
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards a standardized multi-tissue decellularization protocol for the derivation of extracellular matrix materials†
Andreea
Biehl
 ab,
Ana M.
Gracioso Martins
ab,
Zachary G.
Davis
ab,
Daphne
Sze
ab,
Leonard
Collins
c,
Camilo
Mora-Navarro
d,
Matthew B.
Fisher
abe and
Donald O.
Freytes
*ab
ab,
Ana M.
Gracioso Martins
ab,
Zachary G.
Davis
ab,
Daphne
Sze
ab,
Leonard
Collins
c,
Camilo
Mora-Navarro
d,
Matthew B.
Fisher
abe and
Donald O.
Freytes
*ab
aJoint Department of Biomedical Engineering, North Carolina State University & University of North Carolina-Chapel Hill, 4130 Engineering Building III, Campus Box 7115, Raleigh, NC 27695, USA. E-mail: dofreyte@ncsu.edu; Tel: +1 919-513-7933
bComparative Medicine Institute, North Carolina State University, 1060 William Moore Drive, Raleigh, NC 27606, USA
cMolecular Education, Technology and Research Innovation Center (METRIC), North Carolina State University, Dabney Hall, 2620 Yarbrough Drive, Raleigh, NC 27607, USA
dDepartment of Chemical Engineering, University of Puerto Rico-Mayaguez, Route 108, Mayaguez, Puerto Rico, USA
eDepartment of Orthopaedics, University of North Carolina School of Medicine, 102 Mason Farm Road Second Floor, Chapel Hill, NC 27514, USA
First published on 12th December 2022
Abstract
The goal of tissue decellularization is to efficiently remove unwanted cellular components, such as DNA and cellular debris, while retaining the complex structural and molecular milieu within the extracellular matrix (ECM). Decellularization protocols to date are centered on customized tissue-specific and lab-specific protocols that involve consecutive manual steps which results in variable and protocol-specific ECM material. The differences that result from the inconsistent protocols between decellularized ECMs affect consistency across batches, limit comparisons between results obtained from different laboratories, and could limit the transferability of the material for consistent laboratory or clinical use. The present study is the first proof-of-concept towards the development of a standardized protocol that can be used to derive multiple ECM biomaterials (powders and hydrogels) via a previously established automated system. The automated decellularization method developed by our group was used due to its short decellularization time (4 hours) and its ability to reduce batch-to-batch variability. The ECM obtained using this first iteration of a unified protocol was able to produce ECM hydrogels from skin, lung, muscle, tendons, cartilage, and laryngeal tissues. All hydrogels formed in this study were cytocompatible and showed gelation and rheological properties consistent with previous ECM hydrogels. The ECMs also showed unique proteomic composition. The present study represents the first step towards developing standardized protocols that can be used on multiple tissues in a fast, scalable, and reproducible manner.
1. Introduction
Extracellular matrix (ECM) based materials represent an increasingly popular class of biomaterials that promote tissue remodeling, repair, and regeneration at the site of injury while minimizing the risk of adverse reactions. ECM biomaterials are manufactured via removal of cellular components from the native tissue while preserving the main components (e.g., proteins, matrix-bound vesicles etc.) of the extracellular matrix using a process called decellularization. Most remarkably, ECM may be derived from different organs or tissues to recreate specific cellular microenvironments in composition and/or structure.1–3 ECM biomaterials are currently used for both research applications (e.g., substrates for cell growth,4–7 microfluidic cell culture systems,8 ECM-based bio-inks,9–11 drug screening,12–14 cancer studies15–19etc.) and in the clinic as implantable or injectable materials.20–22This breadth of applications calls for new, efficient, and scalable methods of ECM production. Decellularization protocols to date, however, are tissue, organ, and laboratory specific, typically focusing on relatively low quantities of materials when compared to what would be needed for commercial applications. In addition, there is inherent variability in the process that limits the number of batches that can be used. This batch-to-batch variation is a significant limiting factor that has significant cost and quality implications.
Various decellularization protocols for a wide range of tissues and organs exist; however, a single standardized and automated manufacturing process to derive ECM-based biomaterials from multiple tissues remains to be developed.23 These materials represent the raw decellularized form that is used to create ECM-based hydrogels that can be used as cell substrates or as injectable biomaterials. Numerous factors such as the tissue/organ source, the methods and reagents used, and the manual labor aspect of current protocols prevent the standardization of the decellularization process in commercial and laboratory settings. As a result, there is no standard decellularization protocol available for all tissue types and each protocol needs to be adapted to a particular tissue and/or per-lab basis. This knowledge gap negatively impacts the translation of ECM hydrogels and restricts comparisons among research teams.
Our objective was to show proof-of-concept for the derivation of a standardized multi-tissue decellularization protocol to derive ECM substrates (ECM powders) using our previously described automated system.24 By removing the variability inherently introduced when using tissue-specific decellularization protocols, it is possible to perform relevant biological comparisons without the confounding variable introduced by the protocol itself. In addition, by using an automated system, the consistency between batches is improved further enhancing our ability to study tissue specificity and to scale the production as needed. In this study we focus on the decellularization of eight different tissue types (vocal fold lamina propria (VFLP), supraglottic (SG), lung, heart, skin, muscle, tendon, and meniscus) using the same unified protocol and the characterization of the resulting decellularized ECM scaffold and its downstream product, the ECM hydrogel (Fig. 1). Although a true universal protocol will require more extensive characterization of the reagents and times used to derive the ECM powder, to the best of our knowledge, the present study describes the first proof-of-concept protocol using an automated and standardized method to produce ECM substrates allowing for tissue-specific comparisons not possible using established per-lab protocols.
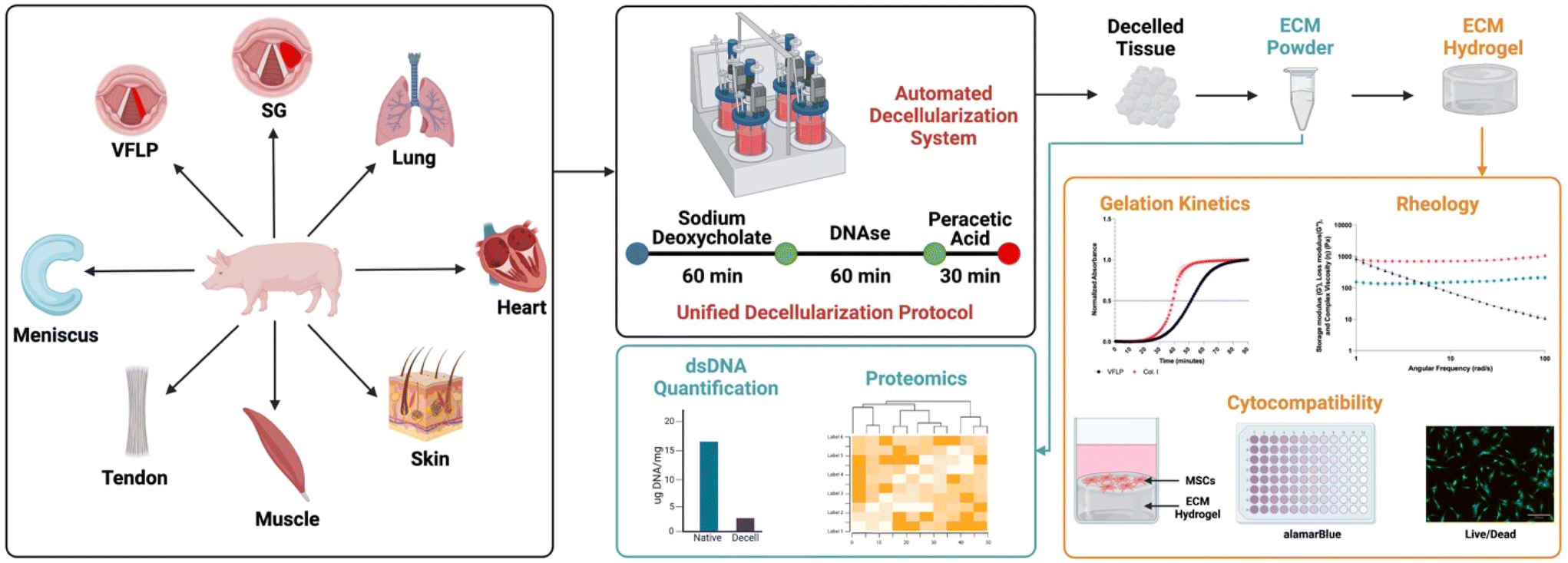 | ||
| Fig. 1 Overview. A new unified protocol and a previously established automated decellularization system were used to derive ECM-based biomaterials from 8 different tissue sources (vocal fold lamina propria (VFLP), supraglottic (SG), lung, heart, skin, muscle, tendon, and meniscus). The resulting ECM scaffolds were further processed into hydrogels. The feasibility of this method was assessed via double stranded DNA (dsDNA) quantification, discovery proteomics, gelation kinetics, rheology, and cytocompatibility assays. | ||
2. Methods
Standardized Tissue Decellularization All decellularizations were performed using an automated decellularization system previously described by our group.24,25 Porcine VFLP, SG, heart, and lung tissues were procured from market weight pigs from Nahunta Pork Outlet (Raleigh, NC). Porcine skin, muscle, and tendon tissues were obtained from 3–6 months old pigs from the College of Veterinary Medicine (CVM) at North Carolina State University. Lastly, porcine menisci were procured from 1 month old pigs from the CVM. The tissues were cleaned of blood, debris, excess fat and connective tissue, and frozen at −80 °C for at least 24 hours. The frozen heart, muscle, and lung were sliced using a commercial meat slicer into approximately 1.5–2.0 mm thick sheets. The heart, muscle, and lung sheets, VFLP, SG, tendon, skin, and meniscus were placed between two flat plates, frozen at −20 °C overnight, and chopped using an Alligator Mini Stainless-Steel Chopper (Amazon, Seattle, WA) into approximately 3.0 mm × 3.0 mm pieces. For each tissue type, approximately 1.0 g was placed in the automated decellularization system and treated with 30 mL each of the solutions listed in Table 1 under constant stirring at 220 rpm. The decellularized ECM scaffolds (VFLP-ECM, SG-ECM, Lung-ECM, Heart-ECM, Skin-ECM, Muscle-ECM, Tendon-ECM, Meniscus-ECM) were then frozen in liquid nitrogen, powdered using a mortar and pestle, and lyophilized overnight.| # | Reagent | Time (minutes) |
|---|---|---|
| 1 | DI water | 5 |
| 2 | DPBS | 5 |
| 3 | DI water | 5 |
| 4 | 4% sodium deoxycholate | 60 |
| 5 | DI water | 5 |
| 6 | DPBS | 5 |
| 7 | DI water | 5 |
| 8 | DNAse (140 Units per mL) | 60 |
| 9 | DI water | 5 |
| 10 | DPBS | 5 |
| 11 | DI water | 5 |
| 12 | 0.1% peracetic acid | 30 |
| 13 | DI water | 5 |
| 14 | DPBS | 15 |
| 15 | DI water | 5 |
2.1 Double stranded DNA (dsDNA) quantification
For all tissue types, approximately 1.0–3.0 mg of lyophilized native and decellularized ECM scaffold were digested in 20 μL stock Proteinase K solution (Qiagen, Hilden, Germany) and 180 μL ATL buffer (Qiagen) overnight at 55 °C. Next, the digested samples were diluted in 800 μL 1× TE buffer (Promega, Madison, WI) and mixed thoroughly. The samples were further diluted (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 for ECM scaffolds and 1:100 for native) using the same buffer. The QuantiFluor® dsDNA System (Promega) was used for the dsDNA quantification according to the manufacturer's instructions. The samples were read using an Infinite M PLEX plate reader (Tecan, Männedorf, Switzerland).
:50 for ECM scaffolds and 1:100 for native) using the same buffer. The QuantiFluor® dsDNA System (Promega) was used for the dsDNA quantification according to the manufacturer's instructions. The samples were read using an Infinite M PLEX plate reader (Tecan, Männedorf, Switzerland).
2.2 Discovery proteomics
320 residues). The two raw data files associated with each biological replicate were loaded into PD as fractions and processed as a single sample. A custom cleavage reagent called Elastase/Tryp was made that combined elastase and trypsin cleavage sites at alanine, isoleucine, lysine, leucine, arginine, and valine at the c-terminus with proline as an inhibitor. A label-free workflow was employed to obtain protein abundance values, and a custom home-built contaminants database was included in the searches to identify presence of human keratin and reagent enzyme peptides. Two SEQUEST HT nodes were put in linear sequence, the first using the custom Elastase/Tryp enzyme (full) and the second using the default trypsin enzyme (full). All other parameters were kept the same for both. SEQUEST HT nodes were set up as follows: maximum of 3 missed cleavage sites; minimum peptide length of 6 amino acids; 5 ppm precursor mass tolerance; 0.02 Da fragment mass tolerance; maximum of 8 equal dynamic modifications, which were oxidation of lysine, methionine, and proline, deamidation of asparagine and glutamine, addition of galactosyl or glucosylgalactosyl to lysine, and conversion of lysine to allysine; static carbamidomethylation of cysteine. Peptides were validated by Percolator after the first search node with q-value set to 0.05 and strict false discovery rate (FDR) set to 0.01. A Spectrum Selector node allowed all peptides with a confidence level “worse than high” to pass to the next SEQUEST node. The final Percolator node q-value was 0.05, and FDR was 0.01. Gene Ontology (GO) functional classification analysis (protein class) was performed using the ‘Gene List Analysis’ tool from https://www.pantherdb.org using Sus scrofa as the organism, and plots were constructed using PRISM or R.26 Associated ECM proteins and sub-units were listed according to Naba et al.27
2.3 Hydrogel preparation
For each ECM type, a digest was prepared at a concentration of 10 mg mL−1 in a ratio of 10:0.6:1 of lyophilized ECM, pepsin (3200–4500 units per mg; MilliporeSigma), and 0.1 M HCl (MilliporeSigma) and placed on a magnetic stir plate at room temperature for approximately 24 hours. The ECM digest was aliquoted and stored at −20 °C. When ready to use, the ECM digests were thawed, and ECM hydrogels at 6 mg mL−1 were prepared by adjusting the pH to 7.3 ± 0.2 using 0.1 M NaOH (MilliporeSigma) and balancing the salt content using 10× DPBS (Genesee Scientific, San Diego, CA) and DI water. Self-assembly was finalized by placing the solution into the incubator for 45 minutes at 37 °C.
A commercially available hydrogel, FibriCol I, Collagen Type I >97% (Advanced Biomatrix, Carlsbad, CA) (Col. I) was used as a control. The hydrogel was prepared according to the manufacturer's instructions by adjusting the salt content with 10× DPBS (Genesee Scientific), neutralizing the solution with 0.1 M NaOH (MilliporeSigma), and adjusting the volume with DI water to a final concentration of 6 mg mL−1. Self-assembly was achieved as described above.
2.4 Gelation kinetics
For each ECM type and Col. I control, 100 μL of the pH and salt balanced solution was pipetted in a well of a 96-well plate in triplicate per batch for a total of 3 batches each and kept on ice until measurement. Absorbance measurements were taken on a Tecan Infinite M PLEX plate reader. The samples were read at 405 nm every 1 minute for a total of 90 minutes at 37 °C. The following equation was used to normalize the data: | (1) |
2.5 Rheology
Rheological measurements were taken using an MCR 92 (AntonPaar, Graz, Austria) with parameters previously established.28,29 Briefly, the ECM digest was salt balanced, pH adjusted, and 1 mL of the final ECM solution was pipetted onto the baseplate at a concentration of 6 mg mL−1. A 25 mm parallel measuring plate (AntonPaar) was lowered to a 1 mm measuring height. The sample was then left at 37 °C for 45 minutes to allow for full self-assembly. A humidifier system was set up to prevent evaporation of the sample during the wait period. After the wait period, an oscillatory frequency sweep from 100–1 rad s−1 with 5% oscillatory shear strain amplitude was conducted to determine the complex viscosity, storage, and loss moduli of n = 3 different digestions for each ECM type, as well as the 6 mg mL−1 Col. I control.2.6 Cell culture conditions
Normal Human Bone Marrow Derived Mesenchymal Stem Cells (hMSCs) (PT-2501, Lonza, Walkersville, USA) were cultured according to the manufacturer's instructions. Briefly, hMSCs were cultured on tissue culture plastic (TCP) treated flasks at an initial seeding density of 5000 cells per cm2. The maintenance media (MSCBM Basal Medium, PT-3238, Lonza) was replaced every two days. hMSCs were passaged at 70–80% confluence using 0.25% trypsin-EDTA (Life Technologies, Carlsbad, CA) at 37 °C for 5 minutes, neutralized using maintenance media, and seeded onto TCP-treated flasks. hMSCs from passage 6 were used in this study.2.7 Cytocompatibility assays
Cell viability was assessed using the Live/Dead assay. Briefly, hMSCs were seeded on top of 0.2 mL ECM (VFLP, SG, lung, skin, muscle, tendon, meniscus) and Col. I control hydrogels at a concentration of 6 mg mL−1 (20000 cells per condition in a 48-well plate). hMSCs cultured on tissue culture plastic (TCP) were used as a secondary control. After 24 and 48 hours in culture, the samples were stained using the Live/Dead Viability/Cytotoxicity Assay (Life Technologies) according to the manufacturer's instructions. The samples were imaged using fluorescence microscopy (Revolve microscope, Echo, San Diego, CA) and counted using ImageJ (U.S. National Institutes of Health, Bethesda, Maryland).
Metabolic activity of hMSCs seeded on top of 0.2 mL of ECM and Col. I control hydrogels at a concentration of 6 mg mL−1 prepared as described above was assessed via the alamarBlue Cell Viability Kit (Invitrogen, Waltham, MA) at 24 and 48 hours in culture according to the manufacturer's instructions. The absorbance measurements were taken on the Infinite M PLEX plate reader. The percentage reduction of alamarBlue reagent of hMSCs was calculated using the following equation:30
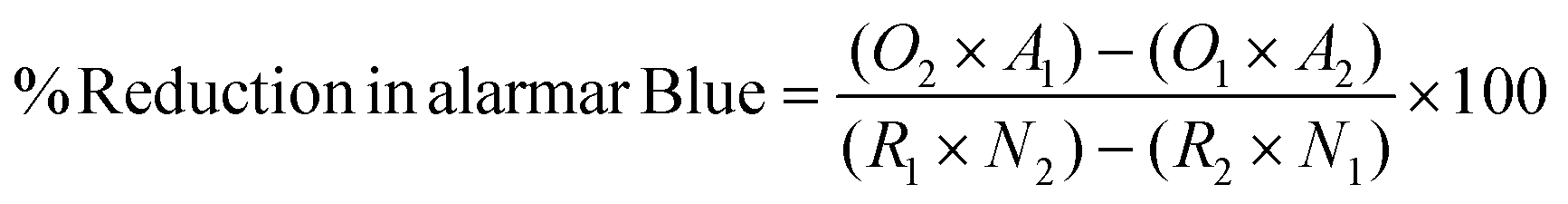 | (2) |
2.8 Statistical analysis
GraphPad Prism 9.0 and R software were used to plot the data and perform statistical analyses. All experiments were performed three independent times. Significance was defined as p < 0.05. For discovery proteomics data processing, Principal Component Analysis (PCA) was conducted with a custom R script (R version 4.1.2, RStudio version 2021.09.2+382) using the ‘prcomp’ function.31,32 Graphs were produced using the ‘ggplot2’,33 ‘Complex Heatmap’,34 and ‘UpSetR’35 packages in R. For the rheological measurements, statistical significance was determined via one-way ANOVA with a Dunnett's multiple test comparison to collagen as the control. For (1) gelation kinetics, (2) Live/Dead assay, and (3) alamarBlue assay, statistical significance was determined via two-way ANOVA with uncorrected Fisher's Least Significant Difference (LSD) when comparing (1) the time to reach 50% gelation between each ECM type and collagen control, (2) the live cell count at 24 vs. 48 hours, and (3) %reduction in alamarBlue when comparing each ECM type vs. collagen control at 24 and 48 hours.3. Results
3.1 Assessment of decellularization efficiency
Fig. 2A shows the automated decellularization system used. The native tissue was transferred to the bioreactor (Fig. 2A.1), placed on a magnetic stir plate (Fig. 2A.2), and treated with decellularization reagents (Fig. 2A.4) automatically fed to the bioreactor via individual peristaltic pumps (Fig. 2A.3). The pumps were controlled via the manufacturer software using a tablet (Fig. 2A.5) programmed to deliver the decellularization reagents and remove waste at specified time points.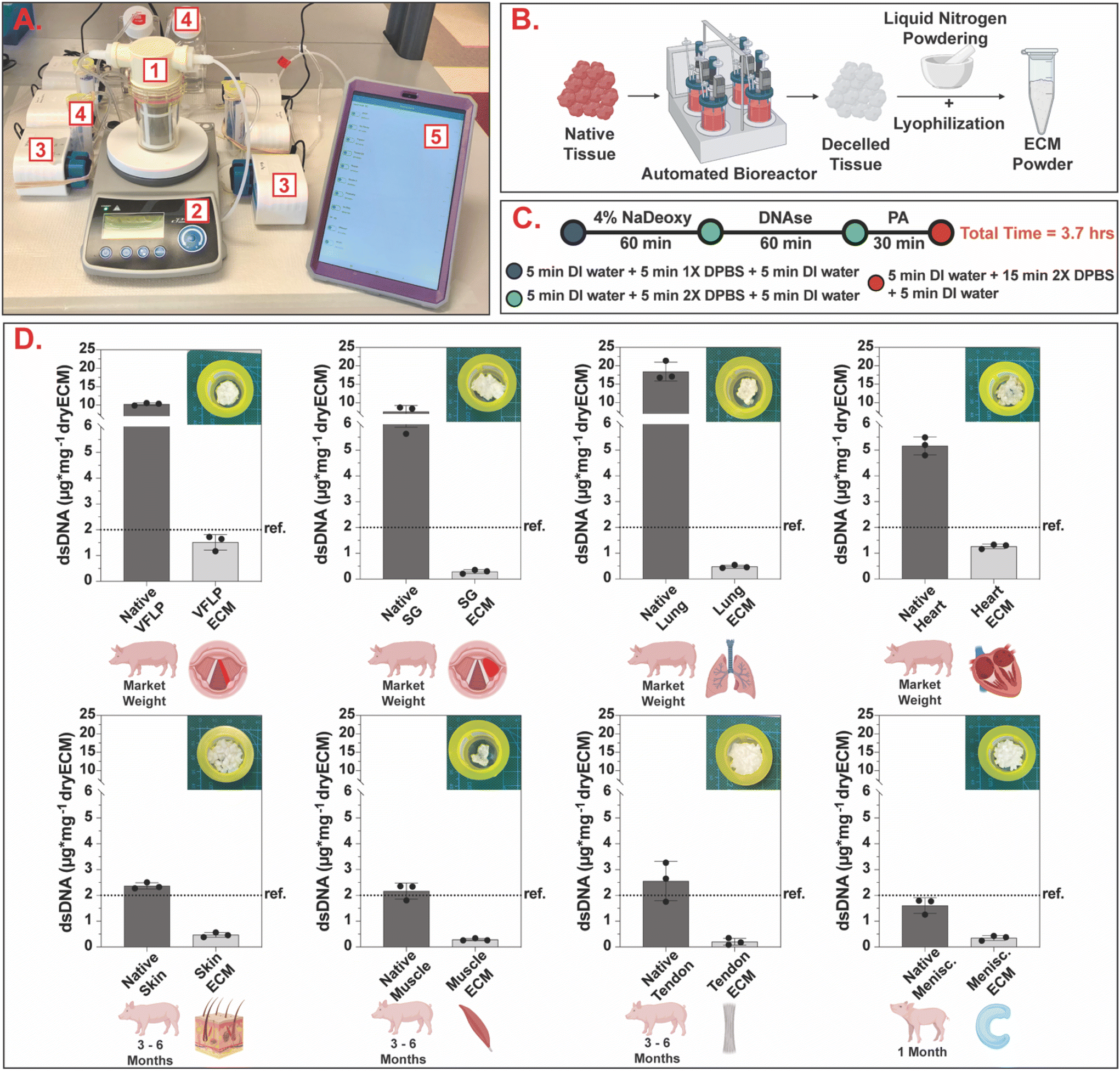 | ||
| Fig. 2 Automated standardized decellularization. (A) Automated decellularization system assembled on the benchtop (1 – bioreactor, 2 – stir plate, 3 – peristaltic pumps, 4 – reagent reservoirs, 5 – tablet for protocol management). (B) Chopped native tissue is placed inside the bioreactor and, after decellularization, the resulting ECM is powdered via the liquid nitrogen method and lyophilized to obtain ECM powder. (C) Schematic showing the decellularization protocol with a total completion time of 3.7 hours (NaDeoxy = sodium deoxycholate, PA = peracetic acid, DI = deionized, DPBS = Dulbecco's phosphate buffered saline). (D) Bar charts showing dsDNA quantification per mg of dry tissue for native and decellularized ECM. For each tissue type, three independent decellularizations were performed. The error bars represent the standard error of the mean (SEM). Ref. = 2 μg mg−1 (Urinary Bladder Matrix – UBM36). | ||
As shown in Fig. 2C, the same decellularization reagents, exposure times, and order in which the tissue was exposed were used for all tissue types. Decellularized ECM scaffolds were obtained in approximately 4 hours. Following decellularization, all samples were characterized via double stranded DNA (dsDNA) quantification to evaluate the efficacy of the unified protocol. The average and standard error of the mean (SEM) for each tissue type for native and decellularized ECM are shown in Fig. 2D and ESI Table 1.† The removal of nuclear content for each tissue type was below the dsDNA value for a commercially available ECM scaffold (urinary bladder matrix (UBM-ECM)) used as a reference for this study.36
3.2 Proteomics
The decellularized ECM scaffolds were characterized via discovery proteomics to identify and evaluate differences in protein composition between each ECM type. As shown in Fig. 3A, 1750 proteins and sub-units were identified in VFLP-ECM (unique = 127, where unique was defined as proteins and sub-units detected only in the specified ECM type in reference to the whole group of 8 ECM types). For the other ECM types, 1343 (unique = 68) were identified in SG-ECM, 2221 (unique = 668) in lung-ECM, 1831 (unique = 445) in heart-ECM, 875 (unique = 105) in skin-ECM, 694 (unique = 58) in muscle-ECM, 493 (unique = 7) in tendon-ECM, and 368 (unique = 16) in meniscus-ECM. All ECM types had 134 proteins and sub-units in common.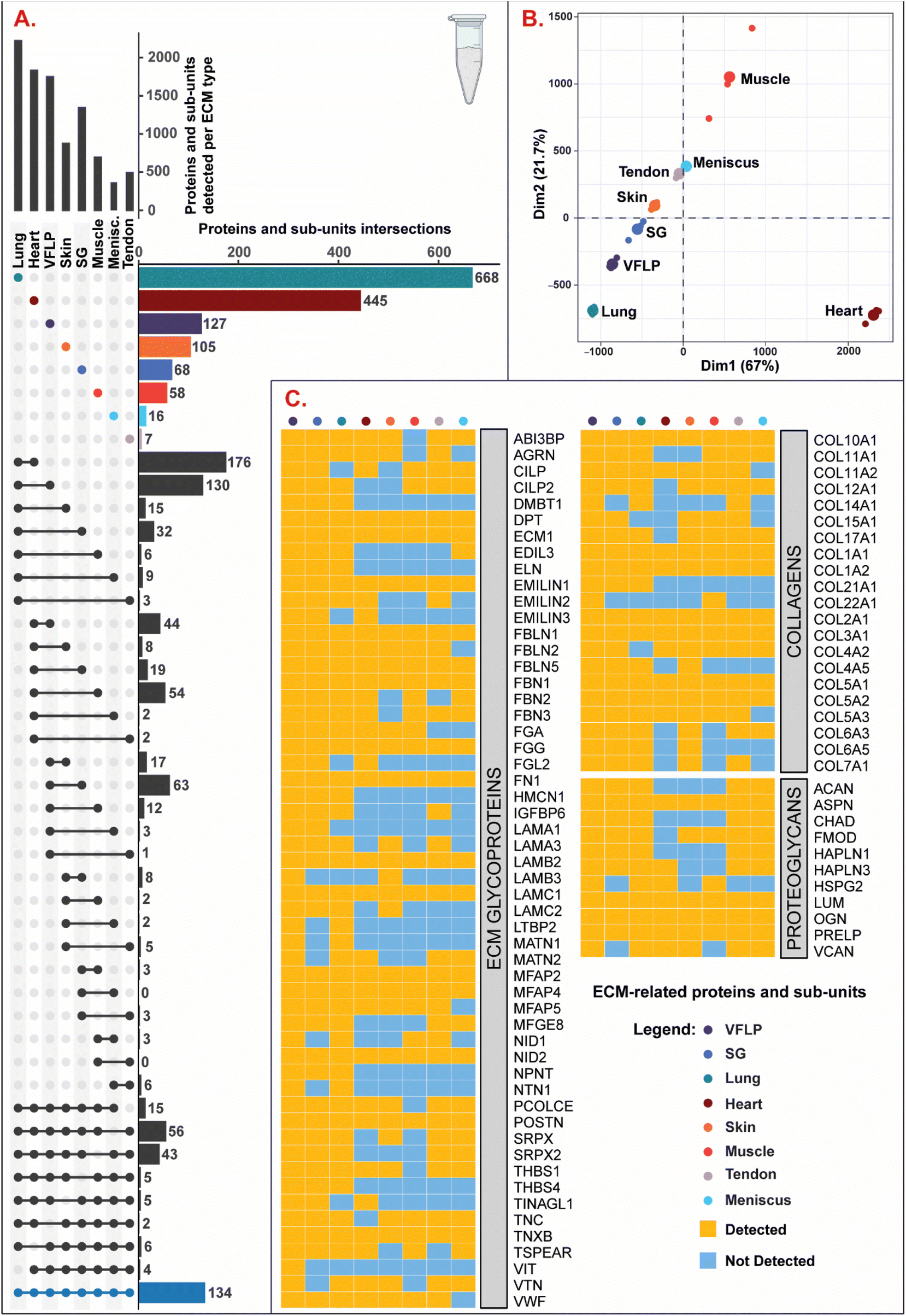 | ||
| Fig. 3 Discovery proteomics. (A) UpSet plot showing the total number of proteins and sub-units detected per condition, the number of unique proteins and sub-units detected per condition, and intersections of multiple conditions. (B) Dimensions 1 and 2 of Principal Component Analysis (PCA) of various ECM types. Each ECM type had n = 3 replicates (small points) with larger points representing the mean coordinates of the replicates. (C) Heatmap showing the detection or absence of collagens, proteoglycans, and ECM glycoproteins per each ECM type. | ||
A PCA plot (Fig. 3B) was used to illustrate the clustering between ECM types based on protein detection and abundance. ECM derived from the respiratory system (SG, VFLP, and lung) were located together in Quadrant III, where lung-ECM, which had the largest number of unique proteins and sub-units, was correspondingly located furthest from the center of the plot. Heart-ECM separated to the bottom left indicating that it had high variance in presence of unique proteins and sub-units along the Dimension 1 axis. The low Dimension 2 value was similar to lung-ECM and reflected the relatively large number of intersecting proteins and sub-units. Interestingly, muscle-ECM had the highest Dimension 2 value indicating that protein content differed from the other ECM types even with fewer unique proteins and sub-units.
In Fig. 3C, the proteins and sub-units were listed and categorized into collagens, proteoglycans, and ECM glycoproteins according to the database from Naba et al.27 For each category, there were several proteins and sub-units that were found in all ECM types such as: (1) collagens: COL1A1, COL1A2, COL2A1, COL3A1, COL5A1, COL5A2, COL10A1, (2) proteoglycans: ASPN, LUM, OGN, PRELP, and (3) ECM glycoproteins: DPT, ECM1, EMILIN1, FBLN1, FBLN5, FBN1, FGG, FN1, LAMB2, LAMC1, MFAP2, MFAP4, NID2, POSTN, TNXB. Of note, VFLP-ECM had the most while muscle-ECM had the least number of identified ECM-related proteins and sub-units. Overall, each ECM type has a unique signature of these proteins and sub-units with varying levels of detection, highlighting tissue-specific protein composition.
3.3 Self-assembly assessment
To evaluate self-assembly, the vial inversion test and gelation kinetics via absorbance measurements were determined for each ECM type and Collagen Type I (Col. I) control prepared at a concentration of 6 mg mL−1 (Fig. 4). Results show that VFLP-, SG-, lung-, skin-, muscle-, tendon-, and meniscus-ECM formed a stable hydrogel (Fig. 4B) and exhibited sigmoidal curves indicative of hydrogel formation similar to Col. I control (Fig. 4C). Heart-ECM failed to form a stable hydrogel (Fig. 4B and ESI Fig. 2†). The time to reach 50% gelation (Table 2) was fastest for meniscus-ECM and slowest for VFLP-ECM and SG-ECM. Lung-, skin-, muscle-, and tendon-ECM had gelation times similar to Col. I hydrogel control (p > 0.05).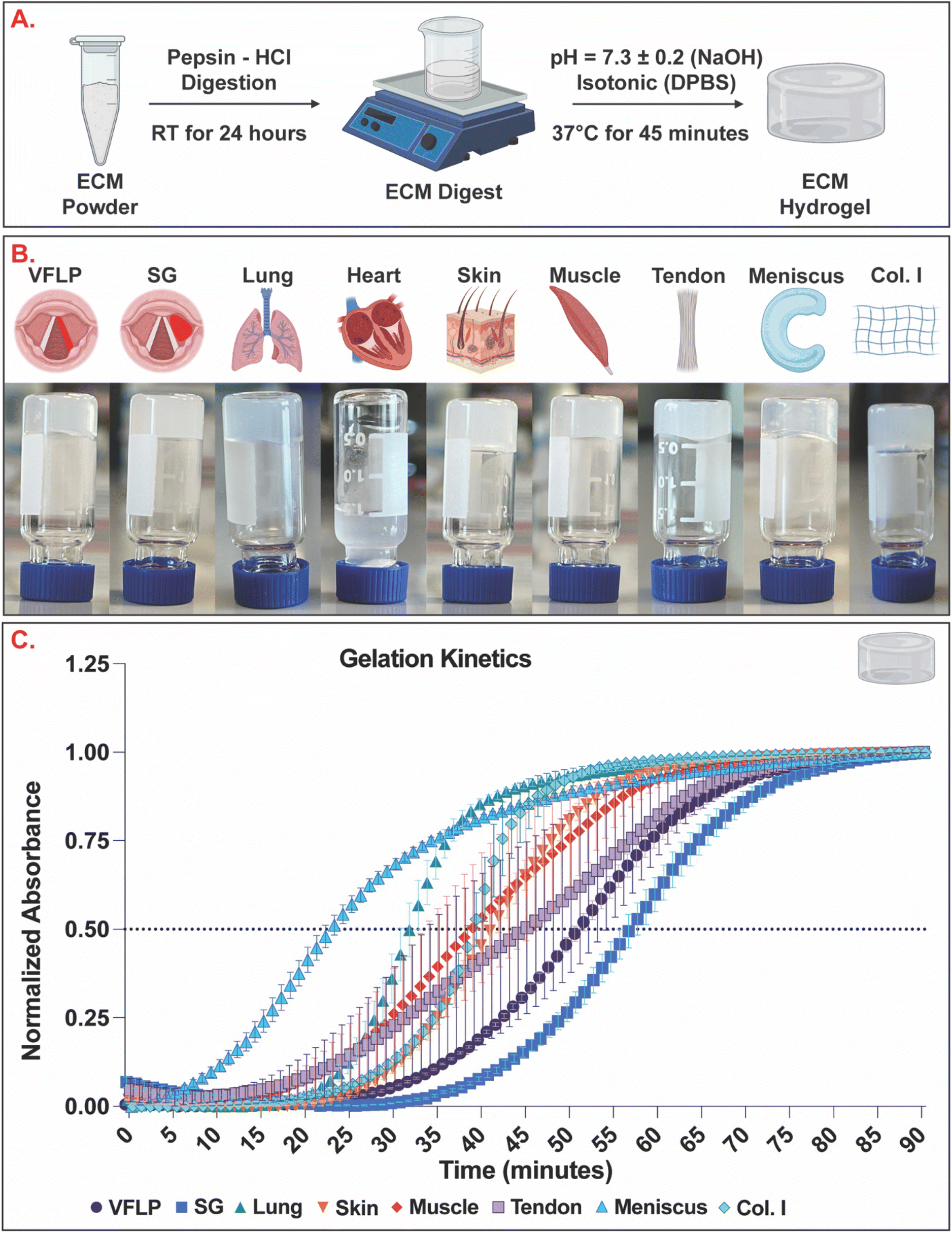 | ||
| Fig. 4 Gelation kinetics. (A) The ECM powder was enzymatically digested and the pH and salt concentration were adjusted to enable self-assembling into ECM hydrogels. (B) Vial inversion assay. (C) Gelation kinetics curves showing the average (of three replicates) normalized absorbance at 405 nm for VFLP-, SG-, lung-, skin-, muscle-, tendon-, and meniscus-ECM, and Col. I control. The error bars represent the standard error of the mean (SEM). | ||
| Tissue category | ECM type | Time (minutes) |
|---|---|---|
| * = statistically significantly different than Col. I control. | ||
| Respiratory | VFLP* | 51.3 ± 0.9 |
| SG* | 57.0 ± 1.5 | |
| Lung | 32.0 ± 1.0 | |
| Dermal | Skin | 41.0 ± 1.2 |
| Musculoskeletal | Muscle | 39.0 ± 5.9 |
| Tendon | 43.7 ± 8.4 | |
| Meniscus* | 22.7 ± 0.9 | |
| Control | Col. I | 39.0 ± 1.0 |
3.4 Rheology
Rheological measurements using oscillatory shear strain were used to determine the complex viscosity, and the storage and loss moduli (Fig. 5). Comparing the complex viscosity at 1 rad s−1, 6 mg mL−1 Col. I hydrogel showed the highest complex viscosity (mean = 1138.7 Pa s, SEM = 174.6); tendon-ECM, skin-ECM, SG-ECM, and VFLP-ECM hydrogels were not significantly different from 6 mg mL−1 Col. I hydrogel (Fig. 5E and F; ESI Table 2†). However, muscle-ECM (p = 0.0008 < 0.05), meniscus-ECM (p = 0.0136 < 0.05) and lung-ECM hydrogels (p = 0.004 < 0.05) were statistically significantly different from 6 mg mL−1 Col.I hydrogel. At 10 rad s−1, tendon-ECM hydrogel had the highest complex viscosity (mean = 95.8 Pa s, SEM = 6.4). Tendon-ECM along with skin-ECM, VFLP-ECM, SG-ECM, and meniscus-ECM hydrogels were not statistically significantly different 6 mg mL−1 Col. I hydrogel (mean = 93.9 Pa s, SEM = 10.7). Lung-ECM (p = 0.0139 < 0.05) and muscle-ECM hydrogels (p = 0.0015 < 0.05) were statistically significantly different from 6 mg mL−1 Col. I hydrogel. For 100 rad s−1, lung-ECM (p = 0.0075 < 0.05), meniscus-ECM (p = 0.0424 < 0.05) and muscle-ECM hydrogels (p = 0.0009 < 0.05) were statistically significantly different from 6 mg mL−1 Col. I hydrogel (mean = 14.7 Pa s, SEM = 1.3), while the other ECMs were not. For all frequencies, muscle-ECM hydrogel had the lowest measured complex viscosities (ESI Table 2†).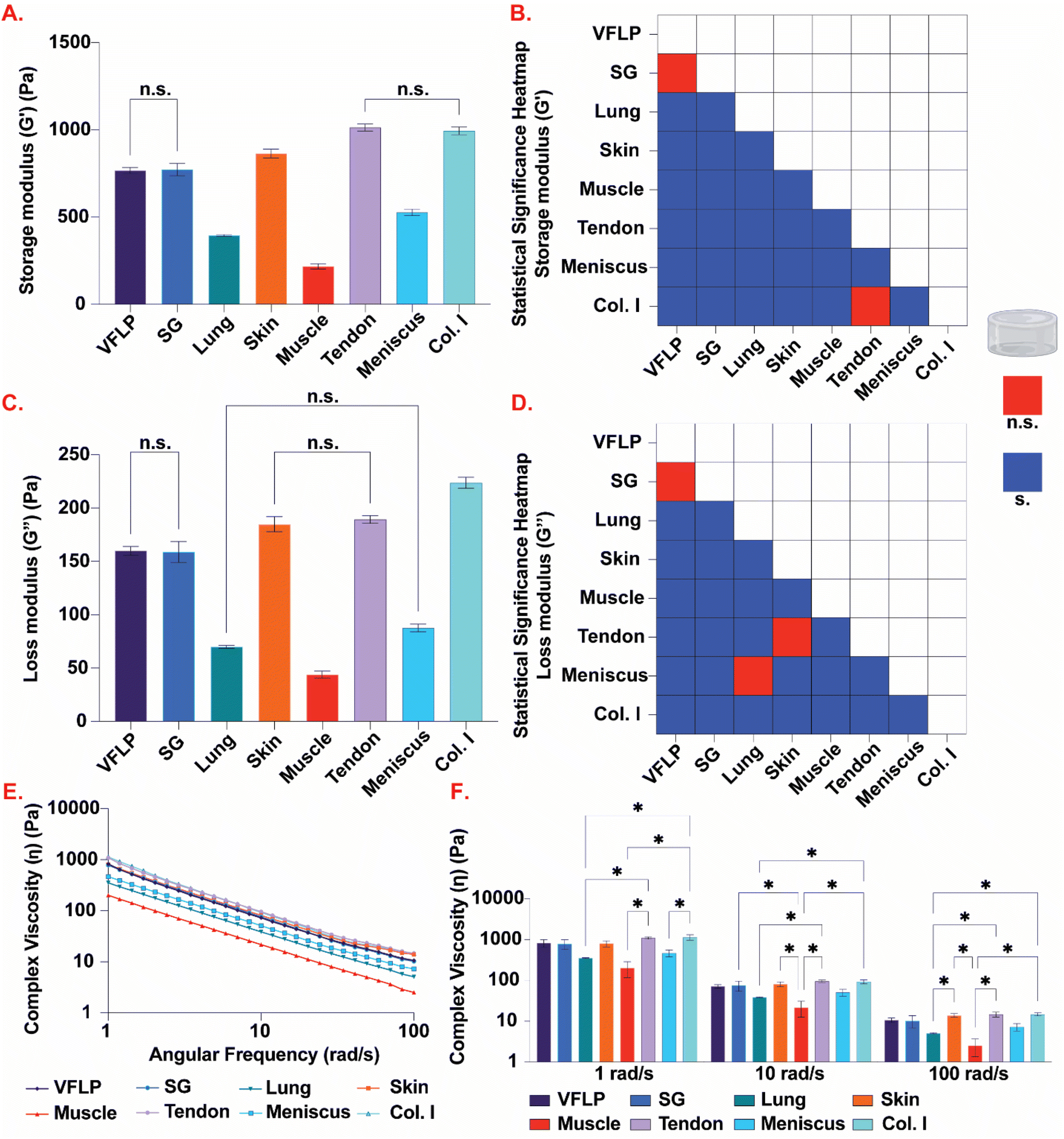 | ||
| Fig. 5 Rheology. (A) Average storage modulus (G′) for VFLP-, SG-, lung-, skin-, muscle-, tendon-, and meniscus-ECM, and Col. I control hydrogels prepared at 6 mg mL−1. (B) Heatmap displaying statistically significant (blue; P < 0.05) and not statistically significant (red; P > 0.05) storage modulus (G′) comparisons among all tested hydrogels. (C) Average loss modulus (G′′) for all tested hydrogels. (D) Heatmap displaying statistically significant (blue; P < 0.05) and not statistically significant (red; P > 0.05) loss modulus (G′′) comparisons among all tested hydrogels. (E) Average complex viscosity (η) for each ECM type and Col. I control plotted over an angular frequency from 1 to 100 rad s−1. (F) Average complex viscosity (η) at specific angular frequencies of 1, 10, and 100 rad s−1. For all plots, the error bars represent the standard error of the mean (SEM). n = 3 each. n.s. = not significant (P > 0.05), s. = significant (P < 0.05), * = statistically significant (P < 0.05). | ||
For the storage modulus, tendon-ECM hydrogel was found to be the stiffest (mean = 1014.0 Pa, SEM = 19.7) and the only hydrogel not statistically significantly different from 6 mg mL−1 Col. I (mean = 994.1 Pa, SEM = 21.9) (Fig. 5A and B, ESI Table 2†). Skin-ECM (p = 0.0001 < 0.05), muscle-ECM (p < 0.0001), lung-ECM (p < 0.0001), VFLP-ECM (p < 0.0001), SG-ECM (p < 0.0001), and meniscus-ECM hydrogels (p < 0.0001) were all statistically significantly different from 6 mg mL−1 Col. I hydrogel. The muscle-ECM hydrogel was found to have the lowest storage modulus (mean = 217.5 Pa, SEM = 15) (Fig. 5A). When comparing the loss modulus of each ECM to 6 mg mL−1 Col. I (mean = 223.6 Pa, SEM = 5.1), all ECMs were found to be statistically significantly different from 6 mg mL−1 Col. I (p < 0.0001 for all comparisons) (Fig. 5C and D, ESI Table 2†). For each group, however, the storage modulus was higher than the loss modulus showing each group did gel before the test was conducted (ESI Fig. 3 and 4†).
3.5 Cytocompatibility assessments
VFLP-, SG-, lung-, skin-, muscle-, tendon-, meniscus-ECM, and Col. I control were self-assembled into hydrogels and hMSCs were grown on top for 24 and 48 hours. hMSCs grown on TCP were used as an additional control. The cytocompatibility was evaluated via fluorescent imaging using the Live/Dead cell viability assay and the metabolic activity was assessed via the AlamarBlue assay. The results showed that all ECM hydrogels are non-cytotoxic for hMSCs. As shown by the Live/Dead assay (Fig. 6A and ESI Fig. 5†), hMSCs were alive in all conditions. Cell proliferation correlated positively with a significant increase in cell culture time from 24 to 48 hours in the case of VFLP-ECM, lung-ECM, muscle-ECM, tendon-ECM, and TCP (p < 0.05). For SG-ECM, skin-ECM, meniscus-ECM, and Col I there was no significant change over time. The metabolic activity results showed constant proliferation at 24 and 48 hours in culture. The %Reduction in alamarBlue was comparable to Col. I control for all ECM hydrogels (p > 0.05). These results indicate that all ECM hydrogels tested are cytocompatible and encourage hMSC attachment and growth.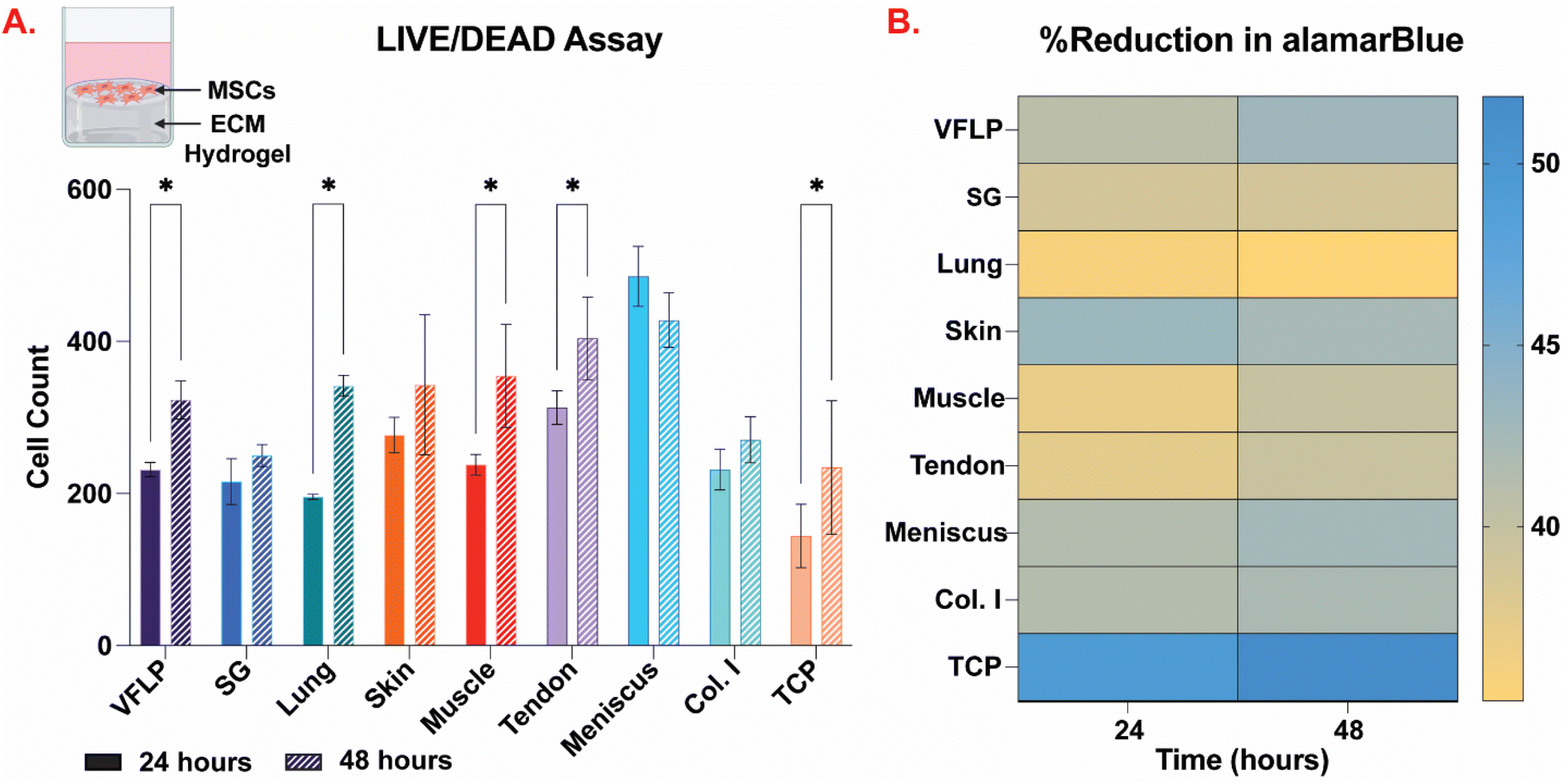 | ||
| Fig. 6 Cytocompatibility assays. (A) Live/Dead cell viability assay quantification using ImageJ (NIH, USA) for human mesenchymal stem cells (hMSCs) grown on VFLP-, SG-, lung-, skin-, muscle-, tendon-, and meniscus-ECM hydrogels, Col. I and tissue culture plastic (TCP) controls at 24 and 48 hours in culture. The error bars represent the standard error of the mean (SEM). * = statistically significant (p < 0.05). n = 3. (B) Metabolic activity of hMSCs grown on the same conditions as described above. n = 3. | ||
4. Discussion
ECM-derived hydrogels fabricated via tissue decellularization and subsequent processing (e.g., powdering, lyophilization, pepsin digestion) are of particular interest for regenerative medicine due to their ability to promote tissue remodeling, reduce fibrosis, and relative safety during clinical use.21,22,37–39 Many ECM scaffolds and materials derived from decellularized tissues and organs (e.g., Oasis®, DermACELL®, Alloderm®, MatriStem® etc.) are commercially available.40 Even though the ECM has a high clinical relevancy, there is a lack of standardization and effective protocol optimization across research labs and industry. A variety of decellularization protocols have been described, and factors such as prolonged exposure times, the labor-intensive aspect of the production process, pre- and post-decellularization steps, tissue/organ source, size, thickness, and/or digestion method negatively impact lot-to-lot variability, limiting fair comparisons between each ECM biomaterial. To address this knowledge gap, the present study took the first step towards developing a standardized and automated decellularization method capable of deriving ECM scaffolds for hydrogel production out of 8 different porcine tissue sources. DNA quantification, discovery proteomics, gelation kinetics, rheology, and cytocompatibility assays were performed to determine the feasibility of this method as a first approach to a unified decellularization protocol (Fig. 1). The goal is to show proof-of-concept that a fast and efficient protocol can be developed using current automated techniques and to highlight the potential limitations of such approach.The system used produces decellularized ECM scaffolds in a powder form in less than 4 hours (Fig. 2) which represents a significant time improvement to current methods which can take from days to weeks.24 The reagents selected for this study represent the first proof-of-concept protocol, but a true unified protocol will require more extensive testing of additional decellularization reagents. Previously, the combination of sodium deoxycholate, DNAse, peracetic acid, and DI water/DPBS washes was successfully applied to VFLP and SG.24,25 Due to its success and significant prior data, this protocol represented the foundation for the six additional tissues decellularized in this study. Table 3 shows a summary explanation for why these reagents were selected and other reagents that will be included in future studies. Additionally, even though not utilized in this study, the automated system allows for the incorporation of an online monitoring system via absorbance spectroscopy25 to further enable in-line monitoring and adjustments that can lead to further protocol optimization for ECM scaffold production. It is important to note that the unified decellularization protocol used in this study could be translated to other automated decellularization systems such as the recently developed open-source apparatus reported by Hamilton et al.41
| Reagent | Justification/role |
|---|---|
| Decellularization reagents used in the current bioreactor | |
| Sodium deoxycholate | Ionic detergent; solubilizes cell and nucleic membranes; protein denaturing |
| DNAse | DNA degradation |
| Peracetic acid | Disinfection |
| Potential additional decellularization reagents | |
| Sodium dodecyl sulfate (SDS) | Ionic detergent; solubilizes cell and nucleic membranes; protein denaturing |
| CHAPS | Zwitterionic detergent; possesses properties of both ionic and non-ionic detergents; mainly used to decellularize thinner tissues |
| Triton X | Non-ionic detergent; tissue delipidation |
| Tween | Non-ionic detergent; tissue delipidation |
| Trypsin | Disrupts tissue ultrastructure; allows for better penetration of subsequent decellularization reagents |
The final dsDNA content present in decellularized tissues is a commonly used end-point measurement to evaluate decellularization efficiency. As shown by Cramer et al., there are commercially available ECMs with higher dsDNA content, however, this does not limit their clinical use.44 In this study, we used the UBM-ECM as a reference for dsDNA removal given its current clinical use.36 As shown in Fig. 2D, all tissues decellularized in this study had a lower dsDNA content than UBM-ECM (less than approx. 2 μg mg−1 dry tissue). Notably, the starting native dsDNA content that we detected in skin, muscle, tendon, and meniscus were close to 2 μg mg−1. However, these values are on the same order of magnitude to values previously reported by other authors for these tissue types: 814 ± 14 ng mg−1 for tendon,45 1598 ± 527 ng mg−1 for muscle,46 0.12 ± 0.019 ng mg−1 for skin,47 12.6 ± 1.7 ng mg−1 for meniscus.48 Even though the starting native dsDNA content is low, the decellularization process is still necessary because any residual cellular material can lead to issues such as in vitro cytotoxicity and in vivo adverse host immune responses.42 The exact concentration of cellular material remaining within the ECM needed to elicit a negative response is yet to be determined and can be based on the ECM type as well as the site of implantation.42,44,49
The protocol described in this study featured a 50 mL bioreactor that allowed for the efficient decellularization of 1 gram of starting native tissue. As a result of the lower amount of starting tissue, the final decellularized ECM yield could be lower when compared to decellularizing sheets or whole organs. Additionally, the use of lower amounts of starting material does not allow for the use of a commercial mill, which would allow for a more consistent particle size and would enable further standardization of the decellularization process. Therefore, future studies will focus on scaling-up the current process from 50 mL bioreactors to industrial scale bioreactors. The scale-up process will require evaluating and adjusting different steps during the process such as the stirring mechanism used, volume of the reactor, concentration of the decellularization reagents, and reagent exposure times. It is important to note that existing methods for hydrogel production require the material to be removed from the decellularization chamber and be processed elsewhere. Future work will also focus on other improvements to standardize this process such as producing ECM hydrogels or other solubilized ECM products inside the bioreactor immediately after the decellularization process.
In comparison to synthetic or single-protein hydrogels (e.g., Type I Collagen, Hyaluronic Acid (HA) etc.), ECM-derived hydrogels can retain a variety of ECM and ECM-related components closely mimicking the native tissue. Analysis via discovery proteomics (Fig. 3) revealed a significant number of proteins and sub-units were retained in the ECM scaffolds after the decellularization process. Even though the same decellularization protocol was applied to all tissue types, each resulting decellularized scaffold shows a unique signature of proteins and sub-units. The data indicates that the standardized protocol is allowing us to rule out the decellularization method as a factor affecting ECM protein composition across materials, enabling insights into tissue specificity based on protein composition. To further characterize and assess the efficiency of the unified protocol, future studies will include comparisons via discovery proteomics between decellularized and native tissues. Future studies will also include quantitative assays to evaluate the amount of targeted proteins. The information gained from these studies will allow us to determine how the different decellularization reagents used are affecting the protein content of the final ECM material. Future studies will also consider how the protein composition of the different tissue types is affected based on the amount of starting material used.
The clinical use of ECMs can sometimes be limited by their immunogenicity due to residual cellular debris present in the scaffold at the end of the decellularization process. Since proteins represent an important source of immunogens, we searched for the detection or absence of 32 potentially immunogenic proteins (ESI Fig. 1†).50–52 Of note, Annexin-A2 (ANXA2) and Actin, cytoplasmic 1 beta-actin (ACTB) were detected in all ECM scaffolds derived via the unified method as well as UBM-ECM used as a reference.39 The highest number of immunogenic proteins and sub-units was detected in UBM (19 proteins and sub-units) followed by VFLP-ECM (14 proteins and sub-units). Since UBM-ECM is currently used clinically, it can be inferred that the immunogenic potential of the ECMs derived by the unified protocol is also low. As reported in the study by Hill et al., the complete removal of cellular debris from decellularized scaffolds is highly unlikely.53 However, successful implementation of ECM scaffolds such as UBM and small intestinal submucosa (SIS) in the clinic suggests that current decellularization protocols adequately remove cellular debris. A limitation of discovery proteins is that it does not provide a quantitative value of proteins present in the ECM scaffolds. Future studies will include immunogenic assays (e.g., Enzyme Linked Immunosorbent Assay (ELISA), immunohistochemistry (IHC) etc.) as described in the “Standard Guide for Evaluating Extracellular Matrix Decellularization Processes”54 to quantify the immunogenic potential of the ECMs derived via the unified protocol. Nevertheless, these preliminary findings support the overall goal of a standardized protocol able to decellularize multiple tissues.
ECM-derived hydrogels are being used in a variety of in vitro and in vivo applications due to their injectability, inherent biological activity, tunable mechanical properties, three-dimensional structure suitable for cell growth, and ability to incorporate drugs and biologically active molecules.22,40 In this study, we used gelation kinetics and rheological measurements to determine the ability of the ECM-derived hydrogels to self-assemble. All hydrogels, with the exception of heart, prepared via this first iteration of an automated and standardized protocol formed a stable hydrogel at 37 °C. As shown in Fig. 4C, the formation of hydrogel is indicated by the sigmoidal curves, which are similar to collagen type I. Additionally, as shown in ESI Fig. 3 and 4,† the storage modulus was higher than the loss modulus throughout the frequency sweep (G′ > G′′), which is another indicator of stable hydrogel formation. Furthermore, tendon-ECM, skin-ECM, SG-ECM, and VFLP-ECM hydrogels showed similar complex viscosities when compared to type I collagen hydrogels. However, skin-ECM and tendon-ECM hydrogels were stiffer than all other ECM hydrogels. It is important to note that skin and tendon tissues are naturally high in type I collagen.55 SG-ECM and VFLP-ECM hydrogels were found to have lower complex viscosities than tendon-ECM or skin-ECM hydrogels. Lung-ECM, muscle-ECM, and meniscus-ECM hydrogels were found to be significantly lower than type I collagen hydrogel. These results show that differences seen in cellular response between tendon-ECM, skin-ECM, SG-ECM, VFLP-ECM, and collagen are not to be attributed to the mechanical properties as these hydrogels will contract and resist flow similarly. However, mechanical differences seen between collagen, lung-ECM, muscle-ECM, and meniscus-ECM hydrogels may contribute to different cellular responses as the latter ECM hydrogel types have lower stiffnesses and would be more susceptible to contraction when stresses are applied by the cells.
The differences in gelation and rheological profiles between each ECM type can also be explained by the tissue-specific protein composition as seen in the discovery proteomics analysis. It has been shown that the presence of glycosaminoglycans (GAGs) and some collagen types (e.g., collagen type V) can affect the in vitro collagen type I self-assembly.56 In this study, heart-ECM did not form a stable hydrogel. Other studies have shown that cardiac ECM can exhibit slow gelation time, fast degradation, and poor mechanical properties. As a result, hydrogels derived from cardiac ECM are often combined with other natural or synthetic biomaterials such as collagen type I or polyethylene glycol (PEG) to improve their mechanical properties.57,58 Future studies will include fine-tuning of the decellularization protocol and the reagents used to achieve appropriate gelation of heart-ECM. Fine-tuning of the decellularization process can be achieved via the online monitoring system previously described,25 which will provide a representative DNA release profile from the tissue in real time for each decellularization reagent used. This information would enable the users to determine the optimal duration of exposure for specific decellularization reagents to protect the ECM while effectively removing the nucleic acid content from the tissue. Lastly, we may need to optimize the digestion process since the protocol used was developed for older, manual methods. Still, this remains a substantial first step towards finding a short and standardized decellularization protocol.
Human mesenchymal stem cells (hMSCs) were seeded on the ECMs that formed a stable hydrogel and their cytocompatibility was evaluated via the Live/Dead and alamarBlue assays (Fig. 6). As shown by these results, the ECM hydrogels derived via the standardized automated method are non-cytotoxic and allowed for hMSC attachment. Cell viability and proliferation studies, in combination with the proteomic analysis, are strong indicators that the proposed automated decellularization method yields tissue-specific ECM-derived hydrogels that are suitable for in vitro cell-based assays. Furthermore, in conjunction with the rheological and gelation studies, these results indicate that the proposed method is suitable for the large-scale production of tissue-specific hydrogels enabling surgical delivery via injection. Future studies will focus on determining the materials immunogenicity in vitro and in vivo.
In summary, these results suggest that the proposed automated method is suitable for the rapid decellularization of a variety of tissue types to obtain cytocompatible and tissue-specific ECM-hydrogels in a scalable and reproducible manner. Furthermore, the modularity of the bioreactor enables further protocol development for the retention of specific proteins at various timepoints within the decellularization steps, as well as the in-line real-time monitoring of dsDNA content via spectrophotometric analysis.25 This study is the first step towards the standardization of automated decellularization processes that could lead to the development of an industry standard for tissue-specific ECM derivatives.
5. Conclusions
This comprehensive study demonstrated the feasibility to derive ECM hydrogels from a variety of tissues using the same protocol in an effort to standardize and scale up production. The bioreactor design and the modularity of the decellularization protocol enables the fine tuning of the decellularization process at precise steps or time points. The proposed method is the first step towards the standardization of a scalable and unified automated decellularization system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health/National Institute on Deafness and Other Communication Disorders (R01DC017139, R01DC017743) and the National Heart, Lung, and Blood Institute (1R01HL147357-01A1). We acknowledge the Comparative Medicine Institute (CMI) at North Carolina State University. We thank the Molecular Education, Technology and Research Innovation Center (METRIC) at North Carolina State University, which is supported by the state of North Carolina, for their contributions. We acknowledge the startup funding support provided to Dr Mora-Navarro by the CAWT under NSF grant OIA-1849243. The figures in this work were created with BioRender.com.References
- A. D. Doyle and K. M. Yamada, Exp. Cell Res., 2016, 343, 60–66 CrossRef PubMed.
- M. Matsusaki and M. Akashi, Polym. J., 2014, 46, 524–536 CrossRef.
- J. S. Choi, J. D. Kim, H. S. Yoon and Y. W. Cho, Tissue Eng., Part A, 2013, 19, 329–339 CrossRef PubMed.
- R. Nakamura, F. Nakamura and S. Fukunaga, Anim. Sci. J., 2014, 85, 706–713 CrossRef PubMed.
- R. P. Bual and H. Ijima, Regener. Ther., 2019, 11, 258–268 CrossRef PubMed.
- D. Jhala and R. Vasita, Polym. Rev., 2015, 55, 561–595 CrossRef CAS.
- P. J. Kluger, S. Nellinger, S. Heine and A.-C. Volz, Curr. Opin. Biomed. Eng., 2020, 6, 410–413 Search PubMed.
- B. M. Gillette, H. Parsa and S. K. Sia, Part 1 Mater Fabr Methods, 2013, 53–79 Search PubMed.
- W. Jeong, M. K. Kim and H.-W. Kang, J. Tissue Eng., 2021, 12, 2041731421997091 Search PubMed.
- S. J. Lee, J. H. Lee, J. Park, W. D. Kim and S. A. Park, Materials, 2020, 13, 3522 CrossRef CAS PubMed.
- J.-Y. Won, M.-H. Lee, M.-J. Kim, K.-H. Min, G. Ahn, J.-S. Han, S. Jin, W.-S. Yun and J.-H. Shim, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 644–649 CrossRef CAS PubMed.
- M. Gerckens, H. Alsafadi, D. Wagner, K. Heinzelmann, K. Schorpp, K. Hadian, M. Lindner, J. Behr, M. Königshoff, O. Eickelberg, A. Yildirim and G. Burgstaller, Pneumologie, 2019, 73, 113–114 Search PubMed.
- A. D. Schwartz, L. E. Barney, L. E. Jansen, T. V. Nguyen, C. L. Hall, A. S. Meyer and S. R. Peyton, Integr. Biol., 2017, 9, 912–924 CrossRef CAS.
- M. V. Monteiro, V. M. Gaspar, L. P. Ferreira and J. F. Mano, Biomater. Sci., 2020, 8, 1855–1864 RSC.
- N. Kaushik, S. Kim, Y. Suh and S.-J. Lee, Arch. Pharmacal Res., 2019, 42, 40–47 CrossRef CAS.
- P. Lu, V. M. Weaver and Z. Werb, J. Cell Biol., 2012, 196, 395–406 CrossRef CAS PubMed.
- V. Poltavets, M. Kochetkova, S. M. Pitson and M. S. Samuel, Front. Oncol., 2018, 8, 431 CrossRef.
- M. Takeda, S. Miyagawa, E. Ito, A. Harada, N. Mochizuki-Oda, M. Matsusaki, M. Akashi and Y. Sawa, Sci. Rep., 2021, 11, 5654 CrossRef CAS.
- A. R. D. Jensen, E. R. Horton, L. H. Blicher, E. J. Pietras, C. Steinhauer, R. Reuten, E. M. Schoof and J. T. Erler, Cancers, 2021, 13, 3331 CrossRef CAS PubMed.
- J. Pak, J. H. Lee, K. S. Park, B. C. Jeong and S. H. Lee, BioRes. Open Access, 2016, 5, 192–200 CrossRef CAS PubMed.
- J. H. Traverse, T. D. Henry, N. Dib, A. N. Patel, C. Pepine, G. L. Schaer, J. A. DeQuach, A. M. Kinsey, P. Chamberlin and K. L. Christman, J. Am. Coll. Cardiol. Basic Trans. Sci., 2019, 4, 659–669 Search PubMed.
- M. T. Spang and K. L. Christman, Acta Biomater., 2018, 68, 1–14 CrossRef CAS PubMed.
- M. J. Hernandez, G. E. Yakutis, E. I. Zelus, R. C. Hill, M. Dzieciatkowska, K. C. Hansen and K. L. Christman, Methods, 2019, 171, 20–27 CrossRef PubMed.
- A. Badileanu, C. Mora-Navarro, A. M. G. Martins, M. E. Garcia, D. Sze, E. W. Ozpinar, L. Gaffney, J. R. Enders, R. C. Branski and D. O. Freytes, ACS Biomater. Sci. Eng., 2020, 6, 4200–4213 CrossRef CAS PubMed.
- C. Mora-Navarro, M. E. Garcia, P. Sarker, E. W. Ozpinar, J. R. Enders, S. Khan, R. C. Branski and D. O. Freytes, Biomed. Mater., 2022, 17, 015008 CrossRef CAS.
- H. Mi, A. Muruganujan, X. Huang, D. Ebert, C. Mills, X. Guo and P. D. Thomas, Nat. Protoc., 2019, 14, 703–721 CrossRef CAS PubMed.
- A. Naba, K. R. Clauser, H. Ding, C. A. Whittaker, S. A. Carr and R. O. Hynes, Matrix Biol., 2016, 49, 10–24 CrossRef PubMed.
- L. S. Gaffney, Z. G. Davis, C. Mora-Navarro, M. B. Fisher and D. O. Freytes, Tissue Eng., Part A, 2022, 28(5–6) DOI:10.1089/ten.tea.2021.0070.
- D. O. Freytes, J. Martin, S. S. Velankar, A. S. Lee and S. F. Badylak, Biomaterials, 2008, 29, 1630–1637 CrossRef PubMed.
- Measuring cytotoxicity or proliferation – alamarBlue Assay Protocol | Bio-Rad, https://www.bio-rad-antibodies.com/measuring-cytotoxicity-proliferation-spectrophotometry-fluorescence-alamarblue.html, (accessed February 23, 2022).
- C. Mora-Navarro, E. W. Ozpinar, D. Sze, D. P. Martin and D. O. Freytes, Biomed. Mater., 2021, 16, 025006 CrossRef PubMed.
- R: The R Project for Statistical Computing, https://www.r-project.org/, (accessed March 9, 2022).
- H. Wickham, ggplot2: Elegant Graphics for Data Analysis, https://ggplot2.tidyverse.org, (accessed March 9, 2022).
- Z. Gu, R. Eils and M. Schlesner, Bioinformatics, 2016, 32, 2847–2849 CrossRef PubMed.
- N. Gehlenborg, UpSetR: A More Scalable Alternative to Venn and Euler Diagrams for Visualizing Intersecting Sets, https://CRAN.R-project.org/package=UpSetR, (accessed March 9, 2022).
- L. Huleihel, G. S. Hussey, J. D. Naranjo, L. Zhang, J. L. Dziki, N. J. Turner, D. B. Stolz and S. F. Badylak, Sci. Adv., 2016, 2, e1600502 CrossRef.
- S. F. Badylak, D. O. Freytes and T. W. Gilbert, Acta Biomater., 2009, 5, 1–13 CrossRef CAS PubMed.
- L. T. Saldin, M. C. Cramer, S. S. Velankar, L. J. White and S. F. Badylak, Acta Biomater., 2017, 49, 1–15 CrossRef CAS PubMed.
- C. Mora-Navarro, A. Badileanu, A. M. G. Martins, E. W. Ozpinar, L. Gaffney, I. Huntress, E. Harrell, J. R. Enders, X. Peng, R. C. Branski and D. O. Freytes, ACS Biomater. Sci. Eng., 2020, 6, 1690–1703 CrossRef CAS PubMed.
- W. Zhang, A. Du, S. Liu, M. Lv and S. Chen, Regener. Ther., 2021, 18, 88–96 CrossRef CAS PubMed.
- A. G. Hamilton, J. M. Townsend and M. S. Detamore, Tissue Eng., Part C, 2022, 28, 137–147 CrossRef CAS.
- P. M. Crapo, T. W. Gilbert and S. F. Badylak, Biomaterials, 2011, 32, 3233–3243 CrossRef CAS PubMed.
- L. J. White, A. J. Taylor, D. M. Faulk, T. J. Keane, L. T. Saldin, J. E. Reing, I. T. Swinehart, N. J. Turner, B. D. Ratner and S. F. Badylak, Acta Biomater., 2017, 50, 207–219 CrossRef CAS PubMed.
- M. C. Cramer and S. F. Badylak, Ann. Biomed. Eng., 2019, 1–22 CAS.
- A. D. Eren, R. Sinha, E. D. Eren, Y. Huipin, S. Gulce-Iz, H. Valster, L. Moroni, J. Foolen and J. de Boer, J. Immunol. Regener. Med., 2020, 8, 100027 CrossRef.
- J. Zhang, Z. Q. Hu, N. J. Turner, S. F. Teng, W. Y. Cheng, H. Y. Zhou, L. Zhang, H. W. Hu, Q. Wang and S. F. Badylak, Biomaterials, 2016, 89, 114–126 CrossRef CAS PubMed.
- R. D. Ventura, A. R. Padalhin, C. M. Park and B. T. Lee, Mater. Sci. Eng., C, 2019, 104, 109841 CrossRef CAS PubMed.
- H. Hanai, G. Jacob, S. Nakagawa, R. S. Tuan, N. Nakamura and K. Shimomura, Front. Cell Dev. Biol., 2020, 8, 581972 CrossRef PubMed.
- Y. Huang, H. Yue, Z. Lian and X. Li, The Decellularization of Whole Organs, in Decellularized Materials, Preparations and Biomedical Applications, Springer, 2021, ch. 5, pp. 253–311 Search PubMed.
- J. Muhamed, T. Anilkumar, A. Rajan, A. Surendran and A. Jaleel, Biomed. Phys. Eng. Exp., 2019, 5, 025003 CrossRef.
- J. M. Aamodt and D. W. Grainger, Biomaterials, 2016, 86, 68–82 CrossRef PubMed.
- U. Boeer, F. F. R. Buettner, M. Klingenberg, G. C. Antonopoulos, H. Meyer, A. Haverich and M. Wilhelmi, PLoS One, 2014, 9, e105964 CrossRef PubMed.
- R. C. Hill, E. A. Calle, M. Dzieciatkowska, L. E. Niklason and K. C. Hansen, Mol. Cell. Proteomics, 2015, 14, 961–973 CrossRef.
- Standard Guide for Evaluating Extracellular Matrix Decellularization Processes, https://www.astm.org/f3354-19.html, (accessed July 10, 2022).
- N. Gallo, M. L. Natali, A. Sannino and L. Salvatore, J. Funct. Biomater., 2020, 11, 79 CrossRef CAS PubMed.
- J. Fernández-Pérez and M. Ahearne, Sci. Rep., 2019, 9, 14933 CrossRef.
- L. Gómez-Cid, M. L. López-Donaire, D. Velasco, V. Marín, M. I. González, B. Salinas, L. Cussó, Á. García, S. B. Bravo, M. E. Fernández-Santos, C. Elvira, J. Sierra, E. Arroba, R. Bañares, L. Grigorian-Shamagian and F. Fernández-Avilés, Int. J. Mol. Sci., 2021, 22, 9226 CrossRef PubMed.
- B. Peña, M. Laughter, S. Jett, T. J. Rowland, M. R. G. Taylor, L. Mestroni and D. Park, Macromol. Biosci., 2018, 18, 1800079 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01012g |
| This journal is © The Royal Society of Chemistry 2023 |