Abstract] OR glioma [Title/Abstract] OR gliosarcoma[Title/Abstract]) AND (“local delivery” [Title/Abstract] OR “drug delivery system” [Title/Abstract] OR “sustained release” [Title/Abstract] OR microsphere*[Title/Abstract] OR microparticle*[Title/Abstract] OR “convection enhanced delivery” [Title/Abstract] OR CED [Title/Abstract] OR hydrogel* [Title/Abstract])) AND (“in vivo” [Title/Abstract] OR “pre-clinical” [Title/Abstract] OR intracranial [Title/Abstract] OR resection[Title/Abstract]).
Wed of science: TI = (glioblastoma OR “brain tumo*” OR glioma OR gliosarcoma) AND AB = (“local delivery” OR “drug delivery system” OR “sustained release” OR microsphere* OR microparticle* OR “convection enhanced delivery” OR CED OR hydrogel*) AND AB = (“in vivo” OR “pre-clinical” OR intracranial OR resection).
Scopus: TITLE-ABS (glioblastoma OR “brain tumo*” OR glioma OR gliosarcoma) AND TITLE-ABS (“local delivery” OR “drug delivery system” OR “sustained release” OR microsphere* OR microparticle* OR “convection enhanced delivery” OR CED OR hydrogel*) AND TITLE-ABS (“in vivo” OR “pre-clinical” OR intracranial OR resection).
2.2 Inclusion and exclusion criteria
After the search, the results of all databases were downloaded to a Microsoft Excel file (Home and Student 2019). Duplicate results from the three databases were removed before screening. The following inclusion and exclusion criteria were used to create a selection framework that was used whilst analyzing titles/abstracts and full text manuscripts.Inclusion criteria were as follows:
• Original research articles written in the English language.
• Articles had at least one in vivo antitumor efficacy study using an orthotopic GBM tumor model without resection of the tumor prior to treatment administration.
• The study had to analyze the local delivery of anticancer agents in comparison to a negative control of either untreated animals, or a vehicle solution.
• Drug preparations used were injectable. The DDS could be drug loaded nano/macro-carriers or scaffolds loaded with either free drugs or nanomedicines.
• Studies must have had quantitative results in terms of the MS time.
Exclusion criteria were as follows:
• Articles that were not available in English.
• Review articles, book chapters and conference proceedings where methodology was insufficient for correct evaluation of the results.
• Articles that had only in vitro studies, clinical studies or did not have a preclinical in vivo antitumor efficacy study.
• Articles that used either gene therapy, monoclonal antibodies, or genetically engineered cells as an antitumor treatment.
• Articles that did not use the orthotopic GBM tumor model in in vivo antitumor efficacy study (e.g., they used a subcutaneous tumor).
• Studies that used a GBM tumor model with resection of the tumor prior to administration of treatment.
• Studies that delivered uninjectable preparations (e.g., wafers), or only delivered liquid formulations of drugs that were not incorporated into a DDS.
• Articles that did not have an appropriate negative control group of either no treatment or empty vehicle control group in in vivo antitumor efficacy study.
• Articles that only researched radiotherapy or only had studies combined with radiotherapy.
• Articles that did not use MS to evaluate in vivo antitumor efficacy.
2.3 Data extraction
Data from included articles was extracted and entered into tables with different categories. The extracted data included the year of publication, first author, drug used, delivery vehicle, cancer model and MS time for each relevant treatment group. The results were listed in the table by in vivo antitumor efficacy studies rather than articles. Many articles had more than one in vivo efficacy study, and all data from different studies which met the inclusion criteria was extracted. Different studies in the same article meant that studies had at least one difference in drug used, delivery vehicle or cancer model. Different drug dosage, times of treatment or injection site were regarded as too similar to represent separately. If two or more of the same studies were in one article, the most appropriate study was chosen and listed in the table according to the design of the in vivo antitumor efficacy study or the optimized study which was focused upon by the authors. Meanwhile, if two different drugs or single drug vs. double drug combination were in one study, the study was listed separately by drug used.For MS time, some data was explicitly written in the article text or tables. However, in some cases the MS data had to be calculated from the Kaplan–Meier survival curves. To achieve this, figures were extracted into Microsoft PowerPoint (Home and Student 2019) and scaled appropriately to calculate the medium survival of the relevant experimental groups.
Percentage change in MS time between the no treatment group (negative control) and the treatment group was also calculated. If the study did not have a no treatment group, the empty vehicle group was used instead as negative control.
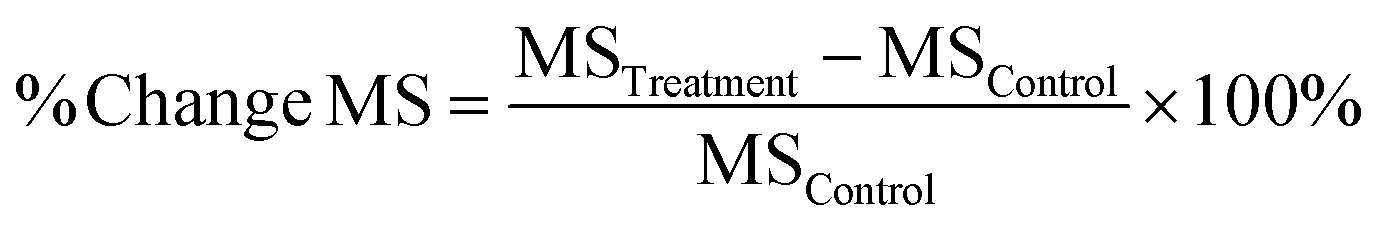 | (1) |
Eqn (1). % Change MS means percentage change in median survival time, MSTreatment means median survival time in treatment group, MSControl means median survival time in the negative control group.
2.4 Statistical analysis
Review Manager software (version: 5.4.1) was used to conduct the meta-analysis to compare the antitumor efficacy between locally administrated DDS and the most appropriate negative control group, locally administrated DDS and locally injected free drug, locally administrated DDS, and systemic administration of either the free drug or the DDS. The MS ratio was used to summarize the MS time data.28 It was used as a method being consistent with the hazard ratio method.29 The survival time ratio was calculated by the following equation. It was log-transformed to give the normal distribution and make the scale symmetric.30,31 The log-transformed MS ratio, the standard deviation (or standard error) and the group size were used as inputs for the meta-analysis. Due to the high heterogeneity (I2 > 50%), the random model was used in the meta-analysis.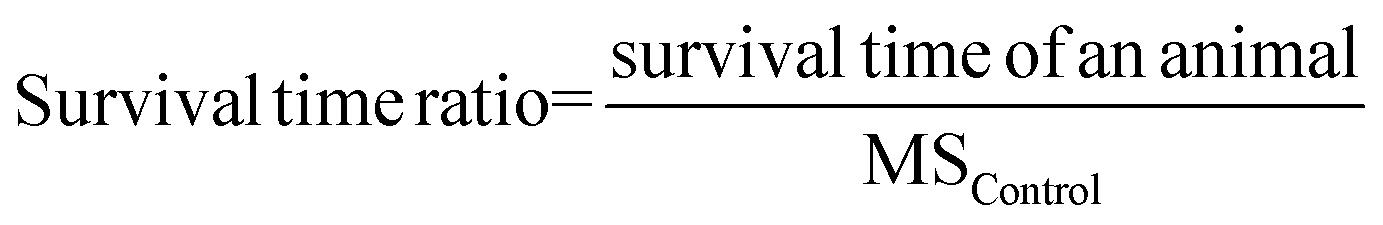 | (2) |
Eqn (2). MSControl means median survival time in the negative control group.
2.5 Quality assessment
The quality of each included article was assessed by a 12-point checklist described previously by Hirst et al.28 One point was allocated to each item and the final score was the sum of all points. The 12-point checklist included: (1) peer-reviewed publication, (2) sample size calculation, (3) random allocation to groups, (4) blinded assessment of outcome, (5) compliance with animal welfare regulations, (6) statement of potential conflict of interests, (7) uniform volume or number of cells inoculated, (8) consistent site of tumor implantation, (9) reported number of animals in which the tumor did not grow, (10) stated number of excluded animals, and given reasons for exclusion, including anomalies, (11) explanation of tumor model used, or multiple glioma models used and (12) presentation of evidence that the chemotherapeutic agent acts directly against tumor.3. Results and discussion
3.1 Results of the systematic search as a bibliographic screening of literature data
Since TMZ was adopted as the gold standard therapy of GBM in 2005, the number of papers related to the development of DDS for GBM has increased considerably. To select the papers that could answer the scientific questions addressed in this review, we performed a systematic search using three search engines and defined inclusions and exclusion criteria.The PRISMA diagram (Fig. 1(A)) shows the screening process conducted in this review. A total of 1854 articles were searched through three databases. 1038 duplicate articles were removed, and 816 articles remained. 520 articles were excluded during the title and abstract screen due to lacking primary data (e.g., review articles), being irrelevant or clearly failing to meet the inclusion criteria. The remaining 296 articles were subject to analysis of the full text. Among these articles, the full text of two articles could not be accessed and no reply was received when the authors were contacted. Finally, 36 articles met all inclusion criteria for a total of 44 individual in vivo antitumor efficacy studies were eligible. These studies were classified into three categories according to what comparison groups were used as follows: 1 – local DDS vs. negative controls (no treatment or vehicle only); 2 – local DDS vs. locally administered free drug; 3 – local DDS vs. systemic administration of either the free drug or DDS. For all studies, a negative control group receiving either no intervention, or vehicle solution only, was required from which the percentage change in MS could be calculated. Data from these studies was extracted and entered into Tables 1, 2 and 3. Fig. 1(B) shows the quantity distribution by the publication year of all included articles in this review. Only two eligible articles were published before the establishment of Stupp protocol in 2005.32,33 However, most of included articles were published after 2008, which indicates an increasing trend of interest for more effective and safer DDS for anti-GBM use.
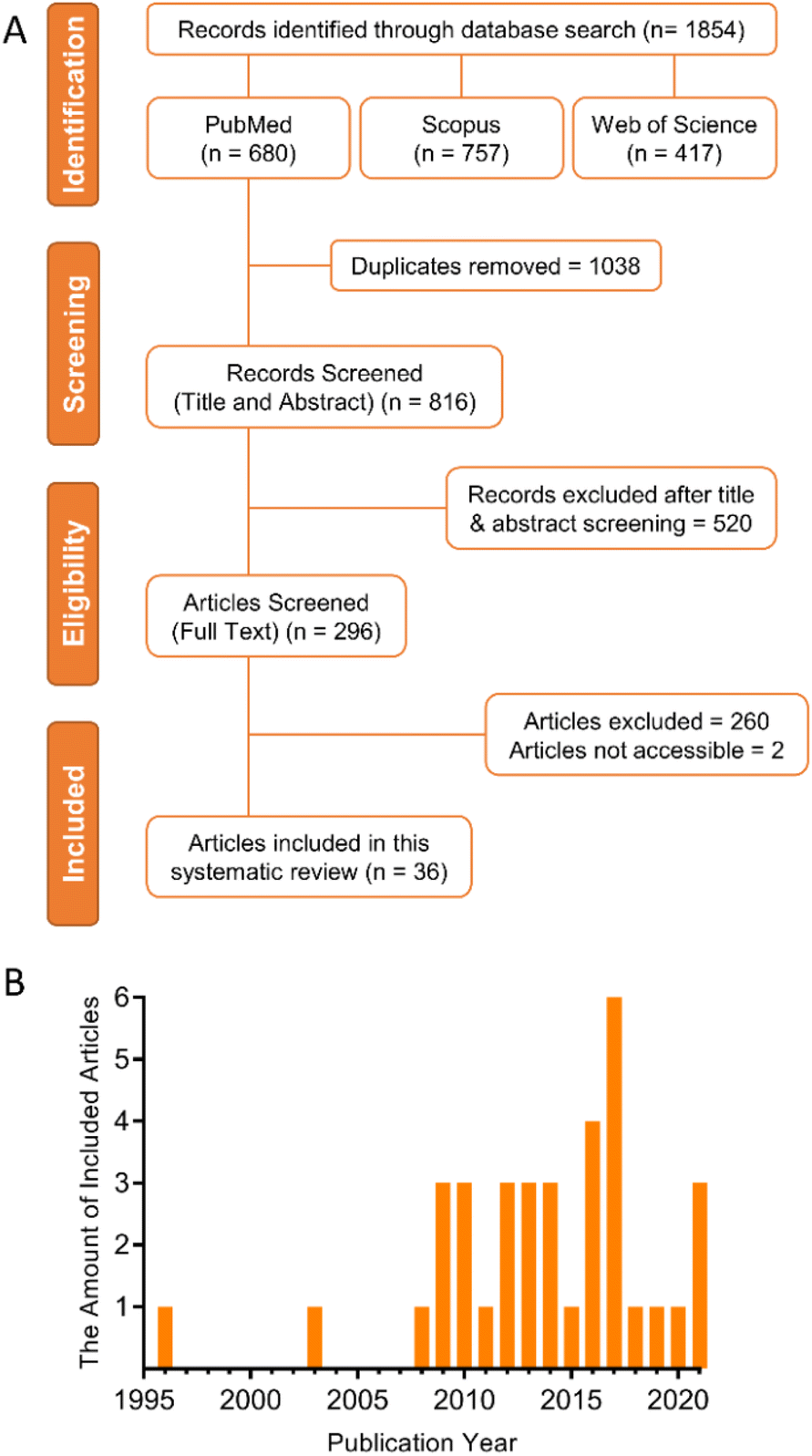 | ||
| Fig. 1 (A) PRISMA diagram showing the screening process of this systematic review. (B) The quantity distribution by publication year of included articles. | ||
| Year | Drug used | Delivery vehicle | Cancer model | Treatment time (days) post-grafting | Number of implanted cells | Drug release patterna | MS no treatment (days) | MS vehicle alone (days) | MS Local delivery via DDS (days) | % Change MS (local DDS vs. control)b | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Abbreviations: MS: median survival time; DDS: drug delivery system; Ref.: reference; GEM: gemcitabine; CPT: camptothecin; EPI: epirubicin; Cu(DDC)2: copper diethyldithiocarbamate; BG: O6-benzylguanine; TMZ: temozolomide; PTX: paclitaxel; DOX: doxorubicin; Fc-diOH: ferrociphenol; TPT: topotecan; GD: gadodiamide; RP: referred to a previous protocol.a The drug release pattern was defined from the results of in vitro drug release studies. The prolonged release was defined as when the DDS released more than 10% of the loaded drugs after the first two days. Studies that did not have drug release data after day two, or the cumulative drug release from day two to the end of the experiment did not exceed 10%, were regarded as rapid release.b No treatment group was chosen as the negative control group to calculate the percentage change of MS. If the study did not have a no treatment group, the empty vehicle group was used instead.c The MS data was measured and calculated from the Kaplan–Meier survival curves (not directly stated in the article).d The calculation of the percentage change in the MS data used at least one MS data not directly stated in the article. | |||||||||||
| 2021 | DOX and GEM | Hydrogel | A25 M cells – female NOD SCID mice | 14 | 100000 |
Rapid | N/A | 61.5 | 79.5 | 54% | 41 |
| 2021 | DOX and GEM | Hydrogel | A25C cells – female NOD SCID mice | 14 | 100000 |
Rapid | N/A | 54.5 | 82.5 | 51% | 41 |
| 2020 | CPT | Polymeric drug | GL261/fluc-DsRed glioma cells – unspecified gender NCr nu/nu mice | 7 | N/A | Prolonged | 21.5c | N/A | 28.5c | 33%d | 42 |
| 2019 | EPI | Nanoparticles | U87MG Lu cells – male nu/nu mice | 2 doses: 5, 12 | 100000 |
Prolonged | N/A | 27 | >60 | 122% | 34 |
| 2017 | Cu(DDC)2 | Liposomes | F98 cells – male Fischer rats | 10 | 10000 |
N/A | N/A | 20.5 | 25 | 22% | 43 |
| 2017 | Panobinostat | Micelles | F98 cells – unspecified gender Wistar rats | 10 | 100000 |
N/A | 24.5 | N/A | >60 | 145% | 35 |
| 2016 | SN-38 | Micelles | U87MG cells – male F344/NJcl-rnu/rnu (nude) rats | 5 | 200000 |
N/A | N/A | 21 | 28 | 33% | 44 |
| 2014 | BG | Nanoparticles | GBM6-luc cells – unspecified gender nude athymic mice | 4 doses: 26, 29, 34, 36 | 20000 |
Rapid | 3 | N/A | 9 | 200% | 36 |
| 2014 | TMZ | Nanoparticles | U87 cells – male athymic nude mice | 2 doses: 4, 7 | 500000 |
Rapid | 19.0c | N/A | 28.0c | 47%d | 40 |
| 2013 | PTX | Hydrogel | 9L cells – female Fischer 344 rats | 0 | RP | N/A | 15 | N/A | 33 | 120% | 37 |
| 2013 | Digoxin | Nanoparticles | BCSCs cells – unspecified gender nude rats | N/A | N/A | Prolonged | 114.5c | N/A | 124.0c | 8%d | 45 |
| 2012 | DOX | Microspheres | BT4Ca cells – male and female BD IX rats | 3 | 8000 | N/A | 19 | N/A | 20 | 5% | 39 |
| 2012 | Irinotecan | Microspheres | BT4Ca cells – male and female BD IX rats | 3 | 8000 | N/A | 19 | N/A | 21 | 11% | 39 |
| 2012 | CPT | Hydrogel | C6 rat glioma cells – male Sprague-Dawley rats | 7 | 1000000 |
RP | 18 | N/A | 26 | 44% | 46 |
| 2010 | Fc-diOH | Lipid nanocapsules | 9L cells – female Fischer 344 rats | 6 | 1000 | N/A | N/A | 25 | 27 | 8% | 47 |
| 2010 | PTX | Hydrogel | 9L cells – female Fischer 344 rats | 0 | 100000 |
RP | 13 | N/A | 31 | 138% | 38 |
| 2009 | TPT and GD | Liposomes | U87MG cells – male nude rats | 2 doses: 5, 8 | 500000 |
N/A | 20 | N/A | 33 | 65% | 48 |
| 2008 | DOX | Liposomes | U251MG cells – male nude (rnu/rnu, homozygous) rats | 7 | 500000 |
N/A | N/A | 42.7 | 64.5c | 51%d | 49 |
| 2008 | DOX | Liposomes | U87MG cells – male Fischer 344/NJc1-rnu/rnu (nude) rats | 7 | 500000 |
N/A | N/A | 15.4 | 17.0c | 10%d | 49 |
| Year | Drug used | Delivery vehicle | Cancer model | Treatment time (days) post-grafting | Number of implanted cells | Drug release patterna | MS no treatment (days) | MS vehicle alone (days) | MS Local free drug delivery (days) | MS Local delivery via DDS (days) | % Change MS (local free drug vs. control)b | % Change MS (local DDS vs. control)b | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abbreviations: MS: median survival time; DDS: drug delivery system; Ref.: reference; MIT: mitoxantrone; TMZ: temozolomide; PTX: paclitaxel; DTX: docetaxel; SQ-Gem: squalenoyl-gemcitabine; DOX: doxorubicin; DI: dithiazanine iodide; BCNU: carmustine; 5-FU: 5-fluorouracil; RP: referred to a previous protocol.a The drug release pattern was defined from the results of in vitro drug release studies. The prolonged release was defined as when the DDS released more than 10% of the loaded drugs after the first two days. Studies that did not have drug release data after day two, or the cumulative drug release from day two to the end of the experiment did not exceed 10%, were regarded as rapid release.b No treatment group was chosen as the negative control group to calculate the percentage change of MS. If the study did not have a no treatment group, the empty vehicle group was used instead.c The MS data was measured and calculated from the Kaplan–Meier survival curves (not directly stated in the article).d The calculation of the percentage change in the MS data used at least one MS data not directly stated in the article. | |||||||||||||
| 2021 | Chlorogenic acid | Gel | C6 cells – male Kunming mice | 2 | 600000 |
Prolonged | N/A | 14.5 | 15 | 18.5 | 3% | 28% | 62 |
| 2021 | MIT | Nanoparticles | Luci+ GL261R132H cells – unspecified gender C57BL/6J mice | 12 | 150000 |
Rapid | 23.0c | N/A | 36.0c | 97.0c | 57%d | 322%d | 53 |
| 2017 | TMZ | Liposomes | CNS-1 cells – male Lewis rats | 5 | 500000 |
N/A | N/A | 11.0c | 15.8 | 19.2 | 44%d | 75%d | 55 |
| 2017 | TMZ | Hydrogel | GBM001 cells – unspecified gender nu/nu mice | 7 | 20000 |
N/A | N/A | 20 | 28 | 38 | 40% | 90% | 57 |
| 2017 | PTX | Gel | C6 cells – male Balb/c mice | 3 | 500000 |
Prolonged | N/A | 15.5 | 18 | 26.5 | 16% | 71% | 56 |
| 2016 | DTX | Cloudy suspension | C6 cells – male Sprague-Dawley rats | 7 | 1000000 |
Rapid | N/A | 23.8 | 33.4 | 70.2 | 40% | 195% | 58 |
| 2016 | SN-38 | Micelles | 9L cells – male Fischer 344 rats | 5 | 10000 |
N/A | N/A | 29 | 28 | 42 | -3% | 45% | 44 |
| 2016 | SQ-Gem | Nanoparticles | RG2 cells – male Fischer 344 rats | 4 | 250000 |
N/A | N/A | 12 | 13.0c | 19.0c | 8%d | 58%d | 59 |
| 2016 | Cisplatin | Polymeric drug | F98EGFR cells – unspecified gender Fischer rats | 14 | 1000 | N/A | 21 | N/A | 76 | 19 | 262% | -10% | 51 |
| 2015 | PTX | Lipid nanocapsules | GL261 cells – female C57BL/6J mice | 12 | 10000 |
N/A | N/A | 28 | 28 | 34 | 0% | 21% | 50 |
| 2014 | DOX | Nanodiamonds | U251MG-Luc cells – unspecified gender NIH-RNU nude rats | 14 | RP | N/A | N/A | 28.4 | 46.6 | 64.6 | 64% | 127% | 52 |
| 2014 | DOX | Nanodiamonds | C6-Luc cells – male Fischer 344 rats | 7 | RP | N/A | N/A | 31.0c | 43.8 | 78.2 | 41%d | 152%d | 52 |
| 2013 | DI | Nanoparticles | GS5 cells – unspecified gender nude rats | 10 | 500000 |
Prolonged | 147 | N/A | 177 | >280 | 20% | 90% | 45 |
| 2013 | PTX | Nanoparticles | U87MG cells – unspecified gender nude rats | 7 | 500000 |
Prolonged | 27 | N/A | 30 | 46 | 11% | 70% | 45 |
| 2010 | Am80 | Micelles | U87MG cells – male Fischer 344 nude rats | 7 | 500000 |
RP | N/A | 14.0c | 15.0c | 15.0c | 7%d | 7%d | 54 |
| 2009 | BCNU | Nanoparticles | C6 cells – male Sprague–Dawley rats | 5 | 1000000 |
Prolonged | 12.9 | N/A | 14.7 | 22.0c | 14% | 71%d | 60 |
| 2009 | DOX | Micelles | 9L cells – male Fischer 344 rats | 7 | 500000 |
N/A | N/A | 16.9 | 19.6 | 36 | 16% | 113% | 61 |
| 2003 | 5-FU | Microspheres | F98 cells – female Fischer F344 rats | 7 | 1,000 | Prolonged | 22.7 | 23.8 | 26.5 | 34.1 | 17% | 50% | 33 |
| 1996 | 5-FU | Microspheres | C6 cells – female Sprague-Dawley rats | 7 | 200000 |
RP | 24.0c | N/A | 30.5c | 39.5c | 27%d | 65%d | 32 |
| Year | Drug used | Delivery vehicle | Cancer model | Treatment time (days) post-grafting | Number of implanted cells | Drug release patterna | MS no treatment (days) | MS vehicle alone (days) | MS Systemic delivery (days) | MS Local delivery via DDS (days) | % Change MS (systemic vs. control)b | % Change MS (local DDS vs. control)b | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abbreviations: MS: median survival time; DDS: drug delivery system; Ref.: reference; TMZ: temozolomide; GemC12: lauroyl-gemcitabine; Fc-diOH: ferrociphenol; RP: referred to a previous protocol.a The drug release pattern was defined from the results of in vitro drug release studies. The prolonged release was defined as when the DDS released more than 10% of loaded drugs after the first two days. Studies that did not have drug release data after day two, or the cumulative drug release from day two to the end of the experiment did not exceed 10%, were regarded as rapid release.b No treatment group was chosen as the negative control group to calculate the percentage change of MS. If the study did not have a no treatment group, the empty vehicle group was used instead.c The MS data was measured and calculated from the Kaplan–Meier survival curves (not directly stated in the article).d The calculation of the percentage change in the MS data used at least one MS data not directly stated in the article. | |||||||||||||
| 2018 | TMZ | Liposomes | U87MG cells – male nu/nu mice | 2 doses: 10, 17 | 100000 |
Prolonged | 25 | N/A | 28.5 | 50 | 14% | 100% | 64 |
| 2017 | GemC12 | Hydrogel | U87MG cells – female NMRI nude mice | 15 | 30000 |
RP | 24 | 28 | 36 | 49 | 50% | 104% | 65 |
| 2013 | Irinotecan | Liposomes | GBM43 cells – female athymic mice | 2 doses: 5, 8 | 300000 |
N/A | 19.0c | N/A | 27.0c | 55.0c | 42%d | 189%d | 66 |
| 2013 | Irinotecan | Liposomes | SF7796 cells – female athymic mice | 20 | 300000 |
N/A | 41.0c | N/A | 48.0c | 58.5c | 17%d | 43%d | 66 |
| 2012 | Fc-diOH | Lipid nanocapsules | 9L cells – female Fischer F344 rats | 6 | 100000 |
N/A | 25 | N/A | 26.5c | 11 | 6%d | -56% | 68 |
| 2011 | TMZ | Microspheres | C6 cells – male Sprague–Dawley rats | 6 | 2000000 |
RP | 19.5 | N/A | 27 | 46.5 | 38% | 138% | 67 |
At this point, the data was split into three subcategories as outlined via the research questions below. While all the studies that met the inclusion criteria are described in the tables, a general discussion of selected papers was carried out to answer the subcategory questions. These are then followed by a meta-analysis of the data and a general discussion on the state of current research in this field.
3.2 Can the MS be improved by administering a DDS directly to the tumor tissue?
All the included studies had a no treatment or vehicle alone negative control group, but 19 of 44 studies had no comparison with local injection of the free drug or systemic administrations. The antitumor effect in these 19 studies, where the DDS was directly injected into the tumor, was variable (Table 1). Five studies prolonged the MS by more than 100% compared to the no treatment or vehicle control group,34–38 whilst others barely showed an increase. In addition, two studies did not reach a significant difference between local DDS treatment group and control group.39Local drug delivery widens the drug choice for the treatment of brain tumors, allowing drug repurposing of drugs that have never been exploited for this therapeutic purpose, but needs to be rationally conceived to effectively result in improved therapeutic outcome and clinical translation. For example, delivering drugs via nano-sized DDSs can increase the therapeutic efficacy and safety of active molecules for GBM.
Nanomedicines, which are commonly used for cancer diagnosis, therapeutic and monitoring, can target and reach tumor tissues by passive targeting (enhanced permeability and retention effect) or active targeting (via ligand receptor or antigen–antibody mechanisms). Singleton et al. designed a water-soluble poloxamer 407 micelle formulation loaded with the pan-histone deacetylase inhibitor Panobinostat.35 The results showed that F98 bearing Fischer344 rats treated with the micelles by CED significantly prolonged the MS compared to untreated rats, and all treated animals survived until the endpoint of the experiment (60 days). This study demonstrated a novel method of delivering a poorly water-soluble drug to brain tumor in a syngeneic infiltrative rat GBM model. In addition, the results of the in vivo toxicity study showed no adverse response after acute striatal infusion of unloaded micelles in healthy rats, showing the safety of this DDS.
Cancer theranostics represents a combined therapeutic and diagnosis approach that can be achieved using nanomedicines, to reduce delays in treatment and ease the subsequent treatment after diagnosis. Bernal et al. prepared a versatile nanoparticle formulation loaded with TMZ.40 This multifunctional platform contained superparamagnetic iron oxide in the shell, which allowed the nanoparticles to be imaged in vivo by magnetic resonance imaging (MRI), and the surface of the nanoparticles was tagged with a fluorescent agent. Even though theranostic agents are often tested following systemic administration, the authors specifically conceived their system for a local administration (<100 nm diameter NPs with negative surface charge, to maximize convection through the extracellular space of brain parenchyma). Then, they exploited the theranostic properties of their nanomedicine to visualize by MRI their distribution in the brain following CED administration distant to the injection site. Moreover, the results of their antitumor efficacy study in the U87 MG xenograft model showed that nanoparticles significantly prolonged the MS of mice by 47% compared to untreated group.
Stephen et al. designed a multifunctional magnetic nanoparticle to deliver O6-benzylguanine by CED thus improving its biodistribution and efficacy.36 This DDS consisted of an iron oxide core which could be used to image the nanoparticles by MRI and evaluate the brain diffusion via CED. The chitosan shell of the nanoparticle contained sulfhydryl groups which were sensitive to a redox environment for localized drug delivery within the cells. The chlorotoxin peptide was linked to the PEG surface for active targeting of GBM cells via MMP2 and Annexin A2 binding. Following CED administration, this DDS produced an excellent volume of distribution within the brain. The results of the antitumor efficacy study in GBM6-Luc xenograft model showed that the mice treated with this DDS combined with oral TMZ significantly prolonged the MS compared to the untreated mice (p < 0.001). Of note, in this study, the author defined the first treatment day as day 0, which was 26 days after tumor inoculation, therefore effecting the percentage change in MS. If the tumor inoculation day was defined as day 0 just as the other studies, the MS would have been 29 days (control group) and 35 days (treatment group), meaning that the percentage change in MS would have been 21%.
In another study, the BT4Ca glioma model was used to evaluate the antitumor efficacy of doxorubicin (DOX) microspheres and irinotecan microspheres administered intratumorally.39 The results showed that the MS was only prolonged by 5% and 11% respectively compared to the no treatment group, and both treatment groups did not reach a statistically significant difference (p = 0.85, 0.33, respectively). The results of another study in this article showed that rats treated with unloaded microspheres or saline boosted the tumor growth.39 The authors speculated that this might have been due to microsphere injection occurring too soon (three days) after tumor inoculation, though clearly more research is needed for better understanding of this. This negative result shows how the time of treatment impacts therapeutic benefit in GBM preclinical orthotopic models. Indeed, following grafting of cells in the brain, it is important to wait for them to form a tumor mass before treating it intratumorally. This will avoid further changes in the tissue architecture and the spreading of tumor cells from the injection site when administering liquids by CED, which would result in more infiltrated tumors and faster tumor growth.
Kikuchi et al. used two different xenograft tumor models to evaluate the antitumor efficacy of DOX liposomes.49 For the U251 MG tumor model, nude rats treated with DOX liposomes had a significantly prolonged MS by 51%. However, for the U87 MG tumor model, nude rats that received DOX liposomes showed an improvement of MS by only 10%, which still had a significant difference (p = 0.016) but minimal therapeutic benefit. This difference in efficacies displayed using these two tumor models might be due – among other factors – to the difference in tumor size at the time of treatment, life span of animals within each model or the different cellular sensitivity to DOX in these two cell lines. The MS of untreated rats inoculated with U251 MG cells was 42.7 days while that of rats inoculated with U87 MG cells was just 15.4 days. In this experimental set-up the life span of the U87 MG model was rather short, perhaps limiting the evaluation of therapeutic efficacy in this experimental setting.
3.3 Does using a DDS improve the efficacy of a locally administered drug?
Similarly to the studies outlined in Table 1, a further nineteen studies analyzed direct injection of a DDS into a tumor model, but this time with the addition of a locally injected free drug comparison group. Separation of these studies (shown in Table 2) allows us to analyze whether the DDS confers any therapeutic benefit compared to the same drug administered without a delivery system. However, first it should be noted that the percentage change in MS between local DDS treatment group and the control group was highly variable, confirming the findings from Table 1.For 89% of the studies, the animals treated locally with a DDS showed an improvement in MS compared to animals treated locally with free drug by the same administration approach. Injecting free drugs intratumorally did not prolong the MS in two studies compared to control group.44,50 By contrast, in a few studies, local free drug administration prolonged the MS greatly.51–53 However, except for two studies,51,54 local DDS treatment was more effective than local free drug treatment.32,33,44,45,50,52,53,55–62
Zhou et al. designed a brain-penetrating PLGA nanoparticle DDS to overcome the infiltrative and heterogeneous nature of GBM and chemotherapy resistance.45 The surface of the nanoparticles could be modified by [18F]NPB4, which allowed the DDS to be tracked by PET imaging. The antitumor efficacy of paclitaxel (PTX)-loaded brain-penetrating nanoparticles was evaluated in the U87 MG model. The results showed that the MS of rats treated with PTX-loaded nanoparticles was significantly longer than the one of animals treated with free PTX or without treatment. However, the authors were aware of the limitations of the model, including the fact that the U87 MG cells are not infiltrative and therefore do not properly mimic human GBMs. Hence, the GS5 cell line – a well-characterized brain cancer stem cell (BCSC) line – which could recapitulate the histopathology of human GBM, was used in subsequent studies. Around 2000 FDA approved compounds were screened to identify drugs which could inhibit the growth of GS5 cells by the thiazolyl blue tetrazolium bromide (MTT) assay. In vitro analysis showed that dithiazanine iodide, an anti-helminthic cyanine dye, effectively inhibited the proliferation of GS5 cells. The authors next evaluated the antitumor efficacy of dithiazanine iodide loaded brain-penetrating nanoparticles in vivo. Rats treated with dithiazanine iodide loaded nanoparticles by CED significantly prolonged the MS compared to untreated rats and rats treated with the free drug by CED.
Chen et al. developed an injectable phospholipid-based gel to deliver PTX locally.56 The viscosity of drug-loaded gel was less than 100cp, which was suitable for injection (for injection, the viscosity should be less than 300cp).63 The gel was added into the dialysis bag and incubated in the artificial cerebrospinal fluid to evaluate the drug release profile in vitro. The results showed that PTX release could be sustained over 30 days. The in vivo efficacy study showed that C6 bearing mice treated with PTX gel significantly prolonged the MS compared to mice treated with saline (p < 0.01) and mice treated with local free PTX (p < 0.05). On the other hand, a different study showed a totally opposite result. Barth et al. used the fifth-generation polyamidoamine dendrimer to conjugate cisplatin and cetuximab.51 Compared with the control group, local free cisplatin prolonged the MS of rats by 262%, while the DDS slightly reduced the MS by 10% in the F98EGFR model. The in vitro results also showed that the DDS were devoid of cytotoxicity perhaps indicating that the cisplatin failed to be released from the bioconjugates.
In summary, a wide range of delivery systems have been analyzed in a variety of rodent models of GBM. It is therefore unsurprising that a broad range of outcomes has been observed, leading us to undertake a meta-analysis of the data as described later on.
3.4 Can local drug administration give higher efficacy than systemic delivery?
Of all the eligible studies included in this review, only six of them compared systemic vs. intratumoral administration of the same drug. In all these studies, outlined in Table 3, the systemic treatment group improved the MS by less than 50% compared to untreated controls, while animals treated with local DDS showed a prolonged MS of more than 100% in four of six studies.64–67 Except for one study,68 local DDSs showed a better antitumor effect than the systemic administration of the same drug.Chen et al. compared the antitumor efficacy of irinotecan liposomes by different routes of delivery.66 Two different types of GBM xenograft models which use cells derived from primary human GBM tissues were used to evaluate the in vivo antitumor efficacy of this DDS. GBM43 cells were shown to be radioresistant and SF7796 cells were derived from a recurrent tumor. The results showed that local delivery of irinotecan liposomes by CED significantly enhanced the antitumor efficacy in both models compared to systemic administration by tail vein (p < 0.001 in GBM43 model and p = 0.048 in SF7796 model). Laine et al. developed an active targeting strategy to deliver ferrociphenol.68 NFL-TBS.40–63 peptide (cell-penetrating peptide) was inserted onto the surface of lipid nanocapsules. The results of the in vivo study in the 9L model showed that the intratumoral delivery by CED significantly reduced the MS, while animals treated with intracarotid administration had increased MS. While this study showed the beneficial effect of active targeting in intracarotid treatment, it also showed that CED administration of ferrociphenol lipid nanocapsules was not safe in this experimental setting leading to severe side effects.
As stated earlier, the current standard of care therapy for GBM is surgical resection followed by the Stupp protocol. TMZ is an alkylating agent able to pass the BBB following oral administration, so it is a good drug candidate to assess whether local delivery can outperform systemic delivery. Lin et al. delivered TMZ-loaded liposomes directly into the tumor by CED.64 The U87 MG-bearing mice treated twice with local TMZ liposomes had a significantly longer MS compared with the control group, while mice treated three times with oral TMZ had no significant improvement in survival. Zhang et al. implanted TMZ poly (D,L-lactide-co-glycolide) (PLGA) microparticles on the surface of the tumor tissue to evaluate the antitumor effect in the C6 glioma model.67 Rats treated with TMZ microparticles locally had a significantly longer MS than rats treated with TMZ orally (p = 0.002). These studies demonstrate that local delivery can enhance antitumor effect and reduce systemic toxicities by decreasing the TMZ concentration in circulating blood.
3.5 meta-Analyses
meta-Analyses of the included studies were carried out to determine the overall effect size for the questions posed previously. Two eligible studies were excluded from the meta-analysis as the survival time of each animal was not available.50,51 A meta-analysis which compared locally administrated injectable DDS with negative control groups across 42 studies was conducted firstly to analyze whether a DDS directly administrated into the tumor tissue could improve the MS via the hazard ratio method.29 For the purpose of this study, the hazard ratio-based “time to event” methodology was used, which gives the MS ratio between the intervention of the local DDS vs. no DDS treatment controls. The diamond in the forest plot (Fig. 2) showed that the total MS ratio (indicated as hazard ratio) was 1.68 (95% confidence interval (CI), 1.63–1.73), which reached a significant difference (p < 0.00001). This result indicated that local administration of injectable DDS was beneficial for the survival time of animals. The heterogeneity in this meta-analysis was at a high degree (I2 = 100%). The reason for this is probably because studies used different model drugs, delivery vehicles, tumor cells, animal models and administration methods. According to Tables 1–3, time of tumor incubation prior to the treatment varied from the same day of implantation to 26 days after implantation; the number of implanted cells varied from 1000 to 2000000. These experiment settings obviously affected the tumor size when starting the treatment, and the survival time of animals without treatment or animals in treatment groups, which would further affect the value of MS ratio. Only 16 of the 44 studies had in vitro drug release results included in the study. Some studies showed that a DDS with a rapid release pattern could prolong the MS of animals treated with the DDS by more than 100% compared with that in the control group,36,53,58 while a sustained-release DDS only slightly increased the MS by less than 30% in some studies.45,62 It might indicate that sustained-release pattern does not necessarily mean a better antitumor efficacy.
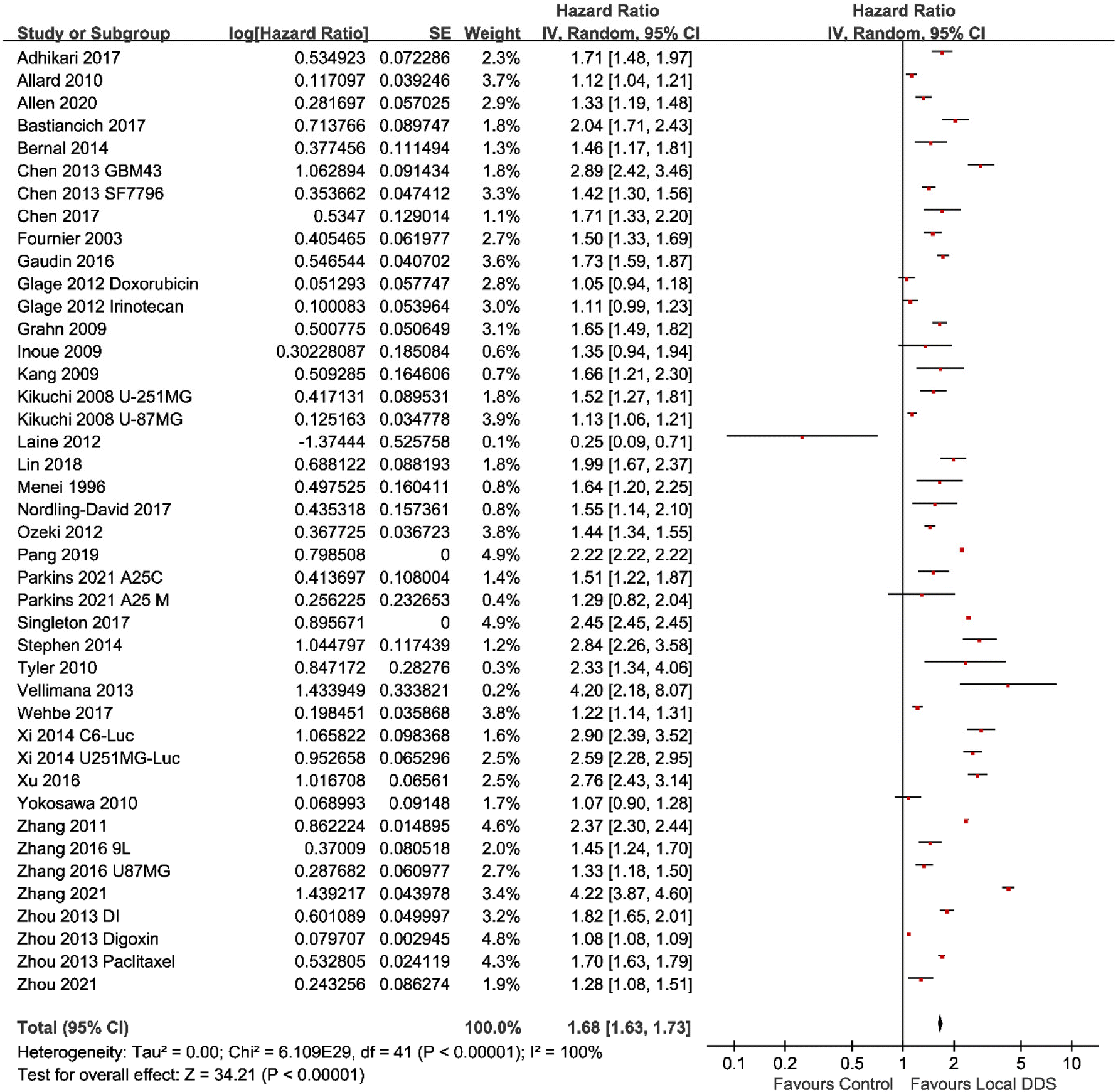 | ||
| Fig. 2 A forest plot of the hazard ratio showing the antitumor efficacy of locally administered injectable DDSs compared with control groups. The table showed the median survival ratio (hazard ratio), the standard error (SE) and 95% confidence intervals (CI) of each study. | ||
Another meta-analysis across seventeen studies was conducted to analyze whether using a DDS improved the efficacy of a locally administered drug. In this case, as a direct comparison was carried out between two therapeutic interventions, the mean difference method was used. Despite a large amount of heterogeneity (I2 = 75%), Fig. 3, shows that local administration of injectable DDS could enhance the antitumor efficacy significantly (mean difference = 0.17; 95% CI, 0.13–0.21; p < 0.00001) compared with locally administrated free drugs.
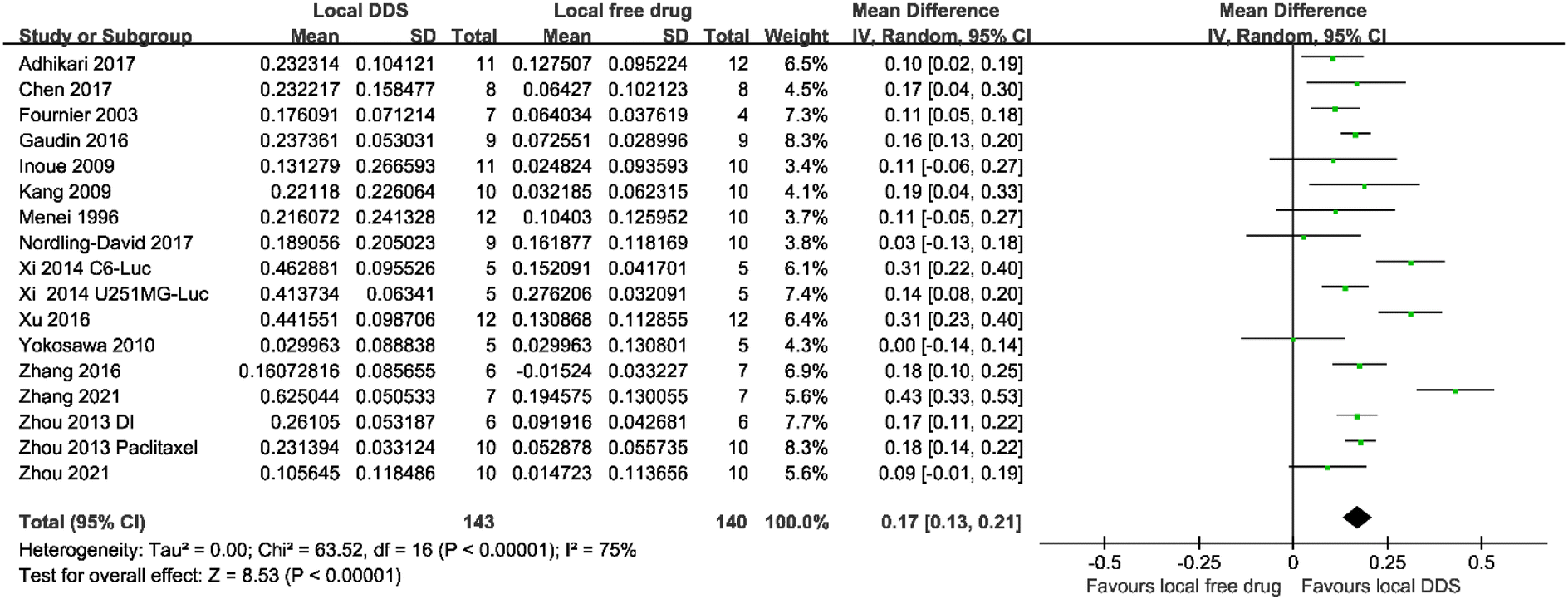 | ||
| Fig. 3 A forest plot showing the antitumor efficacy of locally administered injectable DDSs compared with locally injected free drug. The table showed the log-transformed data of the median survival ratio (Mean), the standard deviation (SD) and the number of animals in each group (Total). The mean difference showed the 95% confidence interval of each study. | ||
Six of all the eligible studies had a systemic treatment group using the same drug as in the local administration. A meta-analysis was conducted across these studies to compare the antitumor efficacy between systemic treatment and local treatment. As shown in Fig. 4, animals locally treated with DDS had a significantly prolonged MS compared with animals systemically treated with either the free drug or the DDS (mean difference = 0.17; 95% CI, 0.07–0.26; p = 0.0007).
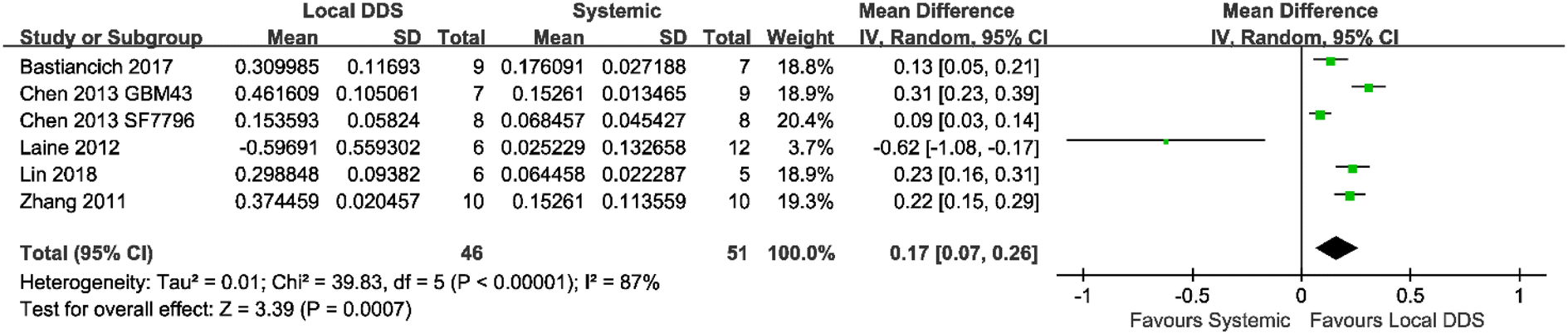 | ||
| Fig. 4 A forest plot showing the antitumor efficacy of locally administered injectable DDSs compared with systemic administration of either the free drug or the DDS. The table showed the log-transformed data of the median survival ratio (Mean), the standard deviation (SD) and the number of animals in each group (Total). Mean difference showed the 95% confidence interval of each study. | ||
3.6 Quality assessment of the included articles
Study quality was assessed by the 12-point checklist in all 36 included articles. The ESI Table S1† shows the full breakdown score of the quality assessment. Except one article, all articles were peer-reviewed publications. None of them mentioned how to calculate the sample size. Most of articles stated the accurate site and the number of inoculated tumor cells for the establishment of cancer model. Only three articles reported the number of animals in which the tumor did not grow and two articles used a blinded assessment for in vivo studies. For greater research transparency and reproducibility, we think that the points on this checklist are important for researchers of future studies to consider. In particular the blinded assessment of outcomes is simple way to remove biases in the study.4. Conclusions
Overall, the results of this study show that local drug delivery within the tumor mass is a promising approach to overcome the intrinsic GBM therapeutic challenges. Bypassing the BBB allows researchers to increase the number of active molecules that can be explored to defeat this devastating tumor and achieve high local drug concentrations thus maximizing their therapeutic effect. While the number of articles describing the use of local treatments for GBM in preclinical models has increased exponentially since Gliadel®'s approval, the clinical trials have been limited leading to no new local treatments approved for GBM since 1997.23 Indeed, the development of local treatments for GBM is challenging, especially for non-resectable tumors where no cavity exists for implantation. Several parameters need to be considered to properly achieve the therapeutic goal. These include a DDS design and formulation development which considers the GBM physiopathology and unique microenvironment, adapted properties for a local application in the brain and a careful characterization of its in vivo safety and efficacy using appropriate preclinical models.69The goal of this systematic review and meta-analysis was to evaluate the therapeutic efficacy of DDS intratumorally delivered via injection in unresected GBM preclinical models. In addition, we asked whether the DDS itself plays a role in outcomes by comparison to locally injected free drug. The meta-analyses showed that whilst there was a high degree of variability across studies, local injection of delivery systems resulted in an improvement of MS compared to control groups and to local injection of free drugs. This study also showed that local drug administration via a DDS could outperform systematic administration of free drugs. These results indicate that local delivery still holds promise for this scope, but efforts should be made to standardize the methods used to test such systems to compare their efficacy.
Indeed, the therapeutic effect observed following intratumoral administration can be subject to many variables and biases. For example, the GBM model chosen for the study can directly impact the outcome as less infiltrative cell lines are likely to respond better to local treatment compared to more aggressive models. Also, the volume injected, the injection rate, the drug dose, and the time of administration – which vary between studies – can make it very difficult to compare data between different groups (or between different investigators working in the same group). Moreover, the tumor size at time of treatment is not always measured and/or reported so it is difficult to replicate the studies and understand the real impact of the treatment in the long-term.
Most of the papers reported in this review used xenograft models using established GBM cell lines to test the efficacy of local treatments. While these models lead to reproducible and reliable experiments with high engraftment and growth rates – and some of them have shown to still mimic the true biological nature of GBM70 – cells are injected in the brain of immunodeficient animals. In recent years, the cross talk between glial cells and immune cells (brain resident macrophages as well as immune cells recruited from the periphery) has been demonstrated to impact tumor cell behavior. Testing local treatments on several models could help our understanding of the real therapeutic efficacy and potential long-term impact, thus partially filling the preclinical-clinical gap.
Whilst the prognosis for GBM is universally poor, there is no local intervention for non-resectable tumors. Stiff wafers such as Gliadel® are not only unsuitable but suffer from rapid release of a monotherapy. The studies in this review show promise in this regard (soft hydrogels and injectable formulations) though it is likely that delivery of multiple drugs acting via differing mechanisms would be beneficial. Key considerations moving forward will be the evaluation of drug penetration to residual GBM cells proliferating deeper into the brain parenchyma,71 obtaining optimal drug release profiles in vivo,23 and multifaceted modes of therapeutic action. However, to date the meta-analysis conducted herein shows clear rationale for the continued development of injectable local drug delivery devices for GBM therapeutics.
Author contributions
Yu Wang: methodology, formal analysis, data curation, writing – original draft; Chiara Bastiancich: writing – original draft, writing – review & editing; Ben Newland: conceptualization, methodology, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Yu Wang is grateful for funding from the China Scholarship Council. Chiara Bastiancich is supported by Fondation ARC pour la recherche sur le cancer (grants n. PDF20190509176 and ARCPJA12020060002222) and Cancéropôle Provence Alpes Côte d'Azur.References
- A. Shergalis, A. Bankhead 3rd, U. Luesakul, N. Muangsin and N. Neamati, Pharmacol. Rev., 2018, 70, 412–445 CrossRef CAS PubMed.
- T. J. Brown, M. C. Brennan, M. Li, E. W. Church, N. J. Brandmeir, K. L. Rakszawski, A. S. Patel, E. B. Rizk, D. Suki, R. Sawaya and M. Glantz, JAMA Oncol., 2016, 2, 1460–1469 CrossRef PubMed.
- T. Yamahara, Y. Numa, T. Oishi, T. Kawaguchi, T. Seno, A. Asai and K. Kawamoto, Brain Tumor Pathol., 2010, 27, 81–87 CrossRef PubMed.
- K. R. Yabroff, L. Harlan, C. Zeruto, J. Abrams and B. Mann, Neuro-Oncology, 2012, 14, 351–359 CrossRef PubMed.
- S. M. Chang, I. F. Parney, W. Huang, F. A. Anderson Jr., A. L. Asher, M. Bernstein, K. O. Lillehei, H. Brem, M. S. Berger, E. R. Laws and the Glioma Outcomes Project Investigators, J. Am. Med. Assoc., 2005, 293, 557–564 CrossRef CAS PubMed.
- M. Weller, M. van den Bent, M. Preusser, E. Le Rhun, J. C. Tonn, G. Minniti, M. Bendszus, C. Balana, O. Chinot, L. Dirven, P. French, M. E. Hegi, A. S. Jakola, M. Platten, P. Roth, R. Ruda, S. Short, M. Smits, M. J. B. Taphoorn, A. von Deimling, M. Westphal, R. Soffietti, G. Reifenberger and W. Wick, Nat. Rev. Clin Oncol., 2021, 18, 170–186 CrossRef PubMed.
- H. Athanassiou, M. Synodinou, E. Maragoudakis, M. Paraskevaidis, C. Verigos, D. Misailidou, D. Antonadou, G. Saris, K. Beroukas and P. Karageorgis, J. Clin. Oncol., 2005, 23, 2372–2377 CrossRef CAS PubMed.
- M. E. Hegi, A. C. Diserens, T. Gorlia, M. F. Hamou, N. de Tribolet, M. Weller, J. M. Kros, J. A. Hainfellner, W. Mason, L. Mariani, J. E. Bromberg, P. Hau, R. O. Mirimanoff, J. G. Cairncross, R. C. Janzer and R. Stupp, N. Engl. J. Med., 2005, 352, 997–1003 CrossRef CAS PubMed.
- R. Stupp, W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M. J. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. G. Cairncross, E. Eisenhauer and R. O. Mirimanoff, N. Engl. J. Med., 2005, 352, 987–996 CrossRef CAS PubMed.
- M. R. Gilbert, M. Wang, K. D. Aldape, R. Stupp, M. E. Hegi, K. A. Jaeckle, T. S. Armstrong, J. S. Wefel, M. Won, D. T. Blumenthal, A. Mahajan, C. J. Schultz, S. Erridge, B. Baumert, K. I. Hopkins, T. Tzuk-Shina, P. D. Brown, A. Chakravarti, W. J. Curran Jr. and M. P. Mehta, J. Clin. Oncol., 2013, 31, 4085–4091 CrossRef CAS.
- A. Brodbelt, D. Greenberg, T. Winters, M. Williams, S. Vernon, V. P. Collins and National Cancer Information Network Brain Tumour Group, Eur. J. Cancer, 2015, 51, 533–542 CrossRef PubMed.
- A. Omuro and L. M. DeAngelis, J. Am. Med. Assoc., 2013, 310, 1842–1850 CrossRef CAS PubMed.
- M. Baumann, W. DuBois, A. Pu, J. Freeman and H. D. Suit, Int. J. Radiat. Oncol., Biol., Phys., 1992, 23, 803–809 CrossRef CAS PubMed.
- B. F. Jordan and P. Sonveaux, Front. Pharmacol., 2012, 3, 94 Search PubMed.
- M. Esteller, J. Garcia-Foncillas, E. Andion, S. N. Goodman, O. F. Hidalgo, V. Vanaclocha, S. B. Baylin and J. G. Herman, N. Engl. J. Med., 2000, 343, 1350–1354 CrossRef CAS.
- M. D. Prados, S. A. Byron, N. L. Tran, J. J. Phillips, A. M. Molinaro, K. L. Ligon, P. Y. Wen, J. G. Kuhn, I. K. Mellinghoff, J. F. de Groot, H. Colman, T. F. Cloughesy, S. M. Chang, T. C. Ryken, W. D. Tembe, J. A. Kiefer, M. E. Berens, D. W. Craig, J. D. Carpten and J. M. Trent, Neuro-Oncology, 2015, 17, 1051–1063 CrossRef CAS PubMed.
- D. Fabian, M. D. P. Guillermo Prieto Eibl, I. Alnahhas, N. Sebastian, P. Giglio, V. Puduvalli, J. Gonzalez and J. D. Palmer, Cancers, 2019, 11, 174 CrossRef CAS PubMed.
- R. Stupp, S. Taillibert, A. Kanner, W. Read, D. Steinberg, B. Lhermitte, S. Toms, A. Idbaih, M. S. Ahluwalia, K. Fink, F. Di Meco, F. Lieberman, J. J. Zhu, G. Stragliotto, D. Tran, S. Brem, A. Hottinger, E. D. Kirson, G. Lavy-Shahaf, U. Weinberg, C. Y. Kim, S. H. Paek, G. Nicholas, J. Bruna, H. Hirte, M. Weller, Y. Palti, M. E. Hegi and Z. Ram, J. Am. Med. Assoc., 2017, 318, 2306–2316 CrossRef CAS PubMed.
- S. Ryu, J. M. Buatti, A. Morris, S. N. Kalkanis, T. C. Ryken, J. J. Olson and A. C. J. G. Committee, J. Neurooncol., 2014, 118, 489–499 CrossRef CAS PubMed.
- W. Wick, T. Gorlia, M. Bendszus, M. Taphoorn, F. Sahm, I. Harting, A. A. Brandes, W. Taal, J. Domont, A. Idbaih, M. Campone, P. M. Clement, R. Stupp, M. Fabbro, E. Le Rhun, F. Dubois, M. Weller, A. von Deimling, V. Golfinopoulos, J. C. Bromberg, M. Platten, M. Klein and M. J. van den Bent, N. Engl. J. Med., 2017, 377, 1954–1963 CrossRef CAS PubMed.
- T. A. Juratli, G. Schackert and D. Krex, Pharmacol. Ther., 2013, 139, 341–358 CrossRef CAS PubMed.
- L. S. Ashby, K. A. Smith and B. Stea, World J. Surg. Oncol., 2016, 14, 225 CrossRef.
- A. Tabet, M. P. Jensen, C. C. Parkins, P. G. Patil, C. Watts and O. A. Scherman, Adv. Healthc. Mater., 2019, 8, e1801391 CrossRef PubMed.
- M. Westphal, D. C. Hilt, E. Bortey, P. Delavault, R. Olivares, P. C. Warnke, I. R. Whittle, J. Jaaskelainen and Z. Ram, Neuro-Oncology, 2003, 5, 79–88 CrossRef CAS PubMed.
- P. C. McGovern, E. Lautenbach, P. J. Brennan, R. A. Lustig and N. O. Fishman, Clin. Infect. Dis, 2003, 36, 759–765 CrossRef CAS PubMed.
- D. A. Bota, A. Desjardins, J. A. Quinn, M. L. Affronti and H. S. Friedman, Ther. Clin. Risk Manage., 2007, 3, 707–715 CAS.
- C. Bastiancich, E. Bozzato, I. Henley and B. Newland, J. Controlled Release, 2021, 337, 296–305 CrossRef CAS PubMed.
- T. C. Hirst, H. M. Vesterinen, E. S. Sena, K. J. Egan, M. R. Macleod and I. R. Whittle, Br. J. Cancer, 2013, 108, 64–71 CrossRef CAS PubMed.
- J. F. Tierney, L. A. Stewart, D. Ghersi, S. Burdett and M. R. Sydes, Trials, 2007, 8, 16 CrossRef PubMed.
- R. J. Simes, Stat. Med., 1987, 6, 11–29 CrossRef CAS PubMed.
- J. P. T. Higgins, T. Li and J. J. Deeks, Choosing effect measures and computing estimates of effect, in Cochrane Handbook for Systematic Reviews of Interventions version 6.3, ed. J. P. T. Higgins, J. Thomas, J. Chandler, M. Cumpston, T. Li, M. J. Page and V. A. Welch, Cochrane, 2022, ch. 6 (updated February 2022) Search PubMed.
- P. Menei, M. Boisdron-Celle, A. Croue, G. Guy and J. P. Benoit, Neurosurgery, 1996, 39, 117–123 CrossRef CAS PubMed ; discussion 123–114.
- E. Fournier, C. Passirani, C. Montero-Menei, N. Colin, P. Breton, S. Sagodira, P. Menei and J. P. Benoit, Cancer, 2003, 97, 2822–2829 CrossRef CAS PubMed.
- H. H. Pang, P. Y. Chen, K. C. Wei, C. W. Huang, Y. L. Shiue, C. Y. Huang and H. W. Yang, Theranostics, 2019, 9, 1752–1763 CrossRef CAS PubMed.
- W. G. Singleton, A. M. Collins, A. S. Bienemann, C. L. Killick-Cole, H. R. Haynes, D. J. Asby, C. P. Butts, M. J. Wyatt, N. U. Barua and S. S. Gill, Int. J. Nanomed., 2017, 12, 1385–1399 CrossRef CAS PubMed.
- Z. R. Stephen, F. M. Kievit, O. Veiseh, P. A. Chiarelli, C. Fang, K. Wang, S. J. Hatzinger, R. G. Ellenbogen, J. R. Silber and M. Q. Zhang, ACS Nano, 2014, 8, 10383–10395 CrossRef CAS PubMed.
- A. K. Vellimana, V. R. Recinos, L. Hwang, K. D. Fowers, K. W. Li, Y. G. Zhang, S. Okonma, C. G. Eberhart, H. Brem and B. M. Tyler, J. Neuro-Oncol., 2013, 111, 229–236 CrossRef CAS PubMed.
- B. Tyler, K. D. Fowers, K. W. Li, V. R. Recinos, J. M. Caplan, A. Hdeib, R. Grossman, L. Basaldella, K. Bekelis, G. Pradilla, F. Legnani and H. Brem, J. Neurosurg., 2010, 113, 210–217 CAS.
- S. Glage, A. L. Lewis, P. Mertens, S. Baltes, P. Geigle and T. Brinker, Clin. Transl. Oncol., 2012, 14, 50–59 CrossRef CAS PubMed.
- G. M. Bernal, M. J. LaRiviere, N. Mansour, P. Pytel, K. E. Cahill, D. J. Voce, S. Kang, R. Spretz, U. Welp, S. E. Noriega, L. Nunez, G. F. Larsen, R. R. Weichselbaum and B. Yamini, Nanomedicine, 2014, 10, 149–157 CrossRef CAS PubMed.
- C. C. Parkins, J. H. McAbee, L. Ruff, A. Wendler, R. Mair, R. J. Gilbertson, C. Watts and O. A. Scherman, Biomaterials, 2021, 276, 120919 CrossRef CAS PubMed.
- J. Allen, J. Wang, O. Y. Zolotarskaya, A. Sule, S. Mohammad, S. Arslan, K. J. Wynne, H. Yang and K. Valerie, J. Controlled Release, 2020, 321, 36–48 CrossRef CAS PubMed.
- M. Wehbe, M. Anantha, M. Shi, A. W. Leung, W. H. Dragowska, L. Sanche and M. B. Bally, Int. J. Nanomed., 2017, 12, 4129–4146 CrossRef PubMed.
- R. Zhang, R. Saito, Y. Mano, A. Sumiyoshi, M. Kanamori, Y. Sonoda, R. Kawashima and T. Tominaga, Drug Delivery, 2016, 23, 2780–2786 CrossRef CAS PubMed.
- J. Zhou, T. R. Patel, R. W. Sirianni, G. Strohbehn, M. Q. Zheng, N. Duong, T. Schafbauer, A. J. Huttner, Y. Huang, R. E. Carson, Y. Zhang, D. J. Sullivan Jr., J. M. Piepmeier and W. M. Saltzman, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11751–11756 CrossRef CAS PubMed.
- T. Ozeki, D. Kaneko, K. Hashizawa, Y. Imai, T. Tagami and H. Okada, Biol. Pharm. Bull., 2012, 35, 545–550 CrossRef CAS PubMed.
- E. Allard, D. Jarnet, A. Vessieres, S. Vinchon-Petit, G. Jaouen, J. P. Benoit and C. Passirani, Pharm. Reson., 2010, 27, 56–64 CrossRef CAS PubMed.
- A. Y. Grahn, K. S. Bankiewicz, M. Dugich-Djordjevic, J. R. Bringas, P. Hadaczek, G. A. Johnson, S. Eastman and M. Luz, J. Neuro-Oncol., 2009, 95, 185–197 CrossRef CAS PubMed.
- T. Kikuchi, R. Saito, S. Sugiyama, Y. Yamashita, T. Kumabe, M. Krauze, K. Bankiewicz and T. Tominaga, J. Neurosurg., 2008, 109, 867–873 CAS.
- G. Lollo, M. Vincent, G. Ullio-Gamboa, L. Lemaire, F. Franconi, D. Couez and J. P. Benoit, Int. J. Pharm., 2015, 495, 972–980 CrossRef CAS PubMed.
- R. F. Barth, G. Wu, W. H. Meisen, R. J. Nakkula, W. Yang, T. Huo, D. A. Kellough, P. Kaumaya, C. Turro, L. M. Agius and B. Kaur, OncoTargets Ther., 2016, 9, 2769–2781 CrossRef CAS PubMed.
- G. Xi, E. Robinson, B. Mania-Farnell, E. F. Vanin, K. W. Shim, T. Takao, E. V. Allender, C. S. Mayanil, M. B. Soares, D. Ho and T. Tomita, Nanomedicine, 2014, 10, 381–391 CrossRef CAS PubMed.
- J. Zhang, C. Chen, A. Li, W. Jing, P. Sun, X. Huang, Y. Liu, S. Zhang, W. Du, R. Zhang, Y. Liu, A. Gong, J. Wu and X. Jiang, Nat. Nanotechnol., 2021, 16, 538–548 CrossRef CAS PubMed.
- M. Yokosawa, Y. Sonoda, S. Sugiyama, R. Saito, Y. Yamashita, M. Nishihara, T. Satoh, T. Kumabe, M. Yokoyama and T. Tominaga, Tohoku J. Exp. Med., 2010, 221, 257–264 CrossRef CAS PubMed.
- M. M. Nordling-David, R. Yaffe, D. Guez, H. Meirow, D. Last, E. Grad, S. Salomon, S. Sharabi, Y. Levi-Kalisman, G. Golomb and Y. Mardor, J. Controlled Release, 2017, 261, 138–146 CrossRef CAS PubMed.
- T. J. Chen, T. Gong, T. Zhao, X. Liu, Y. Fu, Z. R. Zhang and T. Gong, Int. J. Pharm., 2017, 528, 127–132 CrossRef CAS PubMed.
- B. Adhikari, J. Li, M. G. Brandel, D. Futalan, J. Akers, T. Deming, C. C. Chen and B. S. Carter, J. Clin. Neurosci., 2017, 45, 288–292 CrossRef CAS PubMed.
- H. L. Xu, K. L. Mao, C. T. Lu, Z. L. Fan, J. J. Yang, J. Xu, P. P. Chen, D. L. ZhuGe, B. X. Shen, B. H. Jin, J. Xiao and Y. Z. Zhao, Biomaterials, 2016, 107, 44–60 CrossRef CAS PubMed.
- A. Gaudin, E. Song, A. R. King, J. K. Saucier-Sawyer, R. Bindra, D. Desmaele, P. Couvreur and W. M. Saltzman, Biomaterials, 2016, 105, 136–144 CrossRef CAS PubMed.
- C. Kang, X. Yuan, Y. Zhong, P. Pu, Y. Guo, A. Albadany, S. Yu, Z. Zhang, Y. Li, J. Chang and J. Sheng, Technol. Cancer Res. Treat., 2009, 8, 61–70 CrossRef CAS PubMed.
- T. Inoue, Y. Yamashita, M. Nishihara, S. Sugiyama, Y. Sonoda, T. Kumabe, M. Yokoyama and T. Tominaga, Neuro-Oncology, 2009, 11, 151–157 CrossRef CAS PubMed.
- H. Zhou, D. Chen, T. Gong, Q. He, C. Guo, P. Zhang, X. Song, J. Ruan and T. Gong, Eur. J. Pharm. Biopharm., 2021, 166, 103–110 CrossRef CAS PubMed.
- J. J. Xuan, P. Balakrishnan, D. H. Oh, W. H. Yeo, S. M. Park, C. S. Yong and H. G. Choi, Int. J. Pharm., 2010, 395, 317–323 CrossRef CAS PubMed.
- C. Y. Lin, R. J. Li, C. Y. Huang, K. C. Wei and P. Y. Chen, J. Drug Targeting, 2018, 26, 325–332 CrossRef CAS PubMed.
- C. Bastiancich, J. Bianco, K. Vanvarenberg, B. Ucakar, N. Joudiou, B. Gallez, G. Bastiat, F. Lagarce, V. Preat and F. Danhier, J. Controlled Release, 2017, 264, 45–54 CrossRef CAS PubMed.
- P. Y. Chen, T. Ozawa, D. C. Drummond, A. Kalra, J. B. Fitzgerald, D. B. Kirpotin, K. C. Wei, N. Butowski, M. D. Prados, M. S. Berger, J. R. Forsayeth, K. Bankiewicz and C. D. James, Neuro-Oncology, 2013, 15, 189–197 CrossRef CAS PubMed.
- Y. H. Zhang, H. Zhang, J. M. Liu and Z. J. Yue, Med. Oncol., 2011, 28, 901–906 CrossRef CAS PubMed.
- A. L. Laine, N. T. Huynh, A. Clavreul, J. Balzeau, J. Bejaud, A. Vessieres, J. P. Benoit, J. Eyer and C. Passirani, Eur. J. Pharm. Biopharm., 2012, 81, 690–693 CrossRef CAS PubMed.
- C. Bastiancich, A. Malfanti, V. Preat and R. Rahman, Adv. Drug Delivery Rev., 2021, 177, 113951 CrossRef CAS PubMed.
- M. Zalles and R. A. Towner, in Gliomas, ed. W. Debinski, Brisbane (AU), 2021, DOI:10.36255/exonpublications.gliomas.2021.chapter1.
- P. McCrorie, J. Rowlinson, D. J. Scurr, M. Marlow and R. Rahman, Pharmaceutics, 2022, 14, 571 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01534j |
| This journal is © The Royal Society of Chemistry 2023 |