
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanocarriers for the delivery of antibiotics into cells against intracellular bacterial infection
Chao
Wang†
 a,
Yi
Yang†
a,
Yuanyuan
Cao
b,
Kaixin
Liu
a,
Hua
Shi
a,
Xudong
Guo
a,
Wanying
Liu
a,
Rongzhang
Hao
*b,
Hongbin
Song
*a and
Rongtao
Zhao
*a
a,
Yi
Yang†
a,
Yuanyuan
Cao
b,
Kaixin
Liu
a,
Hua
Shi
a,
Xudong
Guo
a,
Wanying
Liu
a,
Rongzhang
Hao
*b,
Hongbin
Song
*a and
Rongtao
Zhao
*a
aChinese PLA Center for Disease Control and Prevention, Beijing, 100071, China. E-mail: zhaorongtao1984@163.com; hongbinsong@263.net
bDepartment of Toxicology and Sanitary Chemistry, School of Public Health, Capital Medical University, Beijing, 100069, China. E-mail: hao@ccmu.edu.cn
First published on 6th December 2022
Abstract
The barrier function of host cells enables intracellular bacteria to evade the lethality of the host immune system and antibiotics, thereby causing chronic and recurrent infections that seriously threaten human health. Currently, the main clinical strategy for the treatment of intracellular bacterial infections involves the use of long-term and high-dose antibiotics. However, insufficient intracellular delivery of antibiotics along with various resistance mechanisms not only weakens the efficacy of current therapies but also causes serious adverse drug reactions, further increasing the disease and economic burden. Improving the delivery efficiency, intracellular accumulation, and action time of antibiotics remains the most economical and effective way to treat intracellular bacterial infections. The rapid development of nanotechnology provides a strategy to efficiently deliver antibiotics against intracellular bacterial infections into cells. In this review, we summarize the types of common intracellular pathogens, the difficulties faced by antibiotics in the treatment of intracellular bacterial infections, and the research progress of several types of representative nanocarriers for the delivery of antibiotics against intracellular bacterial infections that have emerged in recent years. This review is expected to provide a reference for further elucidating the intracellular transport mechanism of nanocarrier-drug complexes, designing safer and more effective nanocarriers and establishing new strategies against intracellular bacterial infection.
Introduction
Intracellular parasitic bacteria (hereafter referred to as intracellular bacteria) cause a wide range of diseases and significantly contribute to chronic, persistent, and latent infections, which seriously threaten public health.1 Intracellular bacteria that invade host cells through various pathways, including the respiratory tract, digestive tract, skin, and mucosa, can use virulence factors to simulate host enzymes or secrete unique proteins to mediate adhesion, invasion, immune escape, and autophagy, enabling their survival and reproduction in host cells.2,3 The intracellular environment provides a preferential niche for bacteria because they are protected not only from host defences but also from antimicrobial therapy. Intracellular bacteria are classified as obligate if they are unable to grow outside a host cell, such as Rickettsia and Chlamydia, or as facultative if they are able to grow either inside or outside of a host cell, such as Mycobacterium tuberculosis (M. tb), Staphylococcus aureus (S. aureus), Salmonella, and Brucella.4 Not only can intracellular bacteria evade the attack of the host immune system, but host cells can also provide a shelter for intracellular bacteria to avoid being killed by antibiotics.5 More importantly, intracellular bacteria can also spread within the host cells from the initial infection site to different tissues, causing diseases such as meningitis,6 osteomyelitis,7 lung infection,8 and endocarditis.9Currently, the main strategy for clinical treatment of intracellular bacterial infections is the use of long-term and high-dose antibiotics. However, more than two-thirds of the known antibiotics have poor efficacy in treating intracellular bacterial infections. Cellular metabolism, active efflux, and an unsatisfactory intracellular distribution of drugs lead to low intracellular bactericidal efficacy, hindering the killing of intracellular pathogenic bacteria, even if high doses are used.10 Lehar et al.11 evaluated the minimum inhibitory concentrations (MICs) of vancomycin (VCM), doxycycline, linezolid, and rifampicin against intracellular and extracellular methicillin-resistant S. aureus (MRSA). The results showed that the MIC of the same antibiotic against intracellular bacteria was 25∼104 times higher than that of extracellular bacteria. Intracellular bacterial infection has become an international public health concern because maintaining intracellular therapeutic concentrations of antibiotics is challenging. Intracellular pathogens are exposed to subtherapeutic concentrations for prolonged periods, which is more likely to result in drug resistance.11 Therefore, developing new safe and efficient approaches to overcome intracellular bacterial infections is crucial.
In recent years, the effective delivery of antibiotics into cells based on nanotechnology for the enhancement of intracellular antibacterial activity has become a hot spot in anti-intracellular bacterial infection research.12–14 Although there are many reviews of nanocarriers, most of the nanocarriers are used to deliver anti-tumor drugs,15 genes16 or CRISPR gene editing systems.17 And those reported nanocarriers used to deliver antibiotics mainly focused on local delivery to improve drug concentration at the infection site18 and overcoming bacterial envelope barrier to promote bacterial internalization of antibiotic.19 The research articles of nanocarriers for effective delivery of antibiotics into host cells to improve the anti-intracellular infection effect is increasing. However, few reviews systematically analysed the typical use and further mechanism of nanocarriers against intracellular infections to broaden their future application. In this review, we provided an overview and comprehensive analysis of the entry mechanism of common intracellular bacteria encountered in clinical practice, the current status of antimicrobial treatment of intracellular bacterial infection, especially, promising novel anti-intracellular bacterial nanocarriers with representative examples. Meanwhile we summarized the novel strategies on how to overcome the problems of poor drug penetration into cells. This review could help understand the intracellular transport mechanism and the progress of nanocarrier-drug complexes, to develop safer and more efficient nanocarriers, and establish new strategies against intracellular bacterial infections.
Common intracellular bacteria
Intracellular bacteria include many clinically important pathogens such as S. aureus, Salmonella, M. tb and Listeria monocytogenes (L. monocytogenes). These pathogens have developed strategies to invade and survive within mammalian host cells. After phagocytosis, the pathogens are delivered to an endosomal compartment that would normally fuse with the lysosomes. However, intracellular pathogens such as M. tb and Salmonella could modify the compartment to prevent the fusion. Other pathogens like L. monocytogenes could escape from endosomal compartment before getting digested followed by replication in the cytosol.20 Failure to eliminate the intracellular pathogens often causes persistent infection, which in turn may lead to chronic infection or life-long latent (slow) infection.21M. tb
Tuberculosis caused by M. tb is one of the most harmful zoonotic infectious diseases. M. tb can escape the surveillance and clearance of the immune system and survive in macrophages for a long time. However, the molecular mechanisms contributing to these effects have not yet been fully elucidated. M. tb is transmitted through droplets containing the bacterium; once the bacteria reach the pulmonary airways, they are recognised by surface receptors on alveolar macrophages. Subsequently, through endocytosis, membrane invagination, and fusion, a phagosome containing M. tb is formed, which secretes virulence factors into the cell and inhibits the host immune response, resulting in persistent intracellular infection.22M. tb has been shown to evade the macrophage killing function by preventing the fusion of phagosomes and lysosomes, and then establishes an ecological niche in macrophages for its reproduction. M. tb can also penetrate phagosome membranes and escape to the cytoplasm.23S. aureus
S. aureus is a common food-borne and nosocomial Gram-positive pathogen. Originally, S. aureus was considered to be an exclusive extracellular pathogen, but recent studies have shown that it can also internalise into various cell types such as keratinocytes and endothelial cells.24 Moreover, S. aureus can invade phagocytic cells that respond to clear bacteria, and spread to tissues and organs through the blood circulation, causing chronic infectious diseases such as osteomyelitis, pneumonia, and endocarditis.2S. aureus invades the host cell through a zipper uptake mechanism, in which the bacterium adheres to the host cell surface and remains in the cell in the form of small colony variants (SCVs).25 When S. aureus changes from a normal state to an SCV, it is more likely to form a bacterial reservoir in the cells, which enables it to skilfully defend itself against the intracellular defence system.25,26 Finally, intracellular S. aureus can not only survive and multiply in the acidic phagolysosomes of the host cell but also escape to the cytoplasmic matrix to further induce cell death and enter the bloodstream, thereby initiating systemic infection.2Salmonella
Salmonella is an intracellular facultative anaerobic pathogen that mainly invades the lymphoepithelial cells or intestinal epithelial cells of the intestinal mucosa, causing diseases such as typhoid, paratyphoid, and gastroenteritis.27 After entering the gastrointestinal tract, Salmonella interacts with intestinal epithelial cells with the help of virulence proteins, triggering the intima folds and actin rearrangement of host cells. Subsequently, the bacteria can be phagocytosed by macrophages.27Salmonella can use the type III secretion system to form Salmonella-containing vacuoles, providing a safe location for the bacteria to multiply and escape the intracellular immune response killing effect.28Salmonella can also mediate the migration of infected macrophages by releasing a variety of virulence factors, which further spread the bacteria to internal organs such as the liver and spleen.L. monocytogenes
L. monocytogenes is a Gram-positive intracellular bacterium that invades phagocytic and non-phagocytic cells primarily through gastrointestinal infection. After invading the body, L. monocytogenes parasitises and replicates in the liver and spleen, and then enters the brain or placenta through the blood to cause meningitis, encephalitis, and sepsis, with a mortality rate of 20–30%.29L. monocytogenes uses fibronectin-binding protein A and Listeria adhesion protein to adhere to and enter host cells through the classical phagocytosis pathway. L. monocytogenes can also invade host cells through a zipper mechanism. Specifically, L. monocytogenes binds to E-cadherin and the hepatocyte growth factor receptor through surface proteins such as internalin In1A and In1B, causing cytoskeleton rearrangement and initiating clathrin-mediated endocytosis.30 Through the action of adhesion virulence factors (internalins) and the secretion of α-hemolysin, L. monocytogenes invades the host intestinal epithelial cells and then escapes from the phagosome into the cytoplasm of the host cell, where it proliferates and differentiates by depriving the host cell of nutrients, thereby causing systemic infection.31Brucella
Brucella is an aerobic facultative intracellular bacterium that can cause brucellosis, miscarriage, and infertility when it invades the reproductive system of animals. Humans can be infected by inhaling aerosolised bacteria or by ingesting or contacting contaminated tissues, resulting in persistent fever, muscle and joint pain, and other symptoms. Additionally, Brucella may cause chronic infections and debilitating diseases such as osteomyelitis, arthritis, neurological symptoms, and endocarditis.32 Macrophages are the main target cells of Brucella. Studies have shown that Brucella can weaken the function of macrophages and partially disable their killing effect and antigen-presenting function, thereby enabling long-term infection in host cells.33 Some Brucella may form Brucella-containing vacuoles in macrophages, which can prevent phagolysosome fusion through rapid acidification and facilitate their reproduction without affecting macrophage survival.34 In addition, the polysaccharides on the Brucella cell membrane can inhibit the apoptosis of phagocytes, which leads to the immune escape and proliferation of Brucella in host cells. Through the bloodstream, endotoxins and a variety of virulence factors are released simultaneously to trigger systemic infection.32Challenges of antibiotics in the treatment of intracellular bacterial infections
Intracellular penetration, accumulation, and distribution are important parameters governing the activity of antibiotics against intracellular bacteria. However, various factors such as the host cell barrier, active efflux of transmembrane transporters, and subcellular conditions have hindered the ability of antibiotics to effectively enter host cells, and weaken the intracellular accumulation and antibacterial ability (Fig. 1). It is difficult to maintain the effective concentration and action time of antibiotics at the intracellular infection site, hindering the clinical treatment of intracellular bacterial infection.35 | ||
| Fig. 1 Challenges of antibiotics in the treatment of intracellular bacterial infections. (a) blockage of antibiotics by cellular barrier, (b) cellular efflux of antibiotics, (C) losing activity under subcellular conditions, (d) different distribution of antibiotics and bacteria, (e) intracellular stress induced transformation into non-replicating form. | ||
Blockage of antibiotic entry into cells
Antibiotics with high hydrophilicity, such as aminoglycosides, have restricted cellular penetration ability and thus failed to kill intracellular bacteria. Although some β-lactam antibacterial drugs can enter cells through passive diffusion, the concentration ratio of the intracellular/extracellular drugs is <1 after equilibrium.36 Therefore, the use of these types of antibiotics to treat intracellular bacterial infections requires a sufficient extracellular drug concentration, which increases the risk of side effects.37Cellular efflux of antibiotics
Active efflux of antibiotics by transmembrane transporters is an important factor affecting the intracellular concentration of antibiotics. Efflux transporters accelerate the efflux of antibiotics to protect cells, but simultaneously reduce the intracellular efficacy of antibiotics.38 For example, quinolones can enter cells by simple passive diffusion with an intracellular/extracellular drug ratio >1. These antibiotics have no specific binding target in cells and can diffuse into different organelles.39 However, the rapid excretion rate of quinolones from cells makes them unable to maintain effective concentrations in the cells for a prolonged period, which limits the ability of these antibiotics to treat intracellular bacterial infections.40Low intracellular bioavailability of antibiotics
Some antibiotics lose their activity under the complex physical and chemical subcellular conditions, such as inactivating enzymes, acidity of lysosomes, and low oxygen concentration. Therefore, their low intracellular active concentration is often subtherapeutic, resulting in low effectiveness against intracellular pathogens, which also provides the right condition for the emergence of antibiotic resistance. For example, the acidic pH in lysosomes weakens the antibacterial activity of the alkalescent azithromycin against intracellular L. monocytogenes and S. aureus.41 Additionally, some antibiotics can bind to negatively charged components in cells. For example, macrolides and glycopeptide antibiotics (e.g. oritavancin) are easily combined with lipoproteins on cell membranes, affecting their antimicrobial efficacy.42Different intracellular distribution of antibiotics and pathogens
The intracellular bactericidal effect of antibiotics is not only related to their intracellular concentration and activity but also their intracellular distribution. Pathogens may inhabit different compartments in host cells. L. monocytogenes proliferates within the cytoplasm,43 whereas Salmonella exploits the late endosomal compartments.44 The subcellular distribution of antibiotics is not uniform, and different subcellular distributions of antibiotics and pathogens may result in the inability of antibiotics to kill intracellular bacteria. For example, oritavancin can effectively kill intracellular S. aureus but does not affect intracellular L. monocytogenes. This is likely due to the co-localisation of oritavancin and S. aureus in the lysosome, whereas L. monocytogenes is located in the cytoplasm.45The dormant character of intracellular bacteria
The stress induced by antibiotics and host oxidative response can cause the transformation of bacteria into a non-replicating metabolic state.46 This prevents many antibiotics from expressing their activity led to the inefficiency against intracellular bacteria. Numerous bacteria like M. tb, Salmonella and S. aureus can persist in a dormant state with reduced metabolism which results in high tolerance to antibiotics over a long period of time.46 For example, Wayne and Sohaskey had discussed the non-replicating nature of M. tb within the host that causes latent infections that are often resistant to conventional treatment.47Poor tissue distribution of antibiotics
Treatment of intracellular bacterial infections also depends on antibiotics entering the cell from the bloodstream and maintaining a certain activity and concentration within the cell. The ability of a drug to do so depends on tissue-related factors (such as perfusion to the tissues and the surface area of the tissue's vascular bed) and drug-related factors (such as lipid solubility, molecular size and plasma protein binding). Actually, different antibiotics have different absorption, distribution and excretion processes after systemic administration in human bodies, which greatly affects the blood concentration and tissue distribution of antibiotics.36 For example, daptomycin and the beta-lactam ceftriaxone, which are highly bound to plasma proteins, hardly diffuse out of capillaries, resulting in their distribution limited to the extracellular milieu. Conversely, other beta-lactam antibiotics and aminoglycosides, which have lower plasma protein-binding, can be rapidly distributed to various tissues and body fluids to reach infection sites.48 In addition, the ability of antibiotics to distinguish between infected and uninfected cells is an important strategy for treatment of intracellular pathogens and reduction of drug side effects, this is also the future development of new antibiotics worthy of research. For example, alveolar macrophages, the host cell susceptible to M. tb, could overexpress mannose receptors, and mannose ligand could be used to modify drugs to improve the ability of antibiotic to target infected cells.49Research and application of nano drug-delivery systems against intracellular bacteria
Given the above challenges related to the use of antibiotics in the treatment of intracellular bacterial infections, antimicrobial peptides (AMPs),50 cell-penetrating peptides,51 and antibacterial nanomaterials52 have received extensive attention as novel antibacterial substances owing to their properties of high membrane permeability and various antibacterial effects. Specifically, progress in nano drug-delivery systems have offered a new direction of anti-intracellular bacteria research. These systems involve one or more antimicrobial adjuncts that are dispersed, absorbed, or encapsulated in nanocarriers (i.e. lipids and macromolecules) for delivering antimicrobial adjuncts into cells to increase the intracellular concentration and antibacterial activity.14,42Compared with traditional drugs, nanocarrier system has several advantages such as high cellular uptake efficiency, long circulation time, and targeting ability.53 As shown in Fig. 2, a nanocarrier system can enter cells by endocytosis, a process in which the cellular membrane engulfs the carrier and splits off to form a self-contained vesicle within the cell. Endocytosis mechanisms include phagocytosis, macropinocytosis, clathrin- and caveolae-mediated endocytosis, and clathrin- and caveolae-independent routes. Besides endocytosis, direct cytosolic delivery is an exciting strategy for the intracellular delivery of biologics, owing to its ability to bypass endosomal entrapment and deliver almost 100% of the encapsulated cargo to the cytoplasm. The routes of direct cytosolic delivery by means including contact release, membrane fusion, direct translocation, and so on.54 In this section, we provide an overview of the current research on several types of recently developed nanocarriers for the delivery of antibiotics into cells against intracellular bacteria (Table 1).
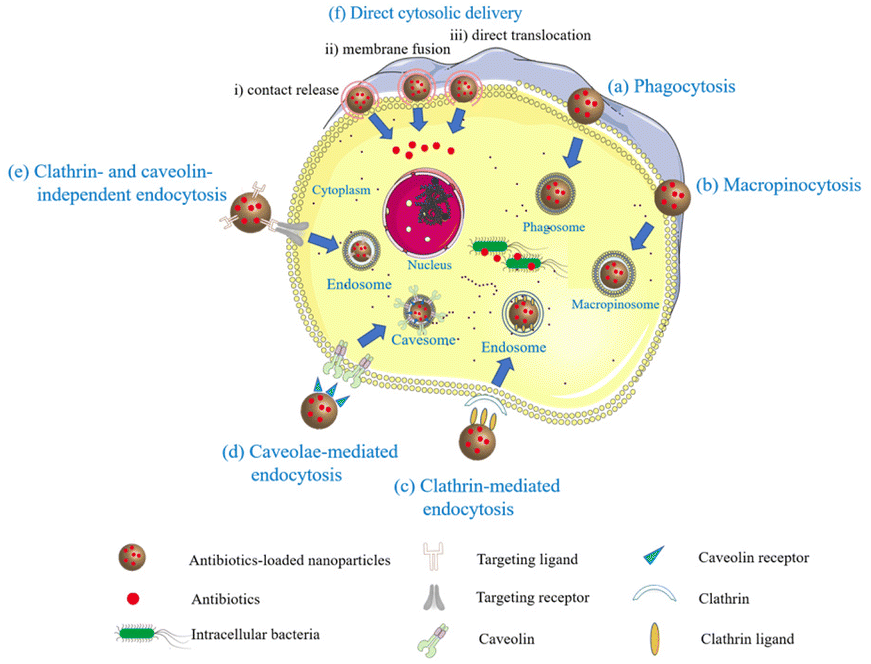 | ||
| Fig. 2 Schematic diagram of the main strategies of nanocarriers for the delivery of antibiotics into cells: (a) phagocytosis, (b) macropinocytosis, (c) clathrin-mediated endocytosis, (d) caveolae-mediated endocytosis, (e) clathrin- and caveolin independent endocytosis, and (f) direct cytosolic delivery: (i) contact release; (ii) membrane fusion; (iii) direct translocation. | ||
| Type of carrier | Examples of encapsulated antibiotics | Representative targeted intracellular bacteria | Advantages | Limitations |
|---|---|---|---|---|
| Liposomes | Penicillin,56 doxycycline,57 vancomycin,13 colistin58 | S. aureus, M. tb, Salmonella | Low immunogenicity; good safety, and membrane fusion ability | Poor storage stability, drug leakage, short half-life, low solubility |
| Solid lipid nanoparticles | Enrofloxacin,60 streptomycin,12 rifampicin49 | Salmonella, M. tb | High stability and good biocompatibility | Loss of high amounts of drug, lack of robust controlled drug release |
| Polymeric nanoparticles | Gentamicin,61 ciprofloxacin,62,66 streptomycin,67 rifampin68 | L. monocytogenes, M. tb, S. aureus | High stability, good biocompatibility, and degradability | Residual organic solvent, hard to expand for large-scale production |
| Inorganic non-metallic nanomaterials | Rifampicin,74,84 gentamicin,75 vancomycin,77 ciprofloxacin78 | S. aureus, M. tb | Large specific surface area, high drug-loading rate, and easy functional modification | Toxic degradation products, complex synthesis processes |
| Metal nanoparticles | Vancomycin,92 gentamicin86 | Pseudomonas aeruginosa, S. aureus, Salmonella | Antibacterial properties, large specific surface area, strong penetrability, and easy surface modification | Possible toxicity, oxidative stress |
| Dendrimers | Azithromycin,70 vancomycin71 | Chlamydia, MRSA | Controllable configuration and flexibility, built-in cavity structure, enriched with active functional groups on the surface | Accumulation in body tissues, possible cytotoxicity |
| Endogenous nanocarriers | Linezolid103 | MRSA | Excellent biocompatibility and low immunogenicity | Hard to obtain high yield of pure nanocarriers |
Liposomes
Liposomes are spherical or spheroid-like vesicles composed of several lipid layers that surround the aqueous space. One of their main advantages is the use of natural nontoxic and biodegradable phospholipids or cholesterol, which have low immunogenicity, good safety, and strong membrane fusion ability.55 Liposomes are one of the most commonly usednanocarriers, and are also widely used to achieve the intracellular delivery of antibiotics. Antibacterial drugs such as VCM, gentamicin, clarithromycin, and rifampicin have been successfully loaded into liposomes to date. Owing to the special structure of liposomes, hydrophilic drugs can be encapsulated in the inner core with hydrophobic drugs placed in the lipid layer. The membrane fusion ability of liposomes has also been used to improve the therapeutic activity of drugs against intracellular pathogens. For example, Zhang et al.56 prepared penicillin-loaded liposome nanoparticles (PenG-PL NPs) and evaluated their inhibitory effect on methicillin-sensitive S. aureus (MSSA) in A549 lung cancer cells. Treatment of the free drug (penicillin concentration, 2.5–20 μg mL−1) to cells (containing 108.49 ± 0.15 colony-forming units [CFU] per mL intracellular bacteria), the intracellular bacteria were reduced by <10%. The PenG-PL NP treatment resulted in significantly higher reduction of intracellular bacteria than the corresponding free drug group, with the highest reduction (67.4 ± 0.6%) observed at a concentration of 20 μg mL−1, which was equivalent to the removal of ≥99.99% (∼108.5 CFU) of bacteria. Franklin et al.57 prepared doxycycline-loaded dipalmitoyl phosphatidylcholine liposomes (DPPC-doxy) and sphingomyelin doxycycline liposomes (sphing-doxy) through a sulfuric acid loading method. They subcutaneously injected 50 mg kg−1 DPPC-doxy and sphing-doxy and 5 mg kg−1 free doxycycline (STD-doxy) in rats. The results showed that the mean in vivo residence time of DPPC-doxy was the highest (111.78 h), followed by sphing-doxy (56.00 h) and STD-doxy (6.86 h). DPPC-doxy and sphing-doxy were also detectable with doxycycline (0.2 μg mL−1) in the rat serum 336 h after administration. Therefore, both DPPC-doxy and sphing-doxy could inhibit the activity of Mycobacterium in J774A.1 macrophages for 24–48 h. Apart from conventional liposome carriers, the composition of surface modification groups can be manipulated to obtain liposomes with a desired stimuli-responsive property. For example, Omolo et al.13 inserted a novel oleic acid-derived quaternary lipid into liposomes as a pH-responsive “switch”, thereby improving the release rate of the loaded VCM in an acidic environment (Fig. 3). Compared with free VCM, the novel liposomes reduced the MIC of VCM against both MSSA and MRSA by 4 times at pH 7.4, and by 8 times and 16 times at pH 6.0, respectively. Additionally, these liposomes reduced the number of MRSA in TPH-1 macrophages and HEK293 cells by 1266.67 times and 704.33 times, respectively, showing excellent pH-responsive properties and intracellular bactericidal activity. The in vivo study also showed that the amount of MRSA in the rats treated with the new liposomes was 189.67 and 6.33 times lower than that of the control group and the free drug group, respectively. Menina et al.58 improved the liposome stability and drug encapsulation efficiency by adjusting the ratio of phospholipids and cholesterol. 30 mol% of cholesterol was utilized to increase the integrity of vesicles by promoting alignment of phospholipids alkyl chains. Three different liposomal formulations (Col-Lip-1, Col-Lip-2, and Col-Lip-3 respectively) were prepared, and then surface-functionalized with a bacteria-derived invasive moiety (S. aureus extracellular adherence protein) to enhance the nanocarriers intracellular delivery of hydrophilic colistin. | ||
| Fig. 3 (A) Opening and closing the gates in the liposome at pH 7.4 and acidic pH. (B) Proposed mechanism of intracellular delivery of VCM and enhancement of antibacterial activity by the pH responsive liposome OA-QL.13 Copyright 2021, Taylor & Francis. | ||
Solid lipid nanoparticles (SLNs)
SLNs are compounds that have properties similar to those of liposomes and polymeric nanoparticles. SLNs are made of biodegradable solid natural or synthetic lipids. Unlike liposomes, SLNs do not have a bilayer structure, but are made of surfactant-stabilised solid matrices. The advantages of SLNs include long-term stability, good biocompatibility, ease of use, and encapsulation of hydrophilic and lipophilic drugs. Uner et al.59 found that 71.1% ± 1.4% of vitamin C palmitate SLNs were not degraded after being stored at 4 °C for 3 months; however, most of the nanoemulsions made of vitamin C were degraded under the same conditions. This indicates that the stability of SLNs is higher than that of nanoemulsions. Currently, SLNs are also used as antibiotic carriers to improve the efficacy against intracellular bacterial infection. The behenic acid SLNs prepared by Xie et al.60 could increase the concentration of enrofloxacin in Salmonella-infected mouse macrophages (RAW264.7) by 27.06–37.71 times. Similarly, the intracellular content of SLNs loaded with enrofloxacin decreased by 27.53% and 46.72% after 0.5 h and 1 h, respectively. These values were much lower than the 53.87% and 78.57% decrease observed by treatment with the free drug. Therefore, the encapsulation of SLNs prolonged the action time of the drug in the cells. After treating the cells for 48 h, the logarithm value (4.15 CFU) of intracellular colonies in the free drug group (enrofloxacin concentration 0.6 μg mL−1) was higher than that (3.80 CFU mL−1) in the SLN-enrofloxacin group (enrofloxacin concentration 0.06 μg mL−1). This indicated that SLNs loaded with enrofloxacin had a far better inhibitory effect on intracellular Salmonella than free enrofloxacin, and 0.6 μg mL−1 SLNs with enrofloxacin could reduce the number of intracellular bacteria by 99.97%. Furthermore, Singh et al.12 successfully prepared oral streptomycin-loaded SLNs (STRS-SLNs) using a cold, high-pressure homogenisation technique. The cellular uptake rate of STRS-SLNs was 60 times higher than that of free streptomycin, and the MIC for intracellular M. tb H37RV (256182) was reduced by 3 times. Oral pharmacokinetics results showed that the drug absorption rate and bioavailability of STRS-SLNs increased by 160–710% compared with that of free drugs. Maretti et al.49 prepared novel solid lipid nanoparticles (SLNas) with surface-modified mannose derivatives and loaded rifampicin (RIF) for anti-intracellular tuberculosis therapy. Mannose relevant ligand could target active drug to alveolar macrophages that overexpress mannose receptors on the cytomembranes. And the alveolar macrophages are the preferred site of M. tb infection. The results showed that mannose functionalized SLNas exhibited a more efficient intracellular capacity (∼80%) using mannose receptor-mediated endocytosis, which was higher than that of non-functionalized SLNas (∼40%) and free RIF (∼20%).Polymeric nanoparticles
Polymeric nanoparticles are prepared from natural or synthetic polymers, which are biodegradable, compatible, and stable. Natural polymers include albumin, collagen, chitosan, haemoglobin, and alginate. Synthetic polymers include poly(amides), poly(amino acids), poly(alk-L-cyanoacrylates), poly(esters), and poly(orthoesters). Among the above, chitosan polymers are used extensively for drug delivery to treat various infectious diseases including intracellular pathogens infection. Chitosan is a natural polycationic alkaline polysaccharide derived from chitin. The primary amine (–NH2) at the C-2 position of the glucosamine residue is conducive to further functional modification. Qiu et al.61 used phosphatidylcholine-chitosan nanoparticles to encapsulate gentamicin, which could effectively destroy biofilms and enter macrophages to kill intracellular L. monocytogenes and Pseudomonas aeruginosa. Gnanadhas et al.62 loaded ciprofloxacin into chitosan-dextran sulfate nanocapsules, which increased the concentration of ciprofloxacin in the blood, lymphoid tissue, spleen, and liver by 63.83%, 149.29%, 81.21%, and 53.81%, respectively. Meanwhile, the half-life period of ciprofloxacin nanocapsules in vivo was prolonged by 411.29% to 3.17 h compared with that of free ciprofloxacin (0.62 h), whereas the plasma clearance rate decreased by 38.96%. In addition, polymeric carriers can be easily modified with specific functionalised groups on the surface by chemical means. For example, specific cellular uptake can occur through receptor-mediated endocytosis, where binding of the ligand-modified polymeric nanocarrier with the cell-surface receptor leads to internalization of the entire nanocarrier–receptor complex. In addition, a polymeric carrier with stimuli responsiveness to temperature, pH, light, heat, and redox enables controllable drug release at the target site, thereby obtaining a better therapeutic effect. For example, the pH-sensitive block copolymer poly(2-(methacryloyloxy)ethylphosphorylcholine)-co-poly(2-(diisopropylamino)ethyl methacrylate) (PMPC-PDPA) can combine specific cellular target in non-professional phagocytic cells (through the affinity of PMPC towards the scavenger receptor B1),63 with effective endosomal and cytosolic drug delivery following internalisation (by the pH-sensitive PDPA).64,65 Fenaroli et al.64 used a PMPC-PDPA block copolymer to prepare pH-sensitive nano-polymer vesicles, which could effectively target macrophages, easily enter tuberculosis-like granuloma tissues, and enhance the bactericidal efficacy towards intracellular Mycobacteria and S. aureus. Furthermore, Elbi et al.66 prepared ciprofloxacin-loaded chitosan nanoparticles (cCNPs) and fucoidan (Fu)-coated cCNPs (Fu-cCNPs). They observed that Fu could improve the cell uptake of nanocarriers by specifically recognizing the macrophage scavenger receptor. The intracellular bactericidal activity was 2 times and 6 times higher than that of cCNPs and bare ciprofloxacin, respectively. Su et al.67 designed a multifunctional diblock copolymer-modified liposome with mannose-mediated targeting, pH responsiveness, and intralysosome drug release properties. The polymer-augmented liposomes (PALs) provide improved cytosolic delivery of streptomycin to alveolar macrophages, an important host cell for intracellular pathogens. Compared with pegylated liposomes, the mannose-targeting capability of the PALs was demonstrated with 2.5 times higher internalization. The pH-sensing functionality enabled PALs to provide enhanced release of streptomycin under endosomal pH condition (70% release in 6 hours) with limited release at physiological pH 7.4 (16%). As a result, the streptomycin-loaded PALs showed a significantly improved intracellular antibacterial activity to kill Francisella tularensis in macrophages, compared with free streptomycin or streptomycin delivered by control PEGylated liposomes (13 and 16 times, respectively) (Fig. 4). Guo et al.68 used nanoprecipitation technology to prepare lipid–polymer hybrid nanoparticles (Rf-LPN). The phospholipid/lipid surface showed a good host cell/biomembrane interaction, and the optimised nanoparticles achieved the highest drug-loading efficiency of 11.7% and an encapsulation rate of 65.7%. The bacterial count of intracellular MRSA treated with Rf-LPN at different concentrations was significantly lower than that of bacteria treated with free rifampicin. When the drug concentration was 10 ng mL−1, the number of intracellular bacteria in the Rf-LPN group decreased by 18.5 times compared with the reduction observed in the free drug group.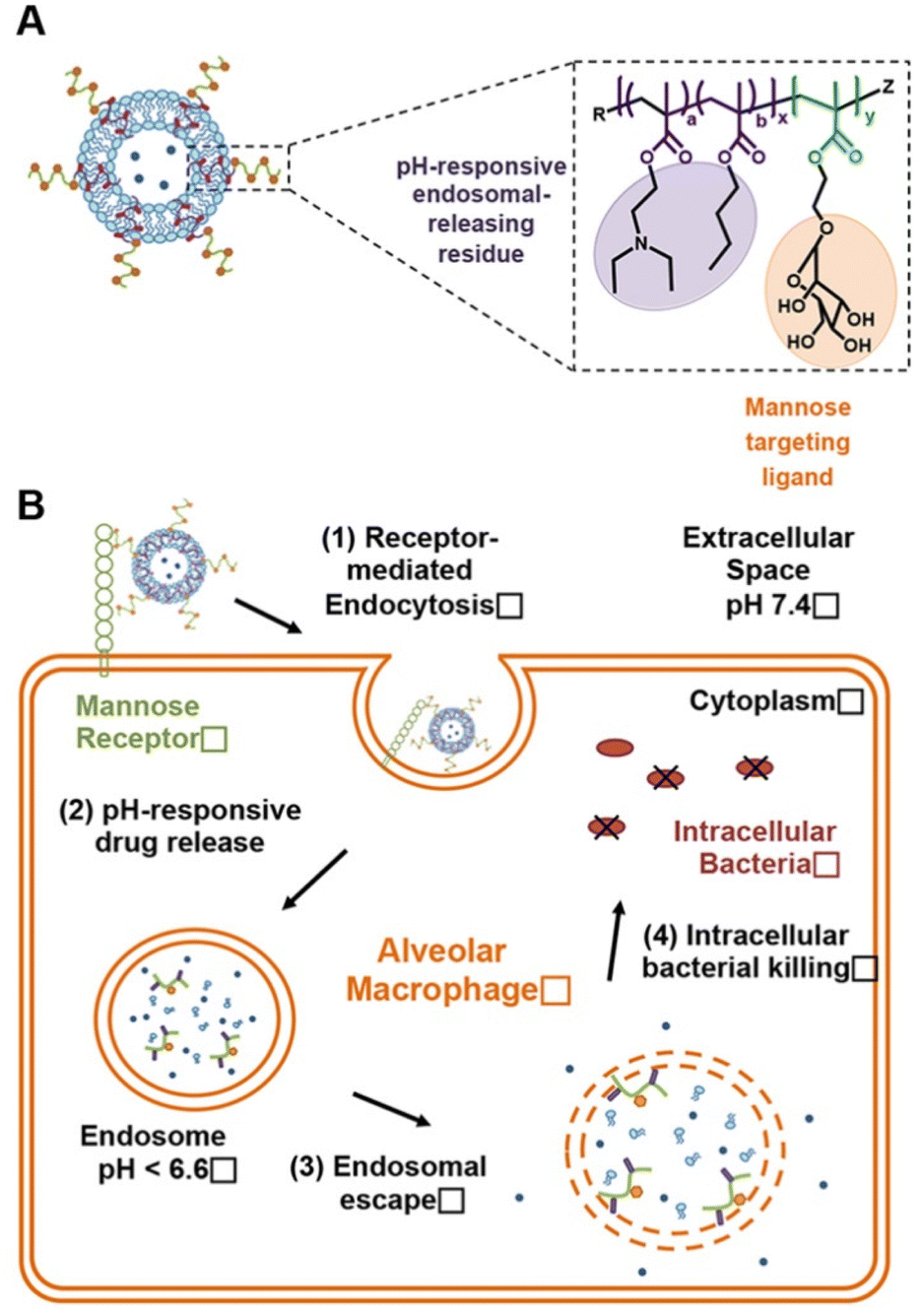 | ||
| Fig. 4 (A) Schematic of the formulation and functions of polymer-augmented liposomes (PALs). PAL was formulated through the hydrophobic effect between the lipid bilayer and hydrophobic block (purple) of the amphiphilic diblock copolymer; (B) envisioned pathway for the cellular uptake of PALs and subsequent intracellular release of the cargo in alveolar macrophages.67 Copyright 2018, Royal Society of Chemistry. | ||
Dendrimers are regularly branched globular macromolecules with a well-defined core–interior–periphery architecture, which are known as “the fourth generation of polymer materials”.69 Mishra et al.70 used fourth-generation hydroxyl-terminated poly (amidoamine) dendrimers for the targeted delivery of azithromycin to intracellular chlamydial inclusion. Dendrimers reduced the area of inclusion by 50% at a drug concentration of 2 ng mL−1, whereas the free drug could not reduce the area of inclusion. Thus, dendritic macromolecules can effectively deliver drugs into inclusion to exert a bactericidal effect. Maji et al.71 used oleic acid-dendrimers (LDH-NPs) to deliver VCM. LDH-NPs have pH-responsive properties and the surface charge changes from negative to positive in an acidic environment. As a result, compared with free VCM, the MIC of LDH-NPs loaded with VCM against MRSA was reduced by 8 times. Under the same MIC, the killing rate of LDH-NPs against intracellular MRSA was 84.19%, which was much higher than that of free VCM (49.26%).
Inorganic non-metallic nanomaterials
Inorganic nanomaterials are a class of nanoparticles with various shapes and particle sizes ranging from 1 to 100 nm, which are synthesised by physical or chemical methods using inorganic materials. Inorganic nanomaterials can bind drugs through electrostatic interactions, hydrophobic interactions, and covalent binding of enzyme-sensitive groups. Inorganic nanomaterials are ideal for drug-delivery platforms owing to their unique physicochemical properties such as facile preparation, good storage stability, and high loading capacity.72 Among them, silicon- and carbon-based nanomaterials are typical representatives of inorganic non-metallic nanomaterials.Silicon-based nanoparticles have been one of the most extensively studied drug carriers given their several advantageous properties such as high surface area, large pore volume, versatile surface chemistry, ease of surface functionalisation, high chemical and thermal stability, and simple synthesis methods.73 Subramaniam et al.74 prepared rifampicin-loaded mesoporous SiO2 nanoparticles, and the drug-loaded nanoparticles showed 28.6% higher intracellular bactericidal efficiency than that of the free drug. Moreover, Yang et al.75 developed a gentamicin-loaded mesoporous silica nanoparticles (MSNs) coated with lipid bilayers and harboring immobilized bacteria-targeting human antimicrobial peptide fragment ubiquicidin (UBI)29–41 (Gen@MSN-LU). It made Gen@MSN-LU target the infection site and kill intracellular S. aureus. The nonspecific uptake of the Gen@MSNs nanoparticles under physiological conditions was prevented by modification their mesoporous channels with bacterial toxin-sensitive liposomes, while the conjugated targeting ligand UBI29–41 allowed nanoparticles to target bacteria in infected tissues.76 Compared with bare gentamicin, the nanocarrier system is more efficient in drug loading, protection from inactivation, overcoming cellular barriers and treating intracellular infections. Hussain et al.77 used nano-mesoporous silica particles to load VCM and a short peptide (CARG) that could target and bind to S. aureus. These particles not only achieved the targeted delivery of drugs in S. aureus-infected animal tissues but also increased the efficacy of antibiotics by approximately 10 times, effectively inhibiting in vivo infection. More importantly, the nanocomposite carrier effectively delivered antibiotics into cells to promote the eradication rate of intracellular bacteria and prevent the occurrence of repeated infections. Despite merits of mesoporous silica nanoparticles (MSN), toxicity and accumulation in tissues remain a limitation for their use. The biomimetic lipid coat around the nanoparticle is a strategy to reduce the toxicity of the mesoporous silica particles and improves biocompatibility. Rajeev J. Mudakavi et al.78 synthesized a lipid coated mesoporous silica nanoparticle (L-MSN) for oral delivery of ciprofloxacin for intracellular elimination Salmonella pathogen. The L-MSN particles exhibited lower cytotoxicity compared to bare MSN particles. It showed 75% cell viability after incubation with 3 mg ml−1 of L-MSN particles, as compared to 50% viability seen in bare MSN group. The L-MSN particles also exhibited controlled release of the antibiotic. Only 30% of the drug released in 30 minutes in L-MSN loaded ciprofloxacin, compared to nearly 90% release in bare MSN particles. More importantly, L-MSN nanoparticles could effectively enter cells and facilitate intracellular transport of antibiotics. In vitro antibacterial activity tests confirmed that L-MSN nanoparticles exhibited improved antibacterial activity in clearing intravacuolar Salmonella infection in RAW 264.7 and HeLa cells. It also showed that intravacuolar targeting of the drug cargo needed lower dose of antibiotic as observed in the in vivo model.
With the ability to form various covalent bonds (sp, sp2, sp3) between atoms, carbon has different crystal structures with distinct physical and chemical properties.79 The application of carbon-based nanoparticles has attracted substantial attention owing to the excellent loading and delivery of various cargos such as drugs and aptamers to living cells. In general, drug molecules are attached to the surface of carbon nanoparticles through covalent and non-covalent bonds.55 Furthermore, carbon-based nanoparticles have been shown to exhibit antimicrobial activity.80 For example, graphene, a single-atom-thick sheet composed of sp2-hybridised carbon atoms, has an ultra-high specific surface area, outstanding mechanical strength, and both sides of the monolithic structure can be loaded with drug molecules. Because of these unique qualities, graphene and graphene oxide applications provide advanced drug transport frameworks and enable the transport of intracellular antimicrobial therapeutics. Li et al.81 studied the interaction between graphene and three types of cells (primary human keratinocytes, human lung epithelial cells, and murine macrophages). The results showed that few-sheet graphene could spontaneously penetrate the cell membrane and enter the cell. Wang et al.82 synthesised aptamer-carboxyfluorescein (FAM)/graphene oxide nanocomposites (GO-nS) and evaluated their molecular probing ability in living cells. The graphene-based nanocomposite not only successfully transported DNA aptamers into cells but also protected the carried oligonucleotides from enzymatic degradation. Additionally, polyethylene glycol-functionalised graphene oxide nanocarriers (PEG-nGOs) prepared by Baek et al.83 successfully delivered single-stranded peptide nucleic acid adsorbed on PEG-nGOs to lung cancer cells through endocytosis without affecting cell viability. The above reports provide a good reference for the development of intracellular drug-delivery vehicles using graphene materials. Moreover, Pi et al.84 modified graphene oxide with PEGylation and mannosylation by an amide reaction, and synthesised a RIF-loaded nanocarrier, Rif@GO-PEG-MAN, that could target macrophages. Rif@GO-PEG-MAN could inhibit 50% of M. tb in macrophages at a low RIF concentration (10 ng mL−1), whereas at a free drug concentration of 50 ng mL−1, only 40% of intracellular M. tb was inhibited, indicating that Rif@GO-PEG-MAN effectively improved the intracellular bactericidal ability of RIF (Fig. 5).
 | ||
| Fig. 5 (A) Schemes for the preparation of GO-PEG-MAN and Rif@GO-PEG-MAN. (B) Proposed endocytosis and drug release mechanism of Rif@GO-PEG-MAN in macrophages for intracellular M. tb treatment.84 Copyright 2019, Elsevier Ltd. | ||
Metal-based nanoparticles
Gold, silver, copper, and other metal-based nanoparticles exhibit a wide range of antibacterial effects on both Gram-positive and Gram-negative bacteria. Since the cell wall surface of Gram-positive and -negative bacteria is negatively charged, the positively charged metal nanoparticles can be tightly adsorbed on the cell membrane surface through electrostatic interaction, resulting in increased membrane permeability to disrupt cell wall function.85 In addition, metal ions can penetrate mammalian cells to generate intracellular ROS or photothermal/photodynamic effects, thereby inducing intracellular antibacterial activity.86,87 As carriers, metal-based nanomaterials have a large specific surface area, strong penetrability, easy surface modification, and advantages in durability and heat resistance. Moreover, metal-based nanomaterials exhibit antibacterial effects, including bacterial cell membrane damage, intracellular damage, and induction of oxidative stress to kill intracellular bacteria. Metal-based nanoparticles deliver antibiotics into cells to exert a synergistic intracellular antibacterial effect.85,87,88Among metal-based nanomaterials, gold nanomaterials are widely used owing to their strong antibacterial ability and excellent biocompatibility.89,90 Gold nanomaterials can be easily tuned to various sizes and shapes, including spheres, rods, cages, and shells, and they are also easy to modify. Gold can disrupt the respiratory chain, decreasing ATPase activity, and reduce the potential of the membrane to cause bacterial cell death.91 In addition, researchers have tried to combine gold nanoparticles with antibiotics to promote the interaction between antibiotics and bacteria, enhance the antibacterial properties of antibiotics, and exhibit a synergistic anti-infection effect.89 For example, researchers have used Au–S bonds to link the cyanamide of VCM with gold ions to synthesise VCM-gold nanoparticles (Van@Au), in which each Van@Au surface could connect to approximately 31 VCM molecules. The free VCM had no antibacterial activity against Escherichia coli (MIC > 128 μg mL−1). In contrast, Van@Au nanoparticles exhibited antibacterial activity against both Gram-negative and Gram-positive bacteria, including multidrug-resistant bacteria such as vancomycin-resistant Enterococci (VRE). The MIC of Van@Au nanoparticles for VRE was 2–4 μg mL−1, indicating the greatly improved antibacterial activity of VCM.92 Mu et al.86 used phosphatidylcholine-modified gold nanoparticles loaded with gentamicin (GPA NPs), which exhibited no cytotoxicity to macrophages. After incubation for 2 h, fluorescence imaging showed that GPA NPs were phagocytosed into the cytoplasm, and their cellular uptake increased in a time-dependent manner. Compared with free gentamicin, the number of Pseudomonas aeruginosa in macrophages was reduced by approximately 103 after treatment with GPA NPs, and the same results were observed in the L. monocytogenes -infected macrophage model.
Silver nanoparticles are another common broad-spectrum antibacterial material, which demonstrate a strong antibacterial effect, including inducing lipid peroxidation and ROS production, hindering cell wall synthesis, increasing membrane permeability, and destabilising ribosomes.93,94
As materials with low toxicity, copper-based nanoparticles, including copper,95 copper oxide,96 and cuprous oxide,97 are widely used in antibacterial therapy. Copper nanoparticles have a variety of bactericidal mechanisms such as destroying cell membranes, damaging bacterial DNA, inhibiting protein synthesis, and blocking different biochemical pathways.98 Recent studies have shown that Ag–Cu alloy metal nanomaterials are more effective in killing bacteria than silver or copper nanoparticles alone, which may be due to the synergistic release of Ag+ and Cu2+ ions, exerting greater damage to bacterial DNA.99,100 Moreover, Abdulrehman et al.99 synthesised Ag–Cu–B (ACB) nanoparticles coupled with cadherin-11 antibody (OBAb) to target osteoblasts infected with intracellular S. aureus. The OBAb-bound ACB nanoparticles were not only effective against extracellular S. aureus but also reduced the number of intracellular S. aureus by 1.32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log at a concentration of 1 mg L−1. However, silver nanoparticles are less stable and prone to aggregation, resulting in reduced antibacterial activity. To improve the dispersion stability of silver nanoparticles in water and enhance their antibacterial efficacy, researchers coated the surface of triangular flake silver nanoparticles (silver nanoplates, Ag NPLs) with one or two layers of gold atoms (Ag@Au1L NPLs and Ag@Au2L NPLs). The gold coating improved the dispersion stability of the silver nanoparticles in a high-salt aqueous solution. The Ag@Au1L NPLs effectively reduced the number of Salmonella typhimurium in RAW 264.7 cells and had no cytotoxicity to these cells. Therefore, the Ag@Au1L NPLs could serve as effective antibacterial agents for intracellular bacterial infection.101
log at a concentration of 1 mg L−1. However, silver nanoparticles are less stable and prone to aggregation, resulting in reduced antibacterial activity. To improve the dispersion stability of silver nanoparticles in water and enhance their antibacterial efficacy, researchers coated the surface of triangular flake silver nanoparticles (silver nanoplates, Ag NPLs) with one or two layers of gold atoms (Ag@Au1L NPLs and Ag@Au2L NPLs). The gold coating improved the dispersion stability of the silver nanoparticles in a high-salt aqueous solution. The Ag@Au1L NPLs effectively reduced the number of Salmonella typhimurium in RAW 264.7 cells and had no cytotoxicity to these cells. Therefore, the Ag@Au1L NPLs could serve as effective antibacterial agents for intracellular bacterial infection.101
Endogenous nanocarriers
Endogenous nanocarriers were recently introduced as a natural delivery system to reduce the body rejection reaction.102 The endogenous nano-based therapeutic system based on cell derivatives3 mainly includes biological endogenous substances such as exosomes, extracellular vesicles, and cell membrane-coated biomimetic nanocomplexes. For example, exosomes denote a family of nanoparticles with a diameter in the range of 30–120 nm that are secreted by most cell types of the body. Owing to its endogenous characteristics, it has evident advantages such as excellent biocompatibility and low immunogenicity. Yang et al.103 developed an exosome drug-delivery system that can deliver linezolid to MRSA-infected macrophages and release the drug in lysosomes. The number of MRSA in macrophages decreased by approximately 0.6, 1.6, and 1.5log after treatment with linezolid-loaded exosomes (drug concentration 20 μg mL−1) for 2, 4, and 24 h, respectively, whereas the free drug could not kill intracellular MRSA.
Conclusion and outlook
In conclusion, the last few years have witnessed great progress in the research of nanocarriers for the delivery of antibiotics into cells in the treatment of intracellular pathogenic infections. Nanocarriers can deliver drugs into cells through phagocytosis, mannose and other receptor-mediated endocytosis pathways, which can improve the availability of drugs in cells and reduce the required dose and toxic side effects of drugs. This provides a new idea for solving the problems faced by conventional antibiotics in the treatment of intracellular bacterial infections.However, there are still issues that need to be overcome to move this field from research to clinical application, such as stability, safety, and production cost. For example, liposomes have poor storage stability; are prone to oxidation, stratification, and drug leakage; and lack active targeting ability, hindering the targeting of infection sites to achieve high-concentration drug accumulation. Additionally, the lipid state of SLNs is complex and the crystal form and particle size during storage are prone to change. Moreover, residual organic solvent and a lack of large-scale production ability may hinder the preparation process of polymeric nanocarriers. Furthermore, inorganic nanomaterials such as mesoporous silicon face challenges of insufficient drug-carrying capacity, toxic degradation products, and complex synthesis processes. One important problem of metal nanoparticles is that toxicity must be carefully examined. More importantly, the unspecific biodistribution of nanoparticles still affects the normal cells, causing a variety of serious side effects. Therefore, the risks of using nanoparticle formulations need to be considered carefully. Firstly, the toxicological effects of nanoparticles need to be analyzed depending on the context of their use (i.e., the route of administration, dose, residence time in the body, material size, and material interaction with the body). However, nanocarriers prepared by different research groups present different particle size, charge, and component proportion due to the lack of standardized and accurate preparation procedure. Therefore, the development of accepted and controllable preparation technology is the premise of obtaining nanocarriers with stable properties and low toxicity. Secondly, the trend of novel nanocarriers is developing multi-functionalized composite nanomaterials. Researchers mostly concern the biological effects rather than the biological mechanism and structure–activity relationship between the components, which hinders the assessment of potential biological toxicity of nanocarriers. Consequently, biological mechanism and structure–activity relationship of nanomaterials needs to be further studied. Thirdly, although most biological application studies have given low or even relatively non-toxic data on the biological toxicity of nanomaterials, these toxicity evaluations are mostly limited to the biological individual level and cell level. In addition, most of the toxicity evaluation methods were based on apparent analysis methods such as observing the morphological changes of the tested organisms/cells, ROS production and intake/enrichment of nanomaterials including metal nanoparticles. However, due to the differences in material dose and particle size, test subjects, measurement methods and test conditions, the evaluation conclusions obtained were poor in comparability. And there is also no clear conclusion on the balance and selection between biological applications and potential toxicological hazards of nanomaterials. The above shortcomings limit the clinical development and application of nanocarriers. Therefore, there is a substantial gap between laboratory research and clinical application. With the development of industrialization and wider application of nanomaterials in the future, it is necessary to establish systematic and standardized toxicological evaluation methods of nanomaterials. Comprehensive studies that integrate knowledge from the fields of microbiology and pharmacology/nanoengineering, as well as further clinical trials are needed to evaluate the safety and efficacy of newly developed nanocarriers.
In addition, researchers should further study the influence of factors such as particle size, surface charge, and dispersion coefficient on the intracellular concentration and action time of antibacterial drugs. For example, the nanoparticles with the smaller size and higher surface area facilitate diffusion of particles into cells.104 Although the internalization of nanoparticles is highly size-dependent by cells, it may not follow the size limits of the commonly defined uptake process, the kinetics of uptake for the same type of nanoparticle varies in the different cell types.105 Besides, the positively charged nanoparticles have high affinity to the negatively charged cell membrane compared to the neutral and electronegative nanoparticles, which facilitates the cell membrane-nanoparticle interaction and the higher uptake by cells.106 Thus, the preparation of the efficient nanocarriers with the optimum size and charge density on their surface for specific cell types to improve their therapeutic efficiency is worthy of further research in the future.107 In addition, the pH-responsive strategy enables carriers to enter the weakly acidic organelles and then be decomposed, releasing the contained drug molecules to kill the pathogenic bacteria in cells.
Notably, some latest controllable preparation nanotechnology to control size and structure of nanomaterials, such as self-assembled DNA nanostructures108 and DNA origami-based nanoprinting,109 would be beneficial to the development of more efficient and low-toxicity nanocarriers. In addition, emerging bioinspired ways such as virus-based nanoparticles110 could be designed as novel functional nanostructures and devices for the delivery of antibiotics, due to their ability to preserve cargo stability and activity by providing the freedom to choose cargo-friendly buffer conditions throughout the encapsulation process. Furthermore, intracellular pathogens exposed to subtherapeutic concentrations for prolonged periods are more likely to result in antibiotic resistance, thus developing alternatives to antibiotics could be a great help in the fight against refractory infection. Nowadays, bacteriophages have been proved to play an important role in combating bacterial infections (phage therapy), making them an important alternative to classical antibiotic strategies.111 Current reviews regarding the application of bacteriophages against intracellular bacteria have also been reported.112,113 Benefit from progress of phage genomics and nano-functionalization technology, the specificity and infectivity of phage are improving. Developing strategies combined with nanocarriers and bacteriophages to improve the treatment of intracellular bacterial infections has great potential. With the continuous development of nanotechnology and the optimisation of nano delivery systems, it is anticipated that therapeutic products will more directly target the bacteria hidden in the cells, which will greatly help in the treatment of infectious diseases.
Author contributions
Chao Wang designed the study and wrote the manuscript. Yi Yang, Yuanyuan Cao and Hua Shi collected the data, Kaixin Liu, Xudong Guo and Wanying Liu searched databases, Rongzhang Hao, Hongbin Song and Rongtao Zhao drafted the work. All authors read and approved the final manuscript.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The authors thank the National Key Research and Development Program of China (No. 2021YFC2301100), the National Natural Science Foundation of China (82202577), and the Key Research and Development Program of Hainan Province (ZDYF2021SHFZ262).References
- W. Feng, G. Li, X. Kang, R. Wang, F. Liu, D. Zhao, H. Li, F. Bu, Y. Yu, T. F. Moriarty, Q. Ren and X. Wang, Adv. Mater., 2022, 34, 2109789 CrossRef
.
- Y. M. Soe, S. Bedoui, T. P. Stinear and A. Hachani, Cell. Microbiol., 2021, 23, e13317 CrossRef CAS PubMed
- S. Subramaniam, P. Joyce, N. Thomas and C. A. Prestidge, Adv. Drug Delivery Rev., 2021, 177, 113948 CrossRef CAS PubMed
- E. Weddle and H. Agaisse, PLoS Pathog., 2018, 14, e1007380 CrossRef PubMed
- A. L. Radtke and M. X. D. O'Riordan, Cell. Microbiol., 2006, 8, 1720–1729 CrossRef CAS PubMed
- A. Anil and A. Banerjee, Front. Cell. Infect. Microbiol., 2020, 10, 590682–590682 CrossRef CAS PubMed
- A. Nasser, T. Azimi, S. Ostadmohammadi and S. Ostadmohammadi, Microb. Pathog., 2020, 148, 104431 CrossRef CAS
- K. Subramanian, B. Henriques-Normark and S. Normark, Cell. Microbiol., 2019, 21, e13077 CrossRef CAS
- A. Oberbach, N. Schlichting, S. Feder, S. Lehmann, Y. Kullnick, T. Buschmann, C. Blumert, F. Horn, J. Neuhaus, R. Neujahr, E. Bagaev, C. Hagl, M. Pichlmaier, A. C. Rodloff, S. Gräber, K. Kirsch, M. Sandri, V. Kumbhari, A. Behzadi, A. Behzadi, J. C. Correia, F. W. Mohr and M. Friedrich, PLoS One, 2017, 12, e0175569 CrossRef PubMed
- T. J. P. Petit and A. Lebreton, Trends Microbiol., 2022, 30, 736–748 CrossRef CAS PubMed
- S. M. Lehar, T. Pillow, M. Xu, L. Staben, K. K. Kajihara, R. Vandlen, L. DePalatis, H. Raab, W. L. Hazenbos, J. H. Morisaki, J. Kim, S. Park, M. Darwish, B.-C. Lee, H. Hernandez, K. M. Loyet, P. Lupardus, R. Fong, D. Yan, C. Chalouni, E. Luis, Y. Khalfin, E. Plise, J. Cheong, J. P. Lyssikatos, M. Strandh, K. Koefoed, P. S. Andersen, J. A. Flygare, M. Wah Tan, E. J. Brown and S. Mariathasan, Nature, 2015, 527, 323–328 CrossRef CAS
- M. Singh, N. Schiavone, L. Papucci, P. Maan, J. Kaur, G. Singh, U. Nandi, D. Nosi, A. Tani, G. K. Khuller, M. Priya, R. Singh and I. P. Kaur, Eur. J. Pharm. Biopharm., 2021, 160, 100–124 CrossRef PubMed
- C. A. Omolo, N. A. Megrab, R. S. Kalhapure, N. Agrawal, M. Jadhav, C. Mocktar, S. Rambharose, K. Maduray, B. Nkambule and T. Govender, J. Liposome Res., 2021, 31, 45–63 CrossRef PubMed
- S. Shin, S. Kwon and Y. Yeo, Pharm. Res., 2022, 39, 1085–1114 CrossRef
- L. Kaur, S. H. Sohal, M. Kaur, S. D. Malhi and S. Garg, Anti-Cancer Agents Med. Chem., 2020, 20, 2012–2024 CrossRef
- E. Keles, Y. Song, D. Du, W.-J. Dong and Y. Lin, Biomater. Sci., 2016, 4, 1291–1309 RSC
- L. Duan, K. Ouyang, X. Xu, L. Xu, C. Wen, X. Zhou, Z. Qin, Z. Xu, W. Sun and Y. Liang, Front. Genet., 2021, 12, 673286–673286 CrossRef CAS PubMed
- W. Gao, Y. Chen, Y. Zhang, Q. Zhang and L. Zhang, Adv. Drug Delivery Rev., 2018, 127, 46–57 CrossRef CAS PubMed
- R. S. Santos, C. Figueiredo, N. F. Azevedo, K. Braeckmans and S. C. De Smedt, Adv. Drug Delivery Rev., 2018, 136–137, 28–48 CrossRef CAS
- D. Samanta, M. Mulye, T. M. Clemente, A. V. Justis and S. D. Gilk, Front. Cell. Infect. Microbiol., 2017, 7, 165–165 CrossRef PubMed
- A. Thakur, H. Mikkelsen and G. Jungersen, J. Immunol. Res., 2019, 2019, 1356540–1356540 Search PubMed
- W. Zhai, F. Wu, Y. Zhang, Y. Fu and Z. Liu, Int. J. Mol. Sci., 2019, 20, 340 CrossRef PubMed
- L. I. Rankine-Wilson, T. Shapira, C. Sao Emani and Y. Av-Gay, Microbiology, 2021, 167, 001041 CrossRef CAS PubMed
- F. Hanses, A. Kopp, M. Bala, C. Buechler, W. Falk, B. Salzberger and A. Schäffler, Endocrinology, 2011, 152, 4148–4157 CrossRef CAS
- S. Zhou, Y. Rao, J. Li, Q. Huang and X. Rao, Microbiol. Res., 2022, 260, 127040 CrossRef CAS
- J. Horn, K. Stelzner, T. Rudel and M. Fraunholz, Int. J. Med. Microbiol., 2018, 308, 607–624 CrossRef PubMed
- F. Du, C. Liao, Y. Yang, C. Yu, X. Zhang, X. Cheng and C. Zhang, Can. J. Vet. Res., 2020, 84, 302–309 Search PubMed
- M. G. Pucciarelli and F. García-Del Portillo, Microbiol. Spectrum, 2017, 5, MTBP-0009-2016 Search PubMed
- R. Figueira and D. W. Holden, Microbiology (Reading), 2012, 158, 1147–1161 CrossRef PubMed
- A. S. Mir, Curr. Protein Pept. Sci., 2021, 22, 620–628 CrossRef PubMed
- J. Pizarro-Cerdá and P. Cossart, Microbiol. Spectrum, 2018, 6, MTBP-0009-2016 Search PubMed
- P. Głowacka, D. Żakowska, K. Naylor, M. Niemcewicz and A. Bielawska-Drózd, Pol. J. Microbiol., 2018, 67, 151–161 CrossRef PubMed
- H. Jiao, Z. Zhou, B. Li, Y. Xiao, M. Li, H. Zeng, X. Guo and G. Gu, Int. J. Mol. Sci., 2021, 22, 3673 CrossRef PubMed
- J. Alves-Silva, I. P. Tavares, E. S. Guimarães, M. M. Costa Franco, B. C. Figueiredo, J. T. Marques, G. Splitter and S. C. Oliveira, Front. Microbiol., 2017, 8, 2217–2217 CrossRef PubMed
- E. Dumont, J. Vergalli, L. Conraux, C. Taillier, A. Vassort, J. Pajović, M. Réfrégiers, M. Mourez and J.-M. Pagès, J. Antimicrob. Chemother., 2019, 74, 58–65 Search PubMed
- P. Tulkens, Eur. J. Clin. Microbiol. Infect. Dis., 1991, 10, 100–106 CrossRef
- F. Lamoth and G. Greub, Expert Rev. Anti-Infect. Ther., 2010, 8, 775–790 CrossRef PubMed
- X.-Z. Li, P. Plésiat and H. Nikaido, Clin. Microbiol. Rev., 2015, 28, 337–418 CrossRef PubMed
- C. Chidiac and Y. Mouton, Infection, 1991, 19(Suppl 7), S365–S371 CrossRef
- X.-Y. Chen, F. Qian, Y.-Y. Wang, Y. Liu, Y. Sun, W.-B. Zha, K. Hao, F. Zhou, G.-J. Wang and J.-W. Zhang, Acta Pharmacol. Sin., 2021, 42, 1930–1941 CrossRef PubMed
- S. Dey, A. Majhi, S. Mahanti, I. Dey and B. Bishayi, Inflammation, 2015, 38, 1050–1069 CrossRef PubMed
- N. Abed and P. Couvreur, Int. J. Antimicrob. Agents, 2014, 43, 485–496 CrossRef PubMed
- K. Ray, B. Marteyn, P. J. Sansonetti and C. M. Tang, Nat. Rev. Microbiol., 2009, 7, 333–340 CrossRef PubMed
- A. Sindhwani, S. B. Arya, H. Kaur, D. Jagga, A. Tuli and M. Sharma, PLoS Pathog., 2017, 13, e1006700 CrossRef
- F. Van Bambeke, S. Carryn, C. Seral, H. Chanteux, D. Tyteca, M.-P. Mingeot-Leclercq and P. M. Tulkens, Antimicrob. Agents Chemother., 2004, 48, 2853–2860 CrossRef
- S. S. Grant and D. T. Hung, Virulence, 2013, 4, 273–283 CrossRef PubMed
- L. Wayne and C. Sohaskey, Annu. Rev. Microbiol., 2001, 55, 139–163 CrossRef PubMed
- M. E. Levison and J. H. Levison, Infect. Dis. Clin. North. Am., 2009, 23, 791–815 CrossRef PubMed
- E. Maretti, L. Costantino, F. Buttini, C. Rustichelli, E. Leo, E. Truzzi and V. Iannuccelli, Drug Delivery Transl. Res., 2019, 9, 298–310 CrossRef PubMed
- A. Bin Hafeez, X. Jiang, P. J. Bergen and Y. Zhu, Int. J. Mol. Sci., 2021, 22, 11691 CrossRef
- A. Brezden, M. F. Mohamed, M. Nepal, J. S. Harwood, J. Kuriakose, M. N. Seleem and J. Chmielewski, J. Am. Chem. Soc., 2016, 138, 10945–10949 CrossRef
- Q. Zhang, W. Wu, J. Zhang and X. Xia, J. Drug Targeting, 2020, 28, 271–281 CrossRef
- C. Wang, B. Wang, S. Zou, B. Wang, G. Liu, F. Zhang, Q. Wang, Q. He and L. Zhang, Biomater. Sci., 2021, 9, 5977–5987 RSC
- W. He, X. Xing, X. Wang, D. Wu, W. Wu, J. Guo and S. Mitragotri, Adv. Funct. Mater., 2020, 30, 1910566 CrossRef
- S. M. Hosseini, M. Taheri, F. Nouri, A. Farmani, N. M. Moez and M. R. Arabestani, Biomed. Pharmacother., 2022, 146, 112609 CrossRef
- C. Zhang, W. Zhao, C. Bian, X. Hou, B. Deng, D. W. McComb, X. Chen and Y. Dong, ACS Appl. Bio Mater., 2019, 2, 1270–1277 CrossRef
- R. K. Franklin, S. A. Marcus, A. M. Talaat, B. K. KuKanich, R. Sullivan, L. A. Krugner-Higby and T. D. Heath, Drug Metab. Dispos., 2015, 43, 1236 CrossRef PubMed
- S. Menina, J. Eisenbeis, M. A. M. Kamal, M. Koch, M. Bischoff, S. Gordon, B. Loretz and C.-M. Lehr, Adv. Healthcare Mater., 2019, 8, 1900564 CrossRef
- M. Uner, S. Wissing, G. Yener and R. J. D. P. Müller, Pharmazie, 2005, 60, 577–582 CAS
- S. Xie, F. Yang, Y. Tao, D. Chen, W. Qu, L. Huang, Z. Liu, Y. Pan and Z. Yuan, Sci. Rep., 2017, 7, 41104 CrossRef PubMed
- Y. Qiu, D. Xu, G. Sui, D. Wang, M. Wu, L. Han, H. Mu and J. Duan, Int. J. Biol. Macromol., 2020, 156, 640–647 CrossRef
- D. P. Gnanadhas, M. Ben Thomas, M. Elango, A. M. Raichur and D. Chakravortty, J. Antimicrob. Chemother., 2013, 68, 2576–2586 CrossRef PubMed
- H. E. Colley, V. Hearnden, M. Avila-Olias, D. Cecchin, I. Canton, J. Madsen, S. MacNeil, N. Warren, K. Hu, J. McKeating, S. Armes, C. Murdoch, M. Thornhill and G. J. M. P. Battaglia, Mol. Pharm., 2014, 11, 1176–1188 CrossRef
- F. Fenaroli, J. D. Robertson, E. Scarpa, V. M. Gouveia, C. Di Guglielmo, C. De Pace, P. M. Elks, A. Poma, D. Evangelopoulos, J. O. Canseco, T. K. Prajsnar, H. M. Marriott, D. H. Dockrell, S. J. Foster, T. D. McHugh, S. A. Renshaw, J. S. Martí, G. Battaglia and L. Rizzello, ACS Nano, 2020, 14, 8287–8298 CrossRef PubMed
- M. Massignani, C. LoPresti, A. Blanazs, J. Madsen, S. P. Armes, A. L. Lewis and G. Battaglia, Small, 2009, 5, 2424–2432 CrossRef CAS
- S. Elbi, T. R. Nimal, V. K. Rajan, G. Baranwal, R. Biswas, R. Jayakumar and S. Sathianarayanan, Colloids Surf., B, 2017, 160, 40–47 CrossRef CAS
- F.-Y. Su, J. Chen, H.-N. Son, A. M. Kelly, A. J. Convertine, T. E. West, S. J. Skerrett, D. M. Ratner and P. S. Stayton, Biomater. Sci., 2018, 6, 1976–1985 RSC
- P. Guo, H. Y. Xue, B. A. Buttaro, N. T. Tran and H. L. Wong, Int. J. Pharm., 2020, 589, 119784–119784 CrossRef CAS PubMed
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS PubMed
- M. K. Mishra, K. Kotta, M. Hali, S. Wykes, H. C. Gerard, A. P. Hudson, J. A. Whittum-Hudson and R. M. Kannan, Nanomedicine, 2011, 7, 935–944 CrossRef CAS PubMed
- R. Maji, C. A. Omolo, N. Agrawal, K. Maduray, D. Hassan, C. Mokhtar, I. Mackhraj and T. Govender, Mol. Pharm., 2019, 16, 4594–4609 CrossRef CAS PubMed
- A.-S. Balderrama-González, H.-A. Piñón-Castillo, C.-A. Ramírez-Valdespino, L.-L. Landeros-Martínez, E. Orrantia-Borunda and H.-E. Esparza-Ponce, Int. J. Mol. Sci., 2021, 22, 12890 CrossRef PubMed
- Z. Gounani, M. A. Asadollahi, J. N. Pedersen, J. Lyngsø, J. Skov Pedersen, A. Arpanaei and R. L. Meyer, Colloids Surf., B, 2019, 175, 498–508 CrossRef CAS PubMed
- S. Subramaniam, N. Thomas, H. Gustafsson, M. Jambhrunkar, S. P. Kidd and C. A. Prestidge, Antibiotics, 2019, 8, 39 CrossRef CAS PubMed
- S. Yang, X. Han, Y. Yang, H. Qiao, Z. Yu, Y. Liu, J. Wang and T. Tang, ACS Appl. Mater. Interfaces, 2018, 10, 14299–14311 CrossRef CAS PubMed
- H. Chen, C. Liu, D. Chen, K. Madrid, S. Peng, X. Dong, M. Zhang and Y. Gu, Mol. Pharm., 2015, 12, 2505–2516 CrossRef CAS
- S. Hussain, J. Joo, J. Kang, B. Kim, G. B. Braun, Z.-G. She, D. Kim, A. P. Mann, T. Mölder, T. Teesalu, S. Carnazza, S. Guglielmino, M. J. Sailor and E. Ruoslahti, Nat. Biomed. Eng., 2018, 2, 95–103 CrossRef PubMed
- R. J. Mudakavi, A. M. Raichur and D. Chakravortty, RSC Adv., 2014, 4, 61160–61166 RSC
- L. Shi, J. Chen, L. Teng, L. Wang, G. Zhu, S. Liu, Z. Luo, X. Shi, Y. Wang and L. Ren, Small, 2016, 12, 4165–4184 CrossRef PubMed
- S. Maleki Dizaj, A. Mennati, S. Jafari, K. Khezri and K. Adibkia, Adv. Pharm. Bull., 2015, 5, 19–23 Search PubMed
- Y. Li, H. Yuan, A. von dem Bussche, M. Creighton, R. H. Hurt, A. B. Kane and H. Gao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12295–12300 CrossRef
- Y. Wang, Z. Li, D. Hu, C.-T. Lin, J. Li and Y. Lin, J. Am. Chem. Soc., 2010, 132, 9274–9276 CrossRef CAS
- A. Baek, Y. M. Baek, H.-M. Kim, B.-H. Jun and D.-E. Kim, Bioconjugate Chem., 2018, 29, 528–537 CrossRef CAS PubMed
- J. Pi, L. Shen, H. Shen, E. Yang, W. Wang, R. Wang, D. Huang, B.-S. Lee, C. Hu, C. Chen, H. Jin, J. Cai, G. Zeng and Z. W. Chen, Mater. Sci. Eng., C, 2019, 103, 109777 CrossRef CAS
- S. Gharpure, A. Akash and B. Ankamwar, J. Nanosci. Nanotechnol., 2020, 20, 3303–3339 CrossRef
- H. Mu, J. Tang, Q. Liu, C. Sun, T. Wang and J. Duan, Sci. Rep., 2016, 6, 18877 CrossRef CAS PubMed
- J. J. Soldevila-Barreda and N. Metzler-Nolte, Chem. Rev., 2019, 119, 829–869 CrossRef CAS PubMed
- J. Kang, M. J. Dietz, K. Hughes, M. Xing and B. Li, J. Antimicrob. Chemother., 2019, 74, 1578–1585 CrossRef CAS PubMed
- X. Zhao, H. Tang and X. Jiang, ACS Nano, 2022, 16(7), 10066–10087 CrossRef CAS PubMed
- S. Sathiyaraj, G. Suriyakala, A. Dhanesh Gandhi, R. Babujanarthanam, K. S. Almaary, T.-W. Chen and K. Kaviyarasu, J. Infect. Public Health, 2021, 14, 1842–1847 CrossRef PubMed
- S. Rosa, C. Connolly, G. Schettino, K. T. Butterworth and K. M. Prise, Cancer Nanotechnol., 2017, 8, 2–2 CrossRef PubMed
- H.-Z. Lai, W.-Y. Chen, C.-Y. Wu and Y.-C. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 2046–2054 CrossRef CAS PubMed
- I. X. Yin, J. Zhang, I. S. Zhao, M. L. Mei, Q. Li and C. H. Chu, Int. J. Nanomed., 2020, 15, 2555–2562 CrossRef CAS PubMed
- T. Bruna, F. Maldonado-Bravo, P. Jara and N. Caro, Int. J. Mol. Sci., 2021, 22, 7202 CrossRef CAS
- Q. Lv, B. Zhang, X. Xing, Y. Zhao, R. Cai, W. Wang and Q. Gu, J. Hazard. Mater., 2018, 347, 141–149 CrossRef CAS PubMed
- G. Rajivgandhi, M. Maruthupandy, T. Muneeswaran, G. Ramachandran, N. Manoharan, F. Quero, M. Anand and J.-M. Song, Microb. Pathog., 2019, 127, 267–276 CrossRef CAS PubMed
- Y. Gurianov, F. Nakonechny, Y. Albo and M. Nisnevitch, Int. J. Mol. Sci., 2019, 20, 439 CrossRef
- B. Bagchi, S. Dey, S. Bhandary, S. Das, A. Bhattacharya, R. Basu and P. Nandy, Mater. Sci. Eng., C, 2012, 32, 1897–1905 CrossRef CAS
- T. Abdulrehman, S. Qadri, S. Skariah, A. Sultan, S. Mansour, J. Azzi and Y. Haik, PLoS One, 2020, 15, e0231276 CrossRef PubMed
- N. M. Zain, A. G. F. Stapley and G. Shama, Carbohydr. Polym., 2014, 112, 195–202 CrossRef
- H. Ichimaru, A. Harada, S. Yoshimoto, Y. Miyazawa, D. Mizoguchi, K. Kyaw, K. Ono, H. Tsutsuki, T. Sawa and T. Niidome, Langmuir, 2018, 34, 10413–10418 CrossRef PubMed
- W. Du, X. Hu, W. Wei and G. Liang, Bioconjugate Chem., 2018, 29, 826–837 CrossRef PubMed
- X. Yang, G. Shi, J. Guo, C. Wang and Y. He, Int. J. Nanomed., 2018, 13, 8095–8104 CrossRef PubMed
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef CAS PubMed
- T. dos Santos, J. Varela, I. Lynch, A. Salvati and K. Dawson, Small, 2011, 7, 3341–3349 CrossRef CAS PubMed
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed
- S. Salatin, S. Maleki Dizaj and A. Yari Khosroushahi, Cell Biol. Int., 2015, 39, 881–890 CrossRef CAS PubMed
- S. Ren, J. Wang, C. Song, Q. Li, Y. Yang, N. Teng, S. Su, D. Zhu, W. Huang, J. Chao, L. Wang and C. Fan, Research, 2019, 2019, 7403580–7403580 CrossRef CAS PubMed
- R. Niu, C. Song, F. Gao, W. Fang, X. Jiang, S. Ren, D. Zhu, S. Su, J. Chao, S. Chen, C. Fan and L. Wang, J. Angew. Chem., 2021, 60, 11695–11701 CrossRef CAS PubMed
- L. Li, C. Xu, W. Zhang, F. Secundo, C. Li, Z. Zhang, X. Zhang and F. Li, Nano Lett., 2019, 19, 2700–2706 CrossRef CAS
- A. Kakasis and G. Panitsa, Int. J. Antimicrob. Agents, 2019, 53, 16–21 CrossRef PubMed
- P. Śliwka, M. Ochocka and A. Skaradzińska, Crit. Rev. Microbiol., 2022, 48, 222–239 CrossRef PubMed
- A. Goswami, P. R. Sharma and R. Agarwal, Crit. Rev. Microbiol., 2021, 47, 461–478 CrossRef PubMed
Footnote |
| † These authors have equal contribution for this work. |
| This journal is © The Royal Society of Chemistry 2023 |