
Recent strategies to develop pH-sensitive injectable hydrogels
Thavasyappan
Thambi
 ,
Jae Min
Jung
and
Doo Sung
Lee
*
,
Jae Min
Jung
and
Doo Sung
Lee
*
School of Chemical Engineering, Sungkyunkwan University, Suwon 16419, Republic of Korea. E-mail: dslee@skku.edu; Fax: +82-31-299-6857; Tel: +82-31-290-7393
First published on 17th January 2023
Abstract
“Smart” biomaterials that are responsive to pathological abnormalities are an appealing class of therapeutic platforms for the development of personalized medications. The development of such therapeutic platforms requires novel techniques that could precisely deliver therapeutic agents to the diseased tissues, resulting in enhanced therapeutic effects without harming normal tissues. Among various therapeutic platforms, injectable pH-responsive biomaterials are promising biomaterials that respond to the change in environmental pH. Aqueous solutions of injectable pH-responsive biomaterials exhibit a phase transition from sol-to-gel in response to environmental pH changes. The injectable pH-responsive hydrogel depot can provide spatially and temporally controlled release of various bioactive agents including chemotherapeutic drugs, peptides, and proteins. Therapeutic agents are imbibed into hydrogels by simple mixing without the use of toxic solvents and used for long-term storage or in situ injection using a syringe or catheter that could form a stable gel and acts as a controlled release depot in a minimally invasive manner. Tunable physicochemical properties of the hydrogels, such as biodegradability, ability to interact with drugs and mechanical properties, can control the release of the therapeutic agent. This review highlights the advances in the design and development of biodegradable and in situ forming injectable pH-responsive biomaterials that respond to the physiological conditions. Special attention has been paid to the development of amphoteric pH-responsive biomaterials and their utilization in biomedical applications. We also highlight key challenges and future directions of pH-responsive biomaterials in clinical translation.
 Thavasyappan Thambi | Thavasyappan Thambi is currently working as a Research Professor at the Graduate School of Biotechnology, Kyung Hee University in South Korea. He received his Ph.D. degree from Sungkyunkwan University, working on the field of microenvironment-specific polymeric nanoparticles for imaging and therapy of intractable diseases and was trained in the Department of Bioengineering, Hanyang University in South Korea. His research is focused on the development of smart hydrogels for biomedical applications. |
 Jae Min Jung | Jae Min Jung was born in Seoul, Korea, in 1988. He received his bachelor's degree (2015) in Chemical Engineering from the University of Seoul. At present, he is pursuing his Ph.D. in Chemistry Engineering at Sungkyunkwan University, South Korea. His research is focused on the development of stimuli-responsive injectable polymeric materials for biomedical applications. |
 Doo Sung Lee | Doo Sung Lee received his Ph.D. in Chemical Engineering from Korea Advanced Institute of Science and Technology. He started his professional career in Sungkyunkwan University in 1984. He became Director of Theranostic Macromolecules Research Center in 2010. He is now a Member of the Korean Academy of Science and Technology and the leader of the Fundamental Science Networking center. Also, he is a Member of the National Academy of Engineering of Korea. His research interest is stimuli-responsive polymeric nanoparticles for biomedical applications and injectable hydrogels for drug and protein delivery. |
Introduction
The term “pH-responsive biomaterials” is used to describe polymeric materials that swell or collapse in response to pH changes from physiological pH to tumor/disease-microenvironmental pH.1–3 Such pH-responsive biomaterials possess acidic or basic functional groups, which can exhibit structural changes at different pH values. Based on the revolutionary idea of poly(2-hydroxyethyl methacrylate) gels in contact lens application, the first pH-responsive biomaterial was synthesized by Professor Kopecek in 1971.4–6 The success of pH-responsive biomaterials in therapeutic applications mainly depends on the surface charge, bioactive ligands, and mechanical cues to the tissue of interest.7 Furthermore, biomaterials should be non-toxic, biocompatible, and slowly bioresorbable or excreted from the body after performing their roles.8–10 More importantly, these foreign materials should not induce local inflammation, and their hydrolysis products should not alter the local pH.11One of the early examples of biodegradable biomaterials was developed by our group in collaboration with Professor Sung Wan Kim in the 1990s.12 This biomaterial is based on urethane-linked PEO–PLLA–PEO copolymers. The free-flowing copolymer sols mixed with therapeutic agents at room temperature form a gel depot at the subcutaneous tissue of animal models. Proteins can be sustainably released from the gel for several days without inducing inflammation at the injection point. However, practical application of this biomaterial as an implantable depot is severely restricted by needle clogging issues.13 On the other hand, surgical implantation of preformed hydrogel depots often results in tissue fibrosis.14 We elegantly resolved this issue by introducing pH-responsive functional polymers in the biomaterials.13 The formed pH- and temperature-responsive copolymers not only solved the clogging issues when implanted into the animals at high concentrations, but also formed nanoparticles at low concentrations and circulated in the bloodstream and site-specifically reached disease sites such as tumors or artery occlusion.15 The introduction of pH-responsive characteristics in the copolymers allows the formation of particulate structures only subcutaneously or in deep tissues due to the change in local pH.16,17 Such pH-responsive copolymers exhibit physicochemical transformation such as swelling, deswelling, degradation and membrane disruption in response to the change in environmental pH.18–20 The conformational changes in the copolymers are attributed to the ionizable acidic or basic pendant residues present in the copolymers.21,22 In addition, the presence of cleavable or labile covalent bonds such as imine, cis-acotinyl, hydrazone, ketal, acetal and orthoesters in the copolymers contributes to the degradation or physicochemical changes.23–26 Upon exposure of copolymers to the appropriate pH and ionic strength, the ionization of functional groups in the copolymers builds up a fixed charge within the copolymers.27–29 This leads to the generation of electrostatic repulsive forces in the copolymer networks that induce pH-dependent swelling and deswelling in the porous hydrogel network as the water is either absorbed or expelled.19,30–32 Owing to these unique characteristic features, hydrogels have been used in various fields of medicine, including oncology, immunology, cardiology and wound healing. The presence of a large volume of water in the hydrogel network provides structural similarity to biological tissues as the network is typically soft with a Young's modulus of 1 kPa to 1 MPa.33–35 This gives excellent biocompatibility to the hydrogels and allows the imbibing of hydrophilic therapeutic agents.36,37
In this review, we describe general strategies used to develop injectable pH-responsive biomaterials for controlled delivery applications. We focus on the fabrication of different pH-responsive biomaterials prepared using either cationic copolymers or anionic polymers. Special attention has been given to the state-of-the-art amphoteric pH-responsive copolymers with the intention of designing strategies that hinder clinical translation and utilization in various biomedical fields. In the last section, we attempt to provide future directions and challenges in the clinical development of injectable pH-responsive biomaterials, including safety considerations, bulk production and cost control, and how these are addressed.
Classification of pH-responsive functional polymers
Among various stimuli-responsive polymers, pH-responsive polymers are an important class of functional polymers that exhibit physicochemical changes such as solubility, chain conformation and surface properties in response to changes in environmental pH.38,39 The term pH-responsive polymers indicates polymers having ionizable basic or acidic residues that can accept protons or release protons in response to changes in pH.40 These polymers usually possess acidic functional groups like carboxylic acids, sulfonic acids, and phosphates in the side chains or basic tertiary groups in the pendant or side chains.41,42 The solubility and chain conformation of pH-responsive polymers can also be modulated using solvents and electrolytes.43 In addition, changes in colloidal properties such as chain extension or collapse of polymer chains, flocculation and the precipitation of copolymers occur in response to environmental pH changes.44 The pH-responsive properties can also contribute to the self-assembly of the copolymers, which leads to the formation of micelles, vesicles, and nanogels.45 In addition, the pH-sensitive properties of the copolymers allow hydrophilic and hydrophobic phase separation and the polyelectrolyte nature of copolymers to be tuned.46 Therefore, pH-responsive copolymers with varying pKa values ranging from pH 1 to 14 have been prepared. Based on the applications, pH-responsive copolymers with suitable ionizable functional groups have been prepared. In addition to synthetic pH-responsive polymers, natural polymers such as chitosan, alginate and hyaluronic acid have been studied because of their pH-responsive characteristics.47pH-Responsive acidic polymers
A pH-responsive acidic polymer consists of weak acids in the side chain of polymers (Fig. 1). At high pH, pH-responsive acidic polymers lose acidic protons and form anionic polyelectrolytes.48 On the other hand, carboxylic acid groups accept protons at low pH and form an uncharged polymer as the acid groups remain unchanged.49 Thus, pH-responsive polymers gain polyelectrolyte nature based on their pKa values. The change in the ionic and non-ionic transition of pH-responsive polymers allows their hydrophobic and hydrophilic nature in aqueous solutions to be tuned. Thus far, numerous pH-responsive acidic polymers have been developed. Among them, poly(acrylic acid) has been extensively used. In addition to the poly(acrylic acid) polymers, pH-responsive polymers with sulfonic acid groups exhibit sharp pH sensitivity and swelling.50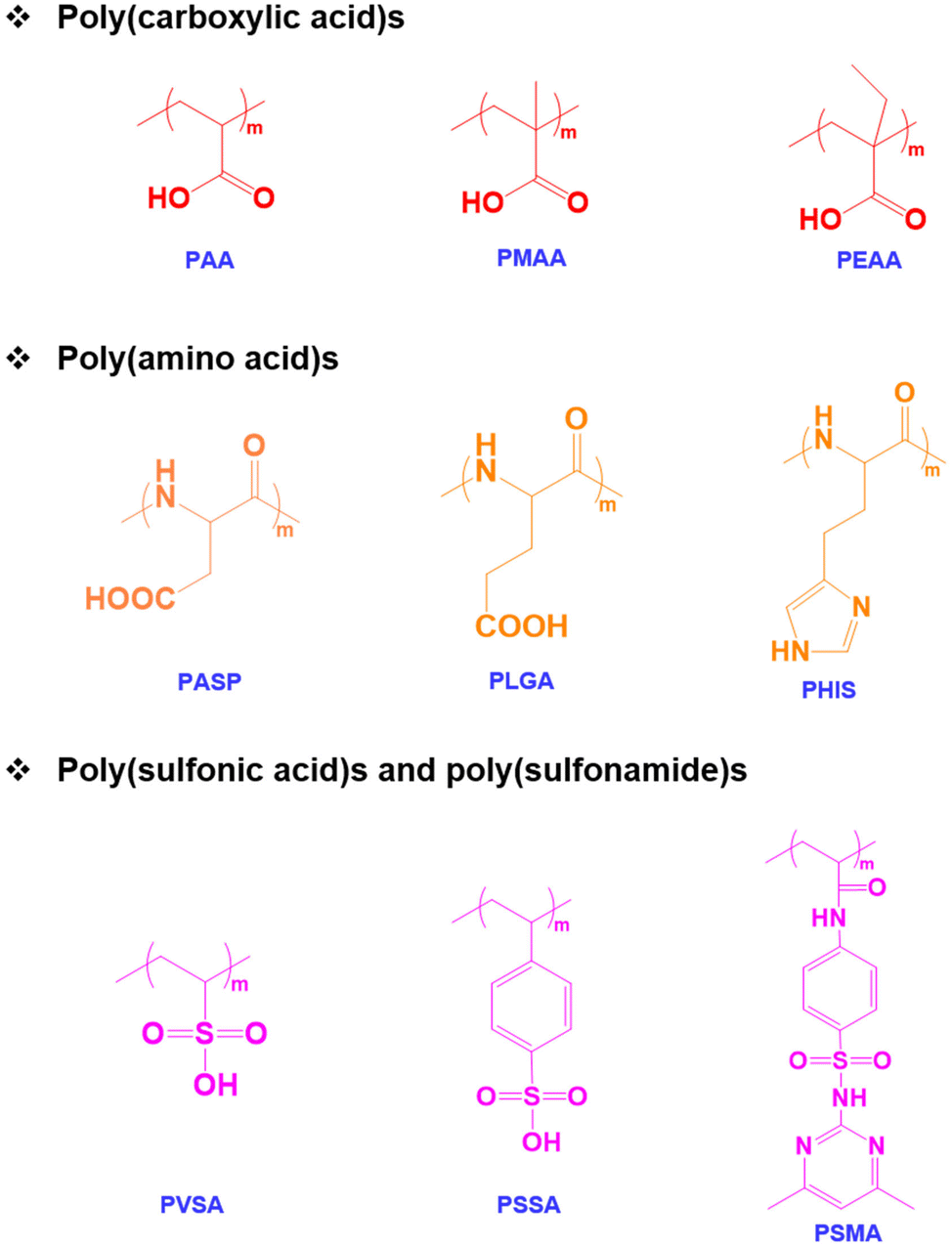 | ||
| Fig. 1 Chemical structure of pH-responsive anionic polymers. | ||
pH-Responsive basic polymers
pH-Responsive basic polymers usually possess amine functional groups in the main backbone or side chain of the polymers.51–53 These weak polybases accept protons at low pH and donate protons at high pH to yield polyelectrolytes. The chemical structures of representative pH-responsive weak polybases, including poly(acrylate)- and poly(vinyl)-based polymers containing primary, secondary, and tertiary amine groups, are shown in Fig. 2. In particular, the poly(dimethylamino)ethyl methacrylate (PDMA) polymer has extensively been used in biomedical applications because of its dual-sensitive nature to both pH and temperature.54,55 In addition, the ionizable tertiary amines in the PDMA polymer can effectively bind with negatively charged therapeutic drugs, peptides, growth factors, and proteins and release them under basic conditions.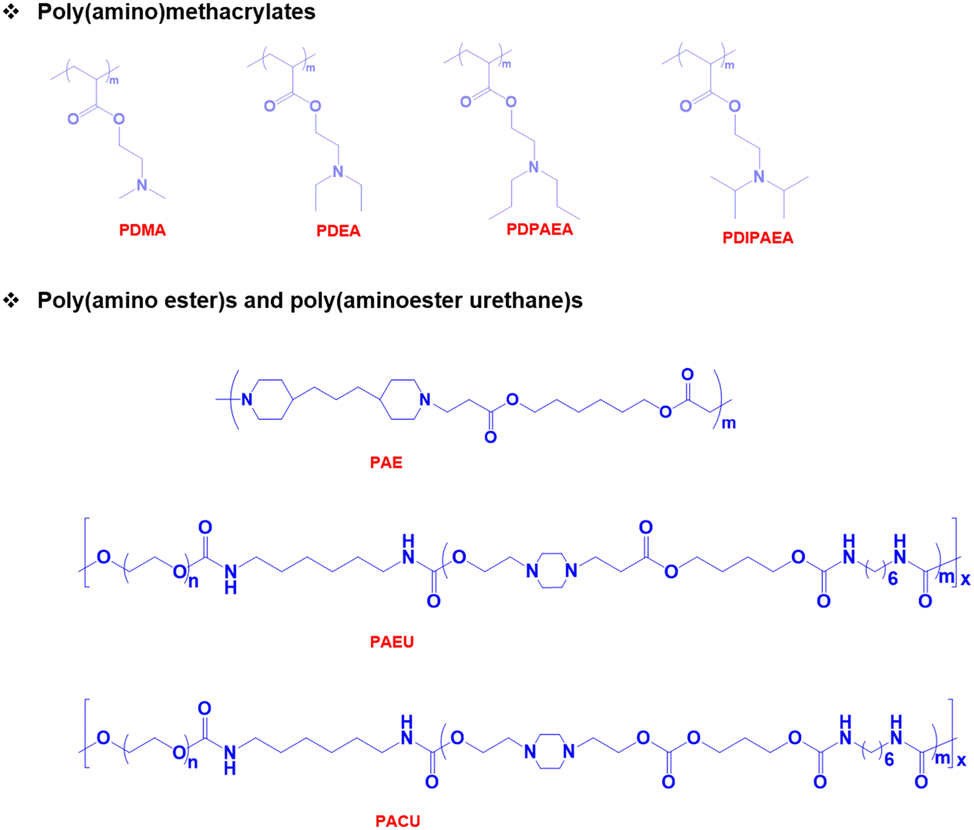 | ||
| Fig. 2 Chemical structure of pH-responsive cationic polymers. | ||
Although these polymers exhibit sharp pH-responsive structural changes, the non-degradable nature of poly(acrylate)-derived weak bases limits their applications. Therefore, researchers have focused on the development of biodegradable pH-responsive copolymers. The classic example of a biodegradable cationic polymer is poly(β-aminoester) (PAE). PAE is often synthesized by the Michael-addition reaction between primary or secondary amines and diacrylate derivatives.56 PAE exhibits a sharp amphiphilic to hydrophilic phase transition at ∼pH 6.5. More importantly, PAE polymers are non-toxic to various cell lines and biodegrade without inducing inflammation at the implantation site.
pH-Responsive amphoteric polymers
Both pH-responsive acidic and basic polymers exhibit a pH-responsive phase transition at high pH and low pH, respectively. However, these polymers are capable of imbibing either cationic or anionic therapeutic agents depending on their surface properties. Interestingly, amphoteric pH-responsive polymers fused with both acidic and basic polymers, which resulted in good swelling in both acidic and basic media.57 Attempts have been made to develop hybrid amphoteric polymers, composed of both synthetic and natural polymers. For instance, chitosan-, alginate-, hyaluronic acid- and starch-based semi-interpenetrating network hybrids have been developed by combining synthetic and natural polymers and the improvement in physical and chemical properties was verified.58 However, some of the semi-interpenetrating network hybrids consist of non-degradable polymers like polyacrylamides and polyacrylates and this limits their application in clinics.In our research group, we developed innovative biodegradable amphoteric copolymers like poly(urethane amino sulfamethazine) copolymers and hyaluronic acid-g-poly(amino urethane) copolymers, which could form cationic and anionic hydrogels in response to the change in pH (Fig. 3).59 The amphoteric copolymers have the ability to load both cationic and anionic therapeutic agents and show good biodegradation in vivo.
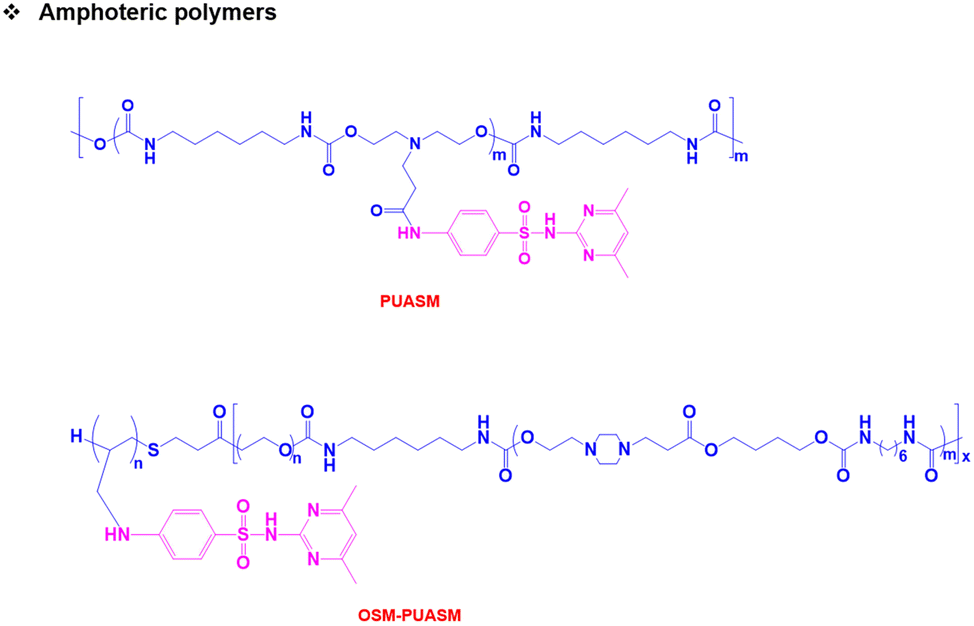 | ||
| Fig. 3 Chemical structure of pH-responsive amphoteric polymers. | ||
One specific class of polymers that could be considered amphoteric polymers is proteins. Thanks to their intrinsic features, proteins possess both positively and negatively charged groups which allow proteins to respond to pH variation in both acidic and basic media.60 In particular, amine and carboxyl groups in the protein backbones will sequentially transform from the ionized to the neutralized state or vice versa. This switch between the two states involves an isoelectric point (pI) of protein which makes up the electrical charge value of proteins in total. Based on the pI value of a certain protein, proteins will have different responses at different pH levels, resulting in multiple applications that can be applied to biomedical applications.
Biomedical application of pH-responsive injectable hydrogels
A pH-responsive injectable hydrogel is a form of in situ-forming hydrogel depot upon injection into different microenvironments (e.g., tumors or other normal tissues).61 It is well known that tumor tissues exhibit low pH characteristics due to the high rate of anaerobic glycolysis.62 As a result, the extracellular pH of tumor tissues is often more acidic than that of normal tissues. Therefore, pH-responsive injectable hydrogel precursors with suitable functional groups can induce a sol-to-gel phase transition due to the association of these functional groups. The stability of the implanted hydrogel is controlled by the internal pH and ionic strength changes. Furthermore, the internal pH can influence the microporous structure and the degree of swelling of the implanted hydrogel. These factors can influence the rate of diffusion and release of the encapsulated therapeutic agents.pH-Responsive injectable hydrogels for protein delivery
Injectable pH-responsive hydrogels are an important class of biomaterials in which a free-flowing sol transforms into immovable semisolids upon administration into warm-blooded animals.63 The classic example of biodegradable pH-responsive injectable hydrogels is PAE. In general, PAE-based copolymers are prepared by the Michael addition reaction between diacrylates and diamines. In our group, we developed a PAE–PEG–PAE triblock copolymer using 4,4′-trimethylene dipiperidine (TMDP) and PEG-diacrylate.64 The PAE–PEG–PAE copolymer without hydrophobic segments effectively dissolved at low pH and transformed to a stable viscoelastic gel upon raising the pH and temperature. The PAE–PEG–PAE copolymer showed good mucoadhesion with mucin derived from pig stomach. The mucoadhesive property was verified by mixing the copolymer and mucin and the viscosity of copolymer–mucin mixtures was examined. The viscosity of the mixture increased as the copolymer concentration increased (from 2.5 wt% to 5.0 wt%) to a fixed mucin concentration (10 wt%). This is due to the increased interactions between the copolymer and mucin. The PAE–PEG–PAE copolymer developed in this study was shown to be rapidly biodegradable (∼7 days) and hence this limits its application in sustained delivery.To improve the stability of PAE-based copolymers, poly(ester)-based copolymers are introduced into PAE copolymers.65 The modified PAE copolymer was prepared by the Michael-type reaction of acrylate–PCL–PEG–PCL–acrylate, piperazine and hexane diacrylate (Fig. 4). The obtained copolymers exhibited a good sol to gel phase transition. This is mainly due to the ionization of the PAE segment in the copolymer at low pH. The hydrophobic and hydrophilic balance in the copolymer can be easily tuned to control the sol to gel phase transition that can cover physiological regions. The obtained pentablock PAE–PCL–PEG–PCL–PAE copolymer exhibited a pH- and temperature-responsive sol-to-gel phase transition and was effectively loaded with insulin due to the possible ionic interaction with cationic PAE segments. An insulin-loaded hydrogel depot can effectively sustain its release due to the controlled degradation of PAE. Precisely, the hydrogel depot released insulin in a sustained manner for 30 days. Nevertheless, the high hydrophobicity of copolymers, due to the presence of PCL, requires a long time for them to be completely eliminated from the body or surgical removal of implanted depots is needed at frequent intervals. This limits the application of pentablock copolymers in the clinic. Therefore, the development of injectable hydrogels that can deliver therapeutic agents in a sustained manner and are biodegraded after performing their role as controlled delivery depots is needed.
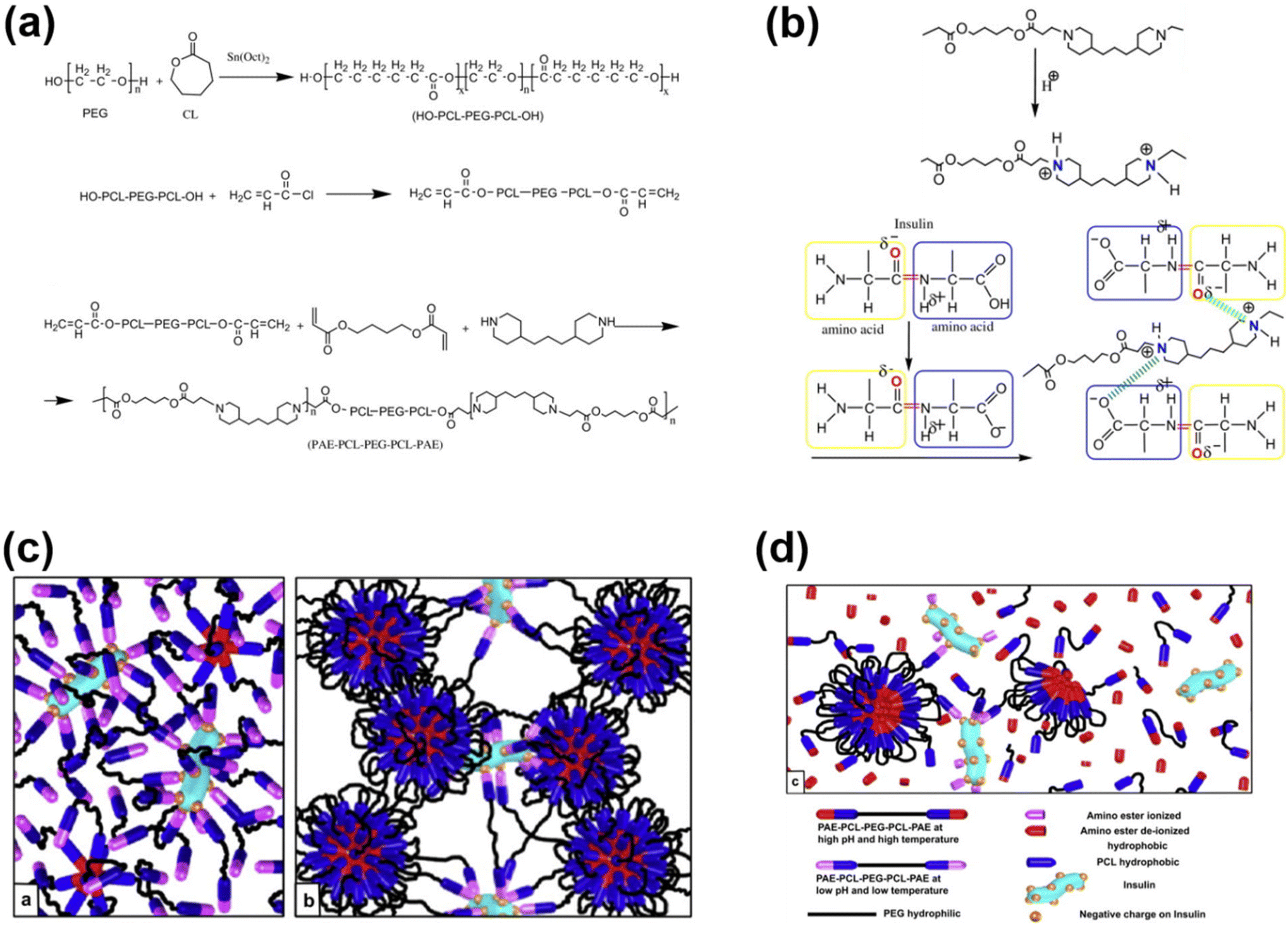 | ||
| Fig. 4 (a) Synthesis route to prepare pH-responsive cationic pentablock copolymers. (b) Ionization of a cationic polymer at low pH and its interaction with anionic residues of insulin. (c and d) Schematic illustration of an insulin-loaded pH-responsive hydrogel and its sustained release characteristics.65 Reproduced from ref. 65 with permission from Elsevier Limited, Copyright 2008. | ||
In an attempt to develop a suitable delivery platform, we synthesized a poly(urethane) and poly(amidoamine) series with different molecular weights ranging from 4 kDa to 20 kDa for the sustained delivery of bioactive agents.66–68 These copolymers show a sharp pH- and temperature-responsive sol-to-gel transition and can effectively imbibe therapeutic proteins by simple mixing. However, the slow biodegradable properties of poly(urethane) and poly(amidoamine) require surgical removal of the hydrogel depot after a certain time. Therefore, we combined the slow degradability of poly(urethane) and rapid degradability of PAE to obtain a novel pH-responsive polymer, poly(β-aminoester urethane) (PAEU) copolymer.69 The PAEU copolymers, composed of degradable urethane and a pH-sensitive tertiary amine, are synthesized by Michael addition polymerization. The presence of cationic amine groups strongly interacts with anionic proteins via strong electrostatic interactions. Furthermore, the strong interactions are further strengthened by the hydrogen bonding interaction between proteins and urethane bonds. A series of PAEU copolymers with a varying molecular weight have been developed by controlling the feed ratio of PEG and PAE and HDI units. Regardless of the molecular weight, the PAEU copolymers exist as free-flowing sols at low pH (pH 6.0) owing to the ionization of PAE, which leads to electrostatic repulsion between PAEU copolymers and allows the copolymers to flow. Interestingly, when the pH was raised to the physiological condition, deionization of PAE units transforms hydrophilic units to hydrophobic units and develops strong inter- and intramolecular interactions resulting in strong gelation. The free-flowing PAEU copolymers at low pH could form strong ionic interactions with hGH. Upon subcutaneous administration, the hGH-loaded PAEU copolymers form a hydrogel depot. The implanted hydrogel depot releases the hGH protein in a sustained manner for 3 days with good control in the initial burst. Furthermore, Singh et al. prolonged the release of hGH for 5 days by developing PAEU hybrids.70 In this study, hGH was first incorporated into the interlayers of cationic layered double hydroxides (LDH) and then loaded into the PAEU copolymer sols at low pH. The pH- and temperature-responsive properties of PAEU copolymers were not affected by LDH incorporation. As a result, the PAEU hybrids formed stable hydrogel depots in the subcutaneous layers of warm-blooded animals and released hGH for 5 days.
The pH-responsive poly(urethane), poly(amidoamine), PAE and PAEU can rapidly alter the pH of surrounding sites as they degrade at acidic or basic pH. In an attempt to neutralize the local pH of the implantation site, we fabricated a pH-responsive poly(amino carbonate urethane) (PACU)-based injectable hydrogel to control the release of hGH and avoid degradation-related local pH change (Fig. 5).71 The PACU copolymers exhibited a sol-to-gel phase transition that covered the physiological window. In vivo biodegradation studies demonstrated the good biodegradation property of the PACU hydrogels without developing inflammation at the injection site. hGH-loaded PACU copolymer sols become stable right after subcutaneous administration and release effective therapeutic level hGH for 4 days.
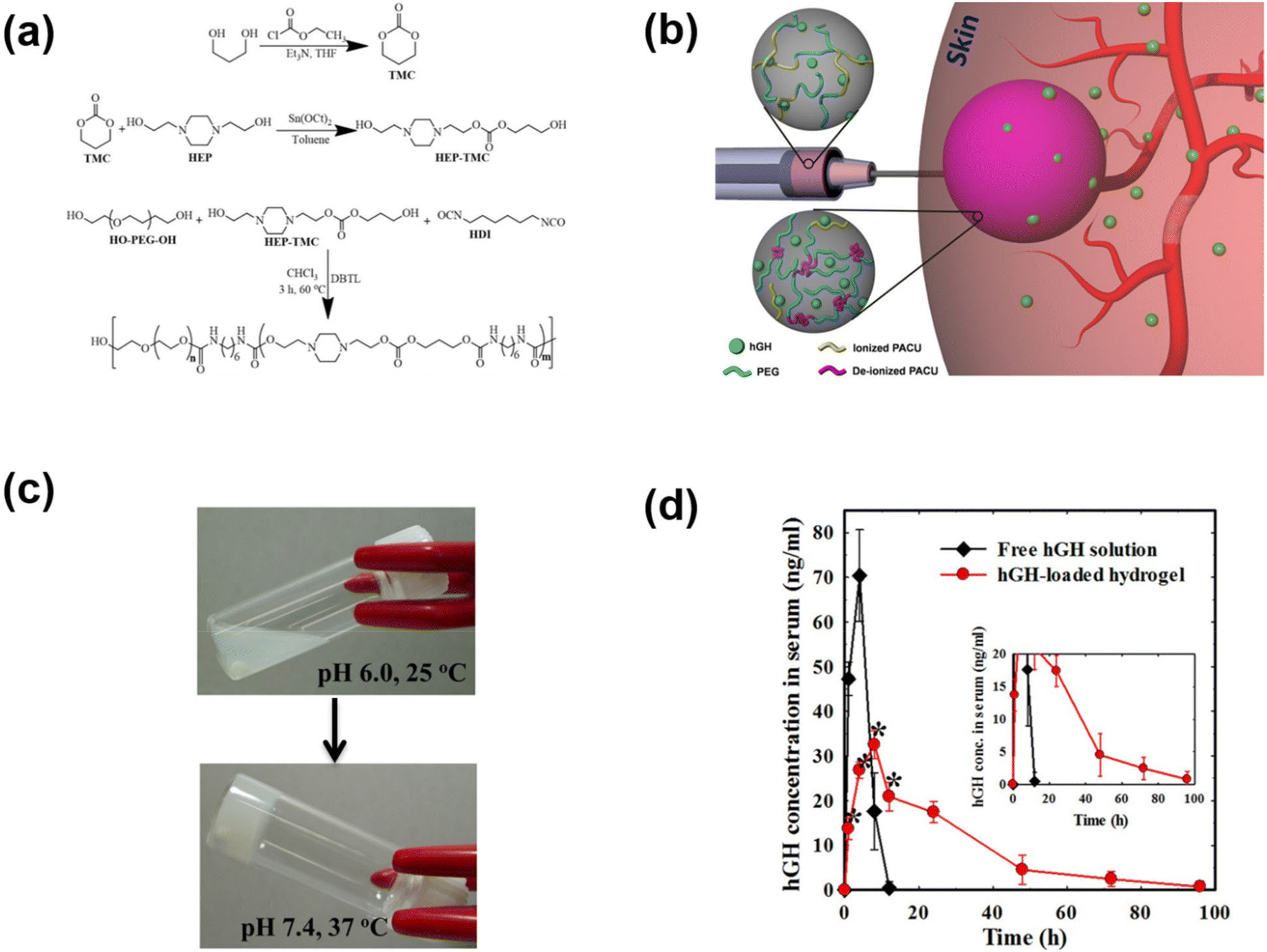 | ||
| Fig. 5 (a) Synthesis route to prepare pH-responsive PACU copolymers. (b) Schematic illustration of the formation of pH-responsive hydrogels in the subcutaneous layers of rats and the sustained release of hGH to blood circulation. (c) Sol-to-gel phase transition of pH-responsive PACU copolymers as a function of pH and temperature. (d) Pharmacokinetic profile of hGH measured after subcutaneous implantation of hGH-loaded PACU hydrogels.71 Reproduced from ref. 71 with permission from Springer Nature Limited, Copyright 2016. | ||
In addition to PAE-based cationic polymers that could effectively control the release of cationic polymers, polypeptides have also been used to develop cationic copolymers by appropriate functionalization of amino acid functional groups. We demonstrated the development of pH-responsive polypeptide-based copolymers for the controlled release of hGH (Fig. 6).72 Firstly, a PBLG–PEG–PBLG triblock copolymer was developed and substituted with diamines to obtain pH-responsive copolymers. The PNLG-based copolymers showed good solubility at low pH and the ionized PNLG copolymers could interact with hGH via electrostatic interactions. The polypeptide copolymer could form a gel in vivo and release hGH for one week. In another study, amine containing sulfamethazine was substituted into the triblock copolymer to obtain an anionic polypeptide.73 The peptide copolymers exhibited solubility at high pH and formed a hydrogel at the physiological pH. Such characteristics allowed cationic protein to be loaded into the hydrogels. Cationic lysozyme was mixed into the anionic copolymers and the gel depot formed in the subcutaneous tissue released the protein over 7 days. In another work, an anionic copolymer hydrogel based on poly(sulfamethazine carbonate urethane) (PSMCU) was developed for the controlled release of cationic proteins. The PSMCU copolymer was found to be non-toxic and completely biodegradable in 5 weeks. Lysozyme-incorporated PSMCU copolymers formed a depot in vivo and controlled the release of lysozyme.
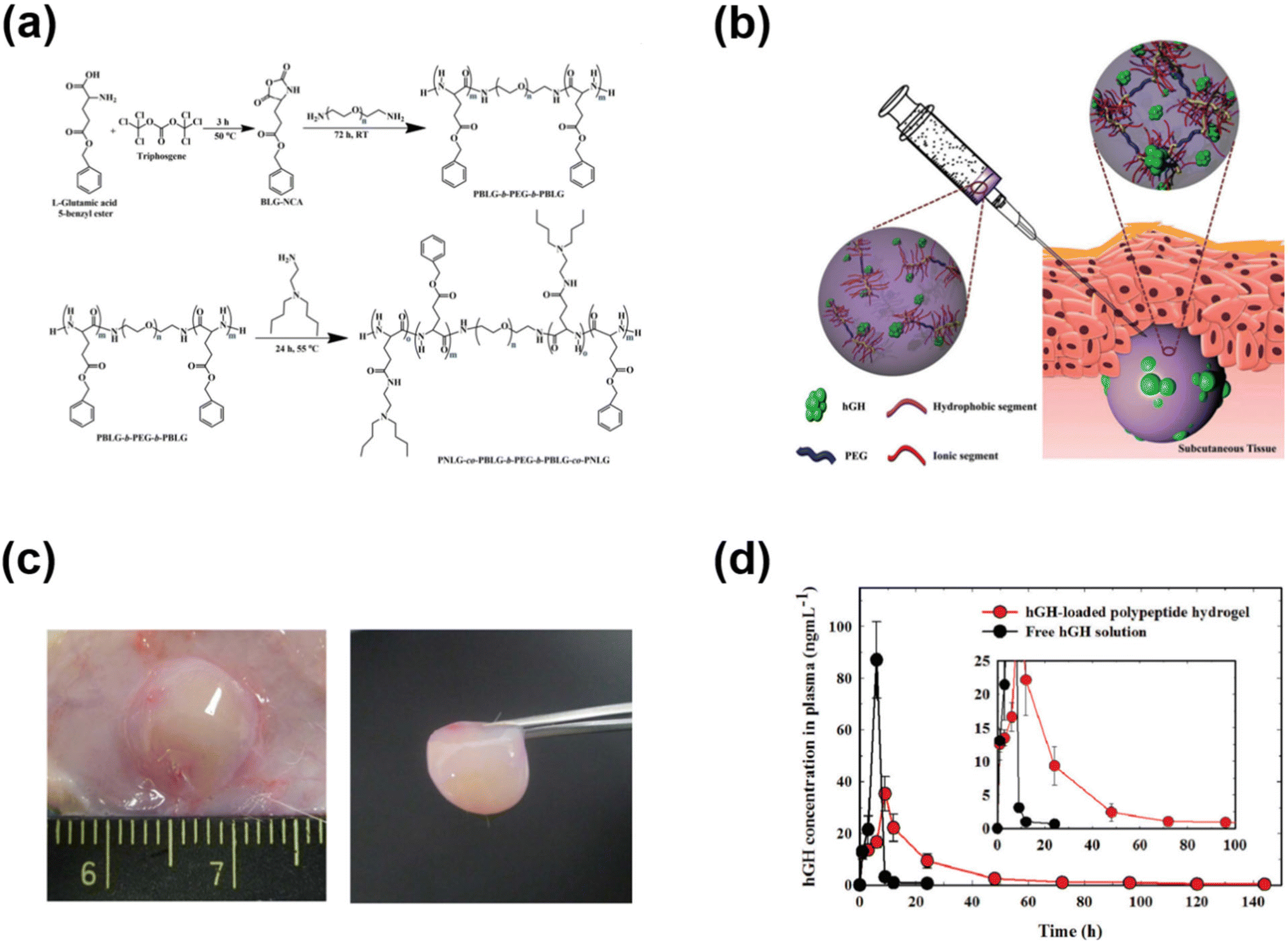 | ||
| Fig. 6 (a) Synthesis route to prepare pH-responsive PNLG copolymers. (b) Schematic illustration of the formation of pH-responsive hydrogels in the subcutaneous layers of rats and the sustained release of hGH to blood circulation. (c) Sol-to-gel phase transition of pH-responsive PNLG copolymers as a function of pH and temperature. (d) Pharmacokinetic profile of hGH measured after subcutaneous implantation of hGH-loaded PNLG hydrogels.72 Reproduced from ref. 72 with permission from The Royal Society of Chemistry, Copyright 2017. | ||
Beside synthetic polymers, natural polysaccharides have been widely engineered and applied for pH-sensitive hydrogels as well.74 In this manner, polysaccharides are often modified or decorated by pH-responsive linkages and/or side groups along the biopolymer chains. For instance, Qi et al. exploited the tunable behavior of the sulfonic acid group with variation of the environmental pH.75 Salecan as a microbial polysaccharide platform was grafted with poly(2-acrylamido-2-methyl-1-propanesulfonic acid) (PAMPS) that contained sulfonic acid moieties. The pKa value of Salecan-g-PAMPS hydrogel was approximately 1.5 and the associating ionization and protonation were clearly revealed at pH 1.2. To be more specific, the protonation of sulfonic anions to form –SO3H groups at lower pKa point augmented H-bonding between protonated sulfonic acid groups and hydroxyl moieties along salecan chains, resulting in a highly restricted polymer matrix and low water capture. On the other hand, higher pH conditions demonstrated an increment in swelling ratio and capture capability, which can be explained by the deionization into –SO3− facilitating hydration and the electrostatic repulsions among highly negatively charged groups. For protein delivery, insulin release was greatly increased in a neutral environment (pH 7.4), simulating normal tissue, as opposed to being markedly decreased under acidic conditions (pH 1.2), such as those found in the human stomach. In parallel, hyaluronic acid was also tailored by oxidizing the polysaccharide chains to introduce aldehyde groups and further modified using 3,3′-dithiobis (propanoic dihydrazide).76 Analogous results showed that hydrogels had a lower size at low pH compared to high pH medium. This phenomenon is associated with the ionization of the HA matrix, especially the dynamic recombination of acylhydrazone bonds. Thanks to its dual stimuli-responsiveness (redox and pH), the hydrogel system offered shape recovery and self-healing properties that have potential applications for various biomedical purposes.
pH-Responsive injectable hydrogels for cancer therapy
In situ forming injectable hydrogels with pH-sensitive properties have promising potential in cancer therapy. The existence of a pH gradient in tumor tissues is a hallmark of cancer; the extracellular pH of tumor tissues ranges from 5.7 to 7.8, whereas the extracellular pH of normal tissue is 7.4.77,78 More importantly, the pH value of cancer cells is found to be 5.4–6.0. Therefore, the pH gradient allows various pH-responsive biomaterials to be developed for chemotherapeutic drug release applications. In general, drug-loaded pH-responsive biomaterials can show slow or no drug release to normal tissues, and the drug release is triggered under acidic pH conditions.Pioneering work on biodegradable, pH-responsive sulfamethazine-based copolymers has been proposed in our laboratory for the controlled release of anticancer agents. The pH-responsive biomaterials were developed by simply conjugating oligo sulfamethazine (OSM) derivatives to the chain-ends of PCLA copolymers.79 The pentablock copolymers obtained in this way exhibited a phase transition upon altering the pH and temperature. The major reason for the change in the sol-to-gel transition is the ionization of the sulfonamide functional group at high pH, and subsequent deionization upon injection under physiological conditions. Subcutaneously implanted pH-responsive copolymers formed drug-eluting hydrogel depots. Paclitaxel (PTX), an antineoplastic drug that induces an anticancer effect by binding to the tubulin inhibitor was loaded into the pH-responsive copolymers and the anticancer properties were examined by subcutaneous injection into the mice implanted with tumors. The PTX-loaded pH-responsive hydrogel eradicated the tumor effectively by single subcutaneous injection.
In another work, sulfamethazine-based pH-sensitive anionic hydrogels have been used to treat hepatocellular carcinoma. In this study, poly(urethane sulfide sulfamethazine) (PUSSM) was used as an anionic block that was copolymerized with PCLA copolymers.80 The anionic PCLA–PUSSM copolymers were found to be soluble at high pH (pH 8.5) and solidified at the physiological pH (pH 7.4). The anthracycline-based chemotherapeutic drug, doxorubicin (DOX), was loaded into the hydrogels via the combination of both hydrophobic and π–π interactions between DOX and sulfamethazine. The strong interaction of DOX with sulfamethazine effectively inhibited the burst release and sustained the drug release. The DOX-loaded copolymers were intra-arterially infused into the hepatocellular carcinoma rabbit model using a microcatheter. The tumor acidic pH conditions allow a hydrogel to form for chemoembolization and effectively inhibit the tumor growth. In other work, anionic pH-responsive PEG–PUSSM copolymers were fabricated as a suitable embolic agent for vascular embolization (Fig. 7).81,82 The PEG–PUSSM copolymer with the radiopaque contrast agent iohexol showed a sol–gel transition when decreasing the pH of the copolymer solution from 8.5 to 7.4. Embolization of the PEG–PUSSM copolymer in rabbit kidney showed the occlusion of arteries, which led to ischemic changes in renal tissues. Among various types of pH-sensitive copolymers, anionic poly(sulfamethazine)-based hydrogels exhibited good adhesive properties (Fig. 8).83 This allowed healing of the cutaneous wounds.
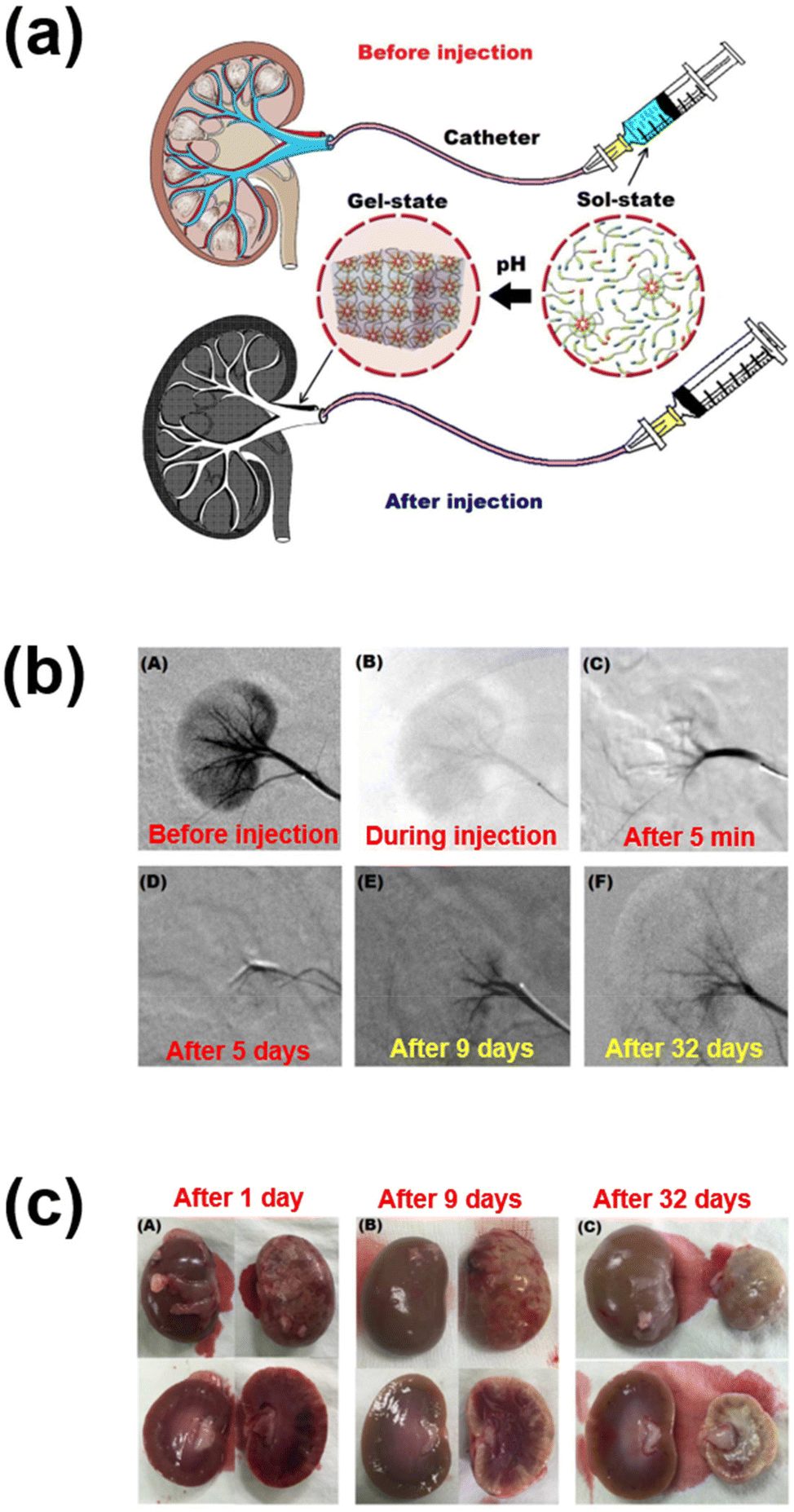 | ||
| Fig. 7 (a) Schematic illustration of renal vessel embolization using pH-responsive PEG–PUSSM copolymers. (b) Digital subtraction angiography images before and after embolization at different time points. (c) Appearance of embolized kidney after injection using pH-responsive copolymers.82 Reproduced from ref. 82 with permission from The Royal Society of Chemistry, Copyright 2016. | ||
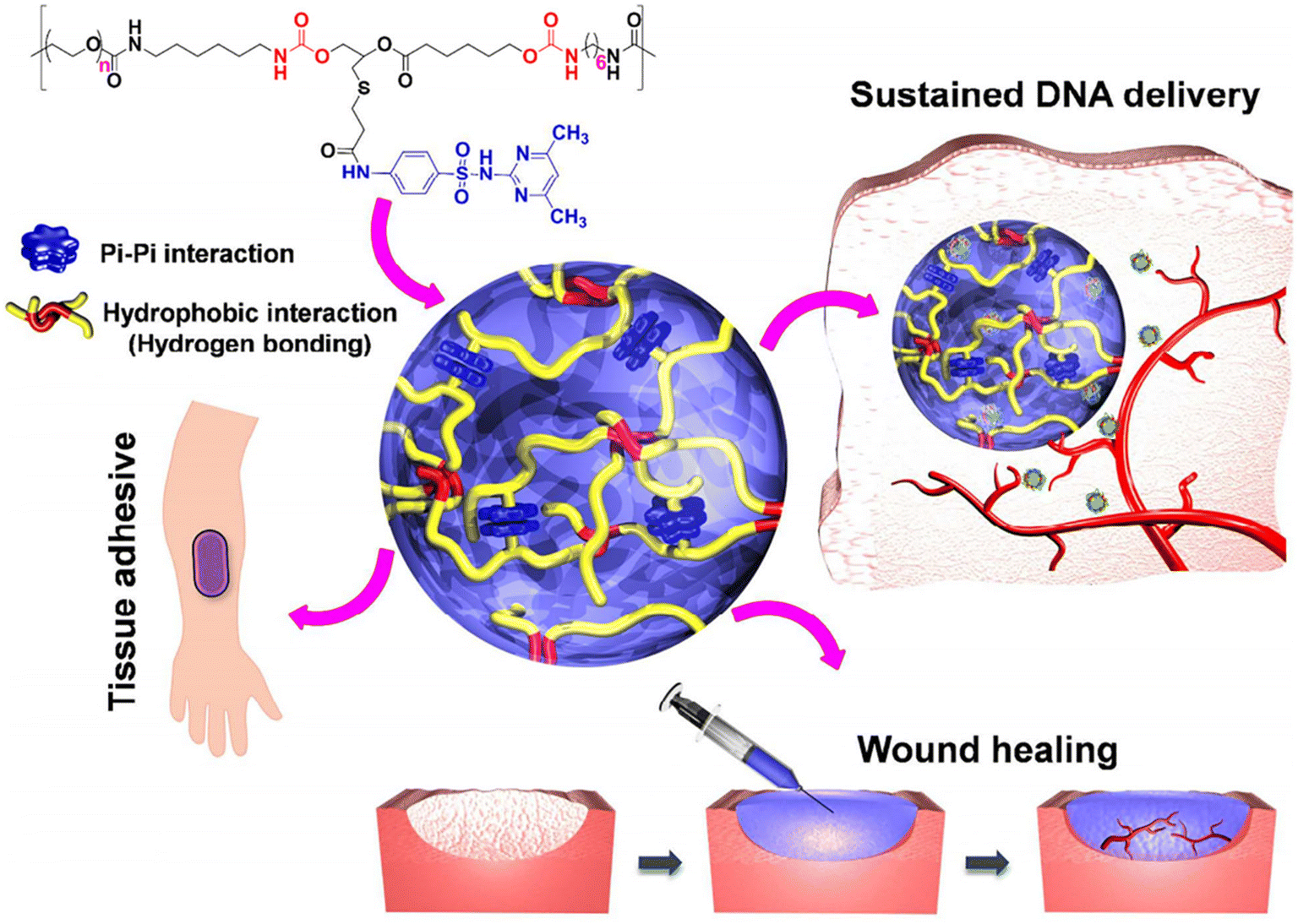 | ||
| Fig. 8 Chemical structure of the PUSMA copolymer and its application in bioadhesive, sustained release and wound healing.83 Reproduced from ref. 83 with permission from The American Chemical Society, Copyright 2018.83 | ||
Physically entrapped chemotherapeutic drugs often diffuse out from the hydrogel network based on its degradation. The degradation pattern of the hydrogel network determines the release profile of encapsulated drugs. Nevertheless, chemical cross-linking of the chemotherapeutics within the hydrogel network can effectively inhibit the premature drug release and therefore the release rate can be determined based on the chemical linkage between the drug and the hydrogel network. Chemotherapeutic drugs linked by non-degradable bonds require the degradation of hydrogel networks to release drugs. Interestingly, drugs linked by labile linkages such as disulfide, ketal, enzyme and azo linkers can trigger the release of drugs upon exposure to appropriate stimuli. Recently, we developed low-pH triggered delivery of tumor-targetable DOX for the treatment of hepatocellular carcinoma.84 To develop the pH-responsive hydrogel network, DOX was conjugated with glucuronic acid and cross-linked with boronic acid cross-linked PCLA copolymers. The boronate ester formed between PCLA and DOX shows pH-dependent assembly and disassembly behavior. Under physiological conditions, DOX was effectively coordinated with the network and triggered release at low pH. To examine the antitumor effect, pH-responsive copolymers were subcutaneously administered into the HepG2 liver tumor xenograft, which effectively suppressed the tumor growth.
In addition to synthetic polymers, polypeptide-based hydrogels are also often employed as local drug delivery depots because of their structural tunability and biocompatibility. A study was conducted by Raza et al. to investigate the capability of chemotherapeutics by exploiting pH-responsive peptide hydrogels as a platform.85 Here, a pH-sensitive octapeptide FEFEFRFK was used as a polymer matrix to encapsulate paclitaxel. At neutral pH, the phenylalanine-based peptide formed a three-dimensional architecture via self-assembly. On the other hand, the hydrogel tended to disassemble in the acidic pH environment of tumors, which governed the burst release of PTX within the treated site. In vivo studies still demonstrated sustained retention up to 96 h and an increment in accumulation of the drug at tumor sites, which eliminated tumor growth and reduced side effects.
Biomedical usage of pH-responsive amphoteric polymers
Unlike traditional pH-responsive polymeric biomaterials that respond to either acidic or basic conditions, we prepared dual-ionic or amphoteric copolymers (e.g., poly(urethane amino sulfamethazine)) through fusion of cationic and anionic copolymers (Fig. 9).59 Owing to the amphoteric characteristics of the copolymers, the resulting particulate network allows encapsulation of both anionic and cationic therapeutics. At high copolymer concentrations (∼20 wt%), the amphoteric copolymers showed closed-loop sol–gel–sol phase transitions, although reversible, in response to changes in pH and temperature with controllable gel regions under physiological conditions. The free-flowing copolymer solutions, under either acidic or basic conditions, mixed with human growth hormone (hGH) were easily administered into the animal models using small needles. The hGH-loaded viscoelastic gel formed in the subcutaneous tissue controllably released hGH without deterioration of the native structure, which implied the biocompatibility of the formed network. The dual-ionic copolymers have also been successfully used to regulate the microenvironment of ischemic regions.86 At high pH (∼pH 8.0), the copolymer exhibited negative charge and formed ionic complex micelles with a positively charged chemoattractant molecule, stromal cell-derived factor-1α (SDF-1α). Systemic administration of ionic complex micelles in a stroke-induced rat model effectively prolonged circulation in the bloodstream. The hydrophilic PEG present in the copolymers provided stealth characteristics to the ionic complex micelles, which allowed long-term circulation. On the other hand, in the infarct region (∼pH 5.5), the copolymer attained positive charge and site-specifically delivered SDF-1α due to the electrostatic repulsion. The controlled delivery, low cytotoxicity, degradability in vivo and easy injectability through subcutaneous or intravenous mode suggest that the dual-ionic or amphoteric pH-responsive copolymers could be promising biomaterials to not only deliver therapeutic agents, but also regenerate tissues. The charge switchable dual-ionic characteristics of the copolymers were also exploited in polyelectrolyte multilayer assembly through iterative adsorption of complementary materials. For instance, the dual-ionic copolymer was used as a triggering layer in polyelectrolyte multilayer assembly on microneedles.87 The positively charged copolymer at low pH was coated on microneedles, and the complementary pair used in this coating was heparin. After the formation of several layers, DNA vaccine-bearing polyplexes were loaded through ionic interactions. The cutaneous implantation of DNA vaccine-loaded microneedle arrays in mice generated antigen-specific robust immune responses. These findings suggest that the dual-ionic characteristics of pH-responsive copolymers can be exploited in versatile biomedical applications.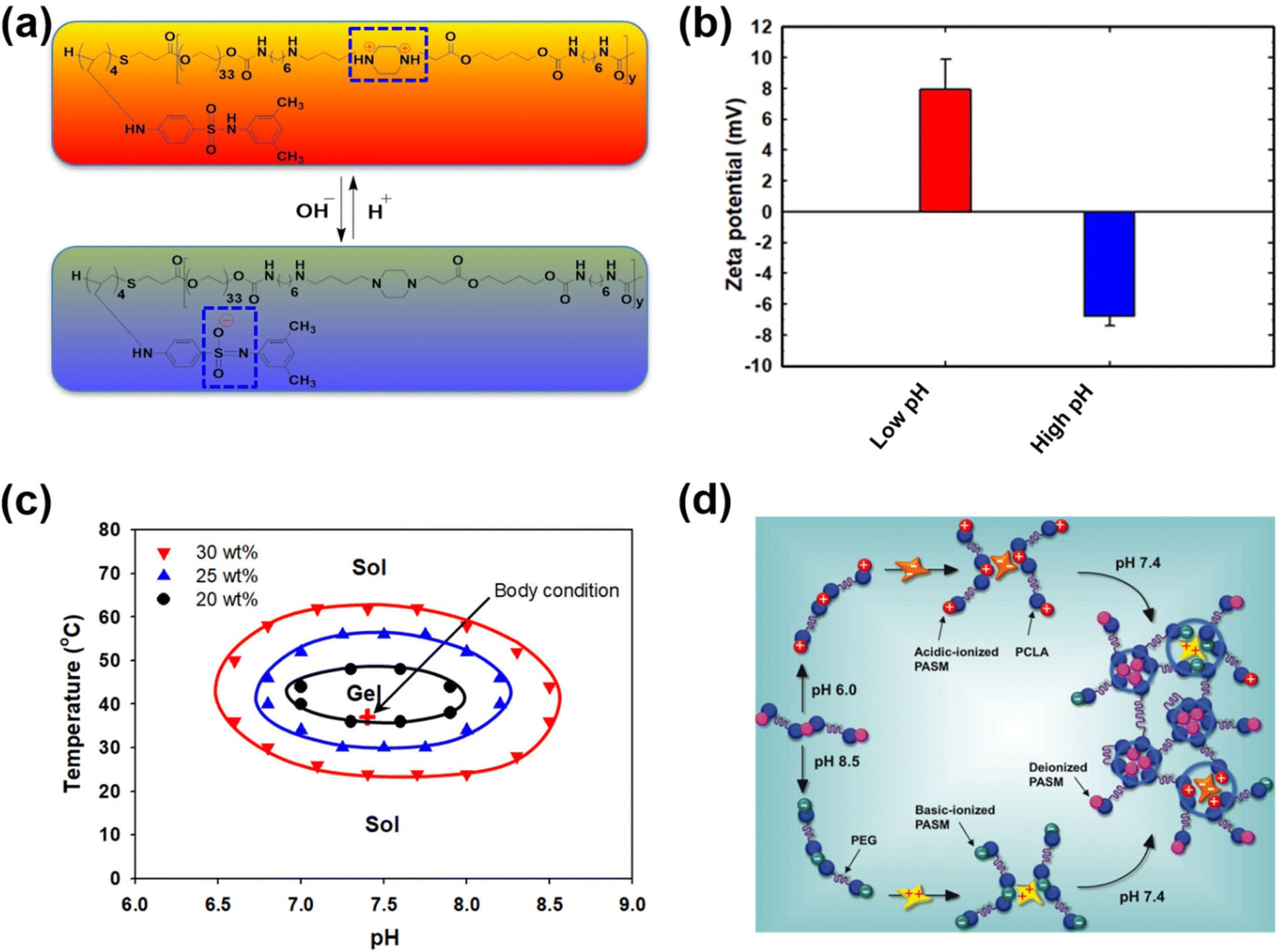 | ||
| Fig. 9 (a) Chemical structure of dual-ionic pH-responsive copolymers under acidic and alkaline pH conditions. (b) Charge switching of dual-ionic pH-responsive copolymers under low- and high-pH conditions. (c) Closed-loop reversible sol–gel–sol phase transition behavior of dual-ionic pH-responsive copolymers in which biological drugs can be mixed under either acidic or basic pH conditions and injected into the body. (d) The charge switching of the copolymer under acidic and basic pH conditions and its ability to interact with appropriate proteins. Under physiological conditions, the pH-responsive copolymers form a hydrogel network and release the biologics in a sustained manner.59 Reproduced from ref. 59 with permission from The Royal Society of Chemistry, Copyright 2012. | ||
The development of pH-responsive amphoteric copolymers by the combination of natural and synthetic polymers could be an interesting approach as the combination minimizes various issues like toxicity and non-biodegradability. An in situ forming injectable biogel (IBG) based on hyaluronic acid–PAEU was used for the controlled delivery of chemotherapeutic agents (Fig. 10).88 The hydrophilic hyaluronic acid effectively covers the PAEU and forms a soft hydrogel network. The soft texture facilitates the adhesiveness and effectively integrates with the subcutaneous tissues, which are bioresorbed after 4 weeks. DOX-loaded IBGs controlled the drug release and prolonged the release for 4 weeks. DOX-loaded IBGs were subcutaneously implanted adjacent to the B16/OVA melanoma xenograft and showed effective inhibition of tumor growth. Similarly, we conjugated PAEU copolymers site-specifically to the sulfhydryl group containing human serum albumin (HSA) (Fig. 11).89 The HSA–PAEU conjugates can effectively improve the circulation half-life of uricase enzyme, a recombinant biological drug. Subcutaneous administration of uricase-loaded HSA–PAEU hydrogels improved the circulation half-life of enzymes. HSA–PAEU hydrogels implanted in the subcutaneous layers of hyperuricemia mice prolonged the release of uricase and normalized the uric acid level in the serum.
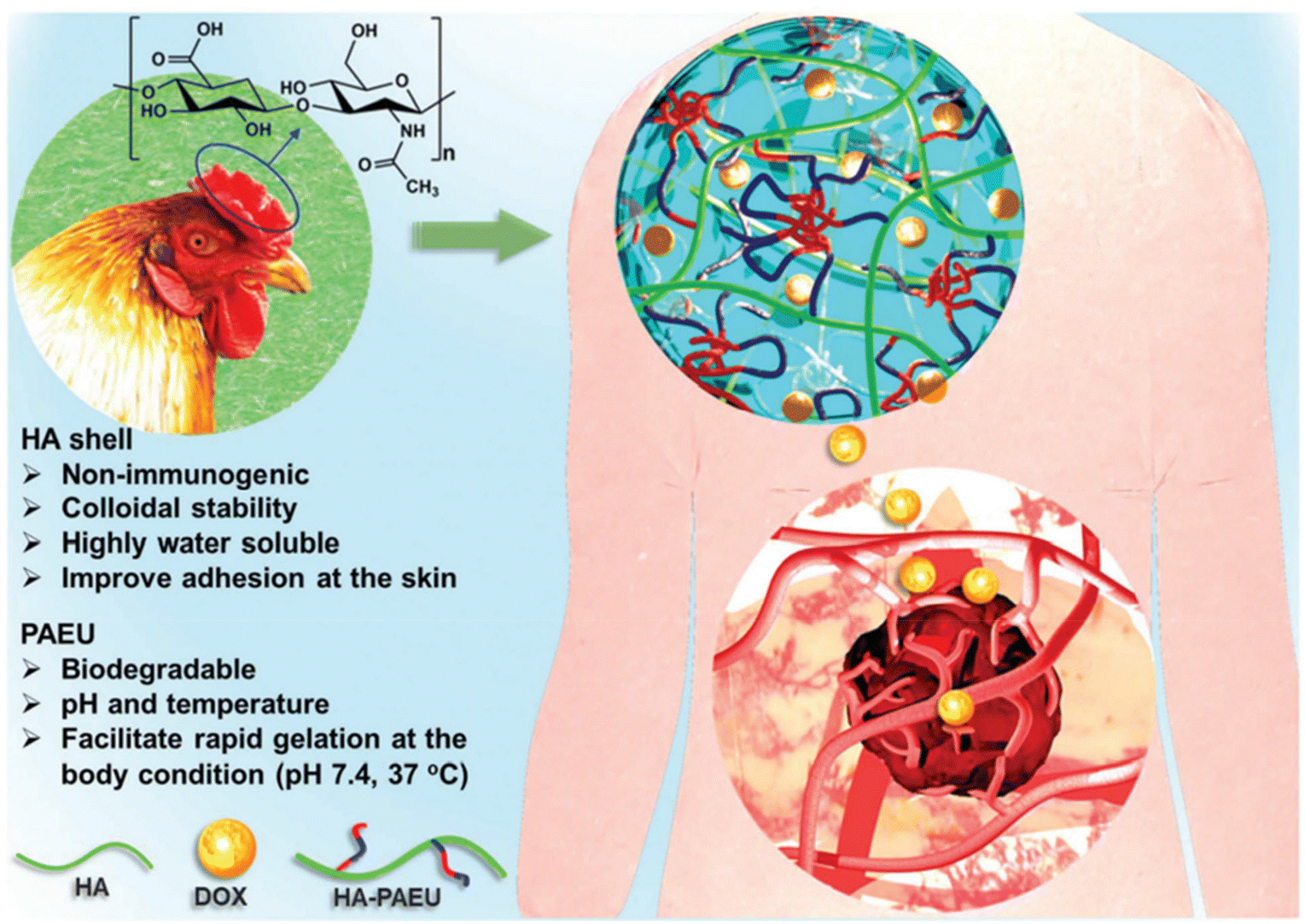 | ||
| Fig. 10 Working principle of IBGs and the sustained release of DOX to effectively infiltrate into the adjacent tumor tissues.88 Reproduced from ref. 88 with permission from The Royal Society of Chemistry, Copyright 2019. | ||
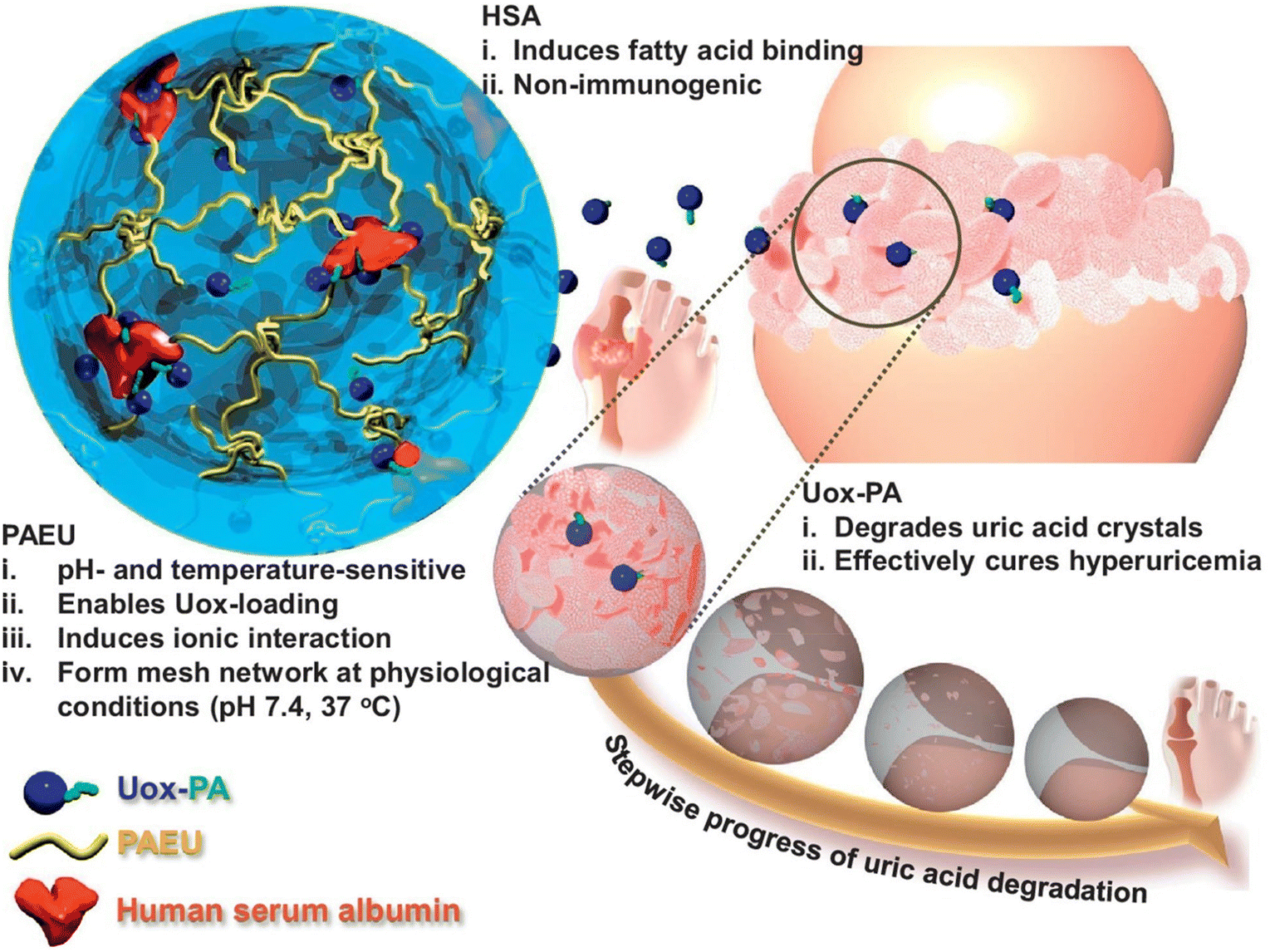 | ||
| Fig. 11 Schematic illustration of formation of a Uox-loaded HSA–PAEU hydrogel depot in hyperuricemia mice. Sustained release of Uox from the hydrogels completely hydrolyzes the uric acid crystals.89 Reproduced from ref. 89 with permission from Elsevier Limited, Copyright 2017. | ||
By their inherent pH-responsiveness, proteins have been recognized as potential candidates for smart hydrogels. A facile design for pH-sensitive hydrogels was reported by Koga et al., in which 4-arm PEG was grafted with peptide segments via a thiol–maleimide click reaction.90 The oligopeptides were synthesized by alternating hydrophobic leucin and ionic glutamic acid and terminated using cysteine. In the minimum 0.5 wt% aqueous solutions, the hybrid star-shaped polymers showed excellent cytocompatibility and a reversible β-sheet to random coil transition. The self-assembly of this star-shaped polymer is of interest as it demonstrates outstanding shear-thinning and self-healing behaviors for injectability. Furthermore, the ionizable blocks of glutamic acid can rapidly adapt to pH change and serve as a desirable polymeric scaffold in response to a pH-oscillation system, which promises great potential for diverse applications.
Conclusion and future perspectives
The last two decades have witnessed enormous exploration of stimuli-responsive biomaterials and their utilization in materials science, nanobiotechnology and pharmaceutics. Despite the efforts and booming number of publications, very few technologies have been commercialized and several others are in the early stages of clinical trials. By using stimuli-responsive biomaterials, on-demand drug release with a high level of control is possible. An ideal stimuli-responsive system can precisely control the drug release in response to stimuli.Among various stimuli-responsive copolymers, pH-responsive polymers have been proved to exhibit outstanding properties as biomaterials. The unique characteristics of dual-ionic pH-responsive biomaterials and their subsequent processing as hydrogels or nanoparticulate structures could be utilized to load biological drugs. Such bioengineered structures are applicable to a variety of biological drugs that require controlled delivery. Simple mixing of biological drugs with copolymers allows the formation of particulate networks and the subsequent release of the drugs, which may unlock the possibility of adapting biological drugs to therapeutic applications. Owing to their elegance and simple operation techniques, dual-ionic pH-responsive biomaterials may be applied to deliver highly immunogenic biological drugs that have failed in clinics. Therefore, we anticipate that pH-responsive biomaterials with superior properties may mitigate the shortcomings of biological drugs and contribute to the development of novel biological drugs. Current research mainly focuses on the delivery of single drug with high-level control of on-demand drug release. Although few reports demonstrated the release of multiple drugs, obtaining the release of different molecules with different release patterns remains a major challenge. The development of a suitable approach to reload the exhausted hydrogel depot using the same or other suitable drugs could be a useful approach. Fortunately, recent advances in biomaterials and the fundamental understanding of medicines offer several possibilities to fabricate an omni-potent hydrogel-based delivery system. Such hydrogel systems improve the efficacy of therapeutics and reduce the product cost, which eventually improve human health care. In addition to the controlled delivery, the tunable mechanical properties of pH-responsive polymeric biomaterials can also be exploited in tissue engineering applications. In the near future, we hope to see more biomaterial products on the market that are fabricated using pH-responsive biomaterials.
Author contributions
TT and JMJ contributed equally to this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2020R1I1A1A01075103).References
- Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Nat. Rev. Mater., 2016, 2, 16075 CrossRef.
- S. Chagri, D. Y. W. Ng and T. Weil, Nat. Rev. Chem., 2022, 6, 320–338 CrossRef PubMed.
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152–167 CrossRef CAS PubMed.
- K. Jindrich, V. Jiri and L. Drahoslav, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 2801–2815 CrossRef.
- K. Joyce, G. T. Fabra, Y. Bozkurt and A. Pandit, Signal Transduction Targeted Ther., 2021, 6, 122 CrossRef CAS PubMed.
- M. Rahmati, E. A. Silva, J. E. Reseland, C. A. Heyward and H. J. Haugen, Chem. Soc. Rev., 2020, 49, 5178–5224 RSC.
- H. Kim, S. G. Kumbar and S. P. Nukavarapu, Curr. Opin. Biomed. Eng., 2021, 17, 100260 CrossRef CAS PubMed.
- A. R. Del Bakhshayesh, N. Asadi, A. Alihemmati, H. Tayefi Nasrabadi, A. Montaseri, S. Davaran, S. Saghati, A. Akbarzadeh and A. Abedelahi, J. Biol. Eng., 2019, 13, 85 CrossRef PubMed.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- D. S. Kohane and R. Langer, Pediatr. Res., 2008, 63, 487–491 CrossRef CAS PubMed.
- K. Battiston, I. Parrag, M. Statham, D. Louka, H. Fischer, G. Mackey, A. Daley, F. Gu, E. Baldwin, B. Yang, B. Muirhead, E. A. Hicks, H. Sheardown, L. Kalachev, C. Crean, J. Edelman, J. P. Santerre and W. Naimark, Nat. Commun., 2021, 12, 2875 CrossRef CAS PubMed.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860 CrossRef CAS PubMed.
- S. P. Schwendeman, R. B. Shah, B. A. Bailey and A. S. Schwendeman, J. Controlled Release, 2014, 190, 240–253 CrossRef CAS PubMed.
- R. Dimatteo, N. J. Darling and T. Segura, Adv. Drug Delivery Rev., 2018, 127, 167–184 CrossRef CAS PubMed.
- A. Roy, K. Manna and S. Pal, Mater. Chem. Front., 2022, 6, 2338–2385 RSC.
- J. Madsen, G. Madden, E. Themistou, N. J. Warren and S. P. Armes, Polym. Chem., 2018, 9, 2964–2976 RSC.
- A. Kasiński, M. Zielińska-Pisklak, E. Oledzka and M. Sobczak, Int. J. Nanomed., 2020, 15, 4541–4572 CrossRef PubMed.
- L. Xu, L. Qiu, Y. Sheng, Y. Sun, L. Deng, X. Li, M. Bradley and R. Zhang, J. Mater. Chem. B, 2018, 6, 510–517 RSC.
- M. Rizwan, R. Yahya, A. Hassan, M. Yar, A. D. Azzahari, V. Selvanathan, F. Sonsudin and C. N. Abouloula, Polymers, 2017, 9, 137 CrossRef PubMed.
- Y. Chen, Y. Gao, L. P. da Silva, R. P. Pirraco, M. Ma, L. Yang, R. L. Reis and J. Chen, Polym. Chem., 2018, 9, 4063–4072 RSC.
- S. L. Canning, T. J. Neal and S. P. Armes, Macromolecules, 2017, 50, 6108–6116 CrossRef CAS PubMed.
- J. Hu, Z. Zheng, C. Liu, Q. Hu, X. Cai, J. Xiao and Y. Cheng, Biomater. Sci., 2019, 7, 581–584 RSC.
- F. Ofridam, M. Tarhini, N. Lebaz, É. Gagnière, D. Mangin and A. Elaissari, Polym. Adv. Technol., 2021, 32, 1455–1484 CrossRef CAS.
- S. Chu, X. Shi, Y. Tian and F. Gao, Front. Oncol., 2022, 12, 855019 CrossRef PubMed.
- W. Wu, L. Luo, Y. Wang, Q. Wu, H.-B. Dai, J.-S. Li, C. Durkan, N. Wang and G.-X. Wang, Theranostics, 2018, 8, 3038–3058 CrossRef CAS PubMed.
- Y. Lu, Q. Lv, B. Liu and J. Liu, Biomater. Sci., 2019, 7, 4963–4983 RSC.
- K. Stubenrauch, I. Voets, G. Fritz-Popovski and G. Trimmel, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1178–1191 CrossRef CAS.
- D. Sprouse, Y. Jiang, J. E. Laaser, T. P. Lodge and T. M. Reineke, Biomacromolecules, 2016, 17, 2849–2859 CrossRef CAS PubMed.
- M. A. Quadir, S. W. Morton, Z. J. Deng, K. E. Shopsowitz, R. P. Murphy, T. H. Epps and P. T. Hammond, Mol. Pharm., 2014, 11, 2420–2430 CrossRef CAS PubMed.
- Q. Shi, H. Liu, D. Tang, Y. Li, X. Li and F. Xu, NPG Asia Mater., 2019, 11, 64 CrossRef CAS.
- B. Gao, Q. Zhang, K. Muhammad, X. Ren, J. Guo, S. Xia, W. Zhang and Y. Feng, Biomater. Sci., 2019, 7, 2061–2075 RSC.
- L. Zhang, S. Hu, L. Zhang, W. Wu, Q. Cheng, J. Li and R. Narain, Biomater. Sci., 2022, 10, 4271–4283 RSC.
- B. Lu, H. Yuk, S. Lin, N. Jian, K. Qu, J. Xu and X. Zhao, Nat. Commun., 2019, 10, 1043 CrossRef PubMed.
- J.-W. Jeong, G. Shin, S. I. Park, Ki J. Yu, L. Xu and J. A. Rogers, Neuron, 2015, 86, 175–186 CrossRef CAS PubMed.
- S. P. Lacour, G. Courtine and J. Guck, Nat. Rev. Mater., 2016, 1, 16063 CrossRef CAS.
- E. Caló and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267 CrossRef.
- F. Wang, J. Chen, J. Liu and H. Zeng, Biomater. Sci., 2021, 9, 3543–3575 RSC.
- T. Yoshida, T. C. Lai, G. S. Kwon and K. Sako, Expert Opin. Drug Delivery, 2013, 10, 1497–1513 CrossRef CAS PubMed.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- W. Tao, J. Wang, W. J. Parak, O. C. Farokhzad and J. Shi, ACS Nano, 2019, 13, 4876–4882 CrossRef CAS PubMed.
- S. Zhuo, F. Zhang, J. Yu, X. Zhang, G. Yang and X. Liu, Molecules, 2020, 25, 5649 CrossRef CAS PubMed.
- Z. Zhao, C. Ding, Y. Wang, H. Tan and J. Li, Biomater. Sci., 2019, 7, 1643–1651 RSC.
- R. Y. H. Tan, C. S. Lee, M. R. Pichika, S. F. Cheng and K. Y. Lam, Polymer, 2022, 14, 1672 CAS.
- K. Qu, Z. Yuan, Y. Wang, Z. Song, X. Gong, Y. Zhao, Q. Mu, Q. Zhan, W. Xu and L. Wang, ChemPhysMater, 2022, 1, 294–309 CrossRef.
- B. H. Morrow, G. F. Payne and J. Shen, J. Am. Chem. Soc., 2015, 137, 13024–13030 CrossRef CAS PubMed.
- J. Du, L. Fan and Q. Liu, Macromolecules, 2012, 45, 8275–8283 CrossRef CAS.
- L. Liu, W. Yao, Y. Rao, X. Lu and J. Gao, Drug Delivery, 2017, 24, 569–581 CrossRef CAS PubMed.
- J. D. Willott, T. J. Murdoch, B. A. Humphreys, S. Edmondson, E. J. Wanless and G. B. Webber, Langmuir, 2015, 31, 3707–3717 CrossRef CAS PubMed.
- T. Swift, L. Swanson, M. Geoghegan and S. Rimmer, Soft Matter, 2016, 12, 2542–2549 RSC.
- F. R. Wibowo, O. A. Saputra, W. W. Lestari, M. Koketsu, R. R. Mukti and R. Martien, ACS Omega, 2020, 5, 4261–4269 CrossRef CAS PubMed.
- H. Tang, W. Zhao, J. Yu, Y. Li and C. Zhao, Molecules, 2018, 24, 4 CrossRef PubMed.
- S. G. Roy and P. De, J. Appl. Polym. Sci., 2014, 131, 41084 CrossRef.
- Z. Li, J. Huang and J. Wu, Biomater. Sci., 2021, 9, 574–589 RSC.
- A. Car, P. Baumann, J. T. Duskey, M. Chami, N. Bruns and W. Meier, Biomacromolecules, 2014, 15, 3235–3245 CrossRef CAS PubMed.
- X. Han, X. Zhang, H. Zhu, Q. Yin, H. Liu and Y. Hu, Langmuir, 2013, 29, 1024–1034 CrossRef CAS PubMed.
- D. M. Lynn and R. Langer, J. Am. Chem. Soc., 2000, 122, 10761–10768 CrossRef CAS.
- K.-T. Huang, P.-S. Hsieh, L.-G. Dai and C.-J. Huang, J. Mater. Chem. B, 2020, 8, 7390–7402 RSC.
- C. Vasile, D. Pamfil, E. Stoleru and M. Baican, Molecules, 2020, 25, 1539 CrossRef CAS PubMed.
- C. T. Huynh, M. K. Nguyen and D. S. Lee, Chem. Commun., 2012, 48, 10951–10953 RSC.
- Y. Wu, D. Y. W. Ng, S. L. Kuan and T. Weil, Biomater. Sci., 2015, 3, 214–230 RSC.
- G. R. Shin, H. E. Kim, J. H. Kim, S. Choi and M. S. Kim, Pharmaceutics, 2021, 13, 1953 CrossRef CAS PubMed.
- M. V. Liberti and J. W. Locasale, Trends Biochem. Sci., 2016, 41, 211–218 CrossRef CAS PubMed.
- J. Huang and X. Jiang, ACS Appl. Mater. Interfaces, 2018, 10, 361–370 CrossRef CAS PubMed.
- M. K. Nguyen, C. T. Huynh and D. S. Lee, Polymer, 2009, 50, 5205–5210 CrossRef CAS.
- D. P. Huynh, M. K. Nguyen, B. S. Pi, M. S. Kim, S. Y. Chae, K. C. Lee, B. S. Kim, S. W. Kim and D. S. Lee, Biomaterials, 2008, 29, 2527–2534 CrossRef CAS PubMed.
- M. K. Nguyen and D. S. Lee, Chem. Commun., 2010, 46, 3583–3585 RSC.
- M. K. Nguyen, C. T. Huynh, G. H. Gao, J. H. Kim, D. P. Huynh, S. Y. Chae, K. C. Lee and D. S. Lee, Soft Matter, 2011, 7, 2994–3001 RSC.
- C. T. Huynh, M. K. Nguyen, D. P. Huynh, S. W. Kim and D. S. Lee, Polymer, 2010, 51, 3843–3850 CrossRef CAS.
- C. T. Huynh, M. K. Nguyen, J. H. Kim, S. W. Kang, B. S. Kim and D. S. Lee, Soft Matter, 2011, 7, 4974–4982 RSC.
- N. K. Singh, Q. V. Nguyen, B. S. Kim and D. S. Lee, Nanoscale, 2015, 7, 3043–3054 RSC.
- V. H. G. Phan, T. Thambi, H. T. T. Duong and D. S. Lee, Sci. Rep., 2016, 6, 29978 CrossRef CAS PubMed.
- M. H. Turabee, T. Thambi, J. S. Lym and D. S. Lee, Biomater. Sci., 2017, 5, 837–848 RSC.
- M. H. Turabee, T. Thambi, H. T. T. Duong, J. H. Jeong and D. S. Lee, Biomater. Sci., 2018, 6, 661–671 RSC.
- Z. Bao, C. Xian, Q. Yuan, G. Liu and J. Wu, Adv. Healthcare Mater., 2019, 8, 1900670 CrossRef PubMed.
- X. Qi, W. Wei, J. Li, G. Zuo, X. Pan, T. Su, J. Zhang and W. Dong, Mol. Pharm., 2017, 14, 431–440 CrossRef CAS PubMed.
- Y. Xu, G. Lu, M. Chen, P. Wang, Z. Li, X. Han, J. Liang, Y. Sun, Y. Fan and X. Zhang, Carbohydr. Polym., 2020, 250, 116979 CrossRef CAS PubMed.
- E. Pérez-Herrero and A. Fernández-Medarde, Acta Pharm. Sin. B, 2021, 11, 2243–2264 CrossRef PubMed.
- I. Druzhkova, M. Lukina, V. Dudenkova, N. Ignatova, L. Snopova, A. Gavrina, L. Shimolina, V. Belousov, E. Zagaynova and M. Shirmanova, Cell Cycle, 2021, 20, 1540–1551 CrossRef CAS PubMed.
- W. S. Shim, J.-H. Kim, K. Kim, Y.-S. Kim, R.-W. Park, I.-S. Kim, I. C. Kwon and D. S. Lee, Int. J. Pharm., 2007, 331, 11–18 CrossRef CAS PubMed.
- Q. V. Nguyen, J. S. Lym, C. T. Huynh, B. S. Kim, H. J. Jae, Y. I. Kim and D. S. Lee, Polym. Chem., 2016, 7, 5805–5818 RSC.
- C. T. Huynh, Q. V. Nguyen, J. S. Lym, B. S. Kim, D. P. Huynh, H. J. Jae, Y. I. Kim and D. S. Lee, RSC Adv., 2016, 6, 47687–47697 RSC.
- Q. V. Nguyen, M. S. Lee, J. S. Lym, Y. I. Kim, H. J. Jae and D. S. Lee, J. Mater. Chem. B, 2016, 4, 6524–6533 RSC.
- T. M. D. Le, H. T. T. Duong, T. Thambi, V. H. Giang Phan, J. H. Jeong and D. S. Lee, Biomacromolecules, 2018, 19, 3536–3548 CrossRef CAS PubMed.
- J. M. Jung, S. H. Kim, V. H. Giang Phan, T. Thambi and D. S. Lee, Biomater. Sci., 2021, 9, 7275–7286 RSC.
- F. Raza, Y. Zhu, L. Chen, X. You, J. Zhang, A. Khan, M. W. Khan, M. Hasnat, H. Zafar, J. Wu and L. Ge, Biomater. Sci., 2019, 7, 2023–2036 RSC.
- D. H. Kim, Y. K. Seo, T. Thambi, G. J. Moon, J. P. Son, G. Li, J. H. Park, J. H. Lee, H. H. Kim, D. S. Lee and O. Y. Bang, Biomaterials, 2015, 61, 115–125 CrossRef CAS PubMed.
- H. T. T. Duong, N. W. Kim, T. Thambi, V. H. Giang Phan, M. S. Lee, Y. Yin, J. H. Jeong and D. S. Lee, J. Controlled Release, 2018, 269, 225–234 CrossRef CAS PubMed.
- T. Thambi, V. H. Giang Phan, S. H. Kim, T. M. Duy Le, H. T. T. Duong and D. S. Lee, Biomater. Sci., 2019, 7, 5424–5437 RSC.
- M. S. Gil, J. Cho, T. Thambi, V. H. Giang Phan, I. Kwon and D. S. Lee, J. Controlled Release, 2017, 267, 119–132 CrossRef CAS PubMed.
- T. Koga, Y. Oatari, H. Motoda, S.-n. Nishimura, Y. Sasaki, Y. Okamoto, D. Yamamoto, A. Shioi and N. Higashi, Biomacromolecules, 2022, 23, 2941–2950 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |