
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An improved, optimised and robust keratin azure assay for accurate assessment of keratinase activity†
Rhona M.
Cowan‡
 a,
Eleanor
Birch‡
a,
Grace
Nisbet§
a,
Chimaeze
Onyeiwu
b,
Clare
Campbell
c,
Ian
Archer¶
d and
Dominic J.
Campopiano
*a
a,
Eleanor
Birch‡
a,
Grace
Nisbet§
a,
Chimaeze
Onyeiwu
b,
Clare
Campbell
c,
Ian
Archer¶
d and
Dominic J.
Campopiano
*a
aSchool of Chemistry, University of Edinburgh, David Brewster Road, King's Buildings, Edinburgh, EH9 3FJ, UK
bJohnstons of Elgin, Newmill, Elgin, Moray, Elgin, IV30 4AF, UK
cPrickly Thistle Scotland Ltd, Evanton Industrial Estate, Beechwood Rd, Evanton, Alness IV16 9XJ, UK
dIndustrial Biotechnology Innovation Centre (IBioIC), Inovo Building, 121 George Street, Glasgow G1 1RD, UK
First published on 6th November 2023
Abstract
Keratin, in the form of coarse sheep wool, has been identified as an undervalued natural resource, which with the appropriate tools (e.g. a keratinase biocatalyst) can be repurposed for various textile and industrial biotechnology applications. For these purposes, we describe a novel method for identifying keratinase activity through the use of α-keratin azure (KA), an anthraquinone dyed substrate. A colourimetric method monitored the keratinase activity of Proteinase K (PK), which degrades the KA substrate and releases soluble products that are observed at 595 nm. Initially, the azure dye standard, Remazol Brilliant Blue R (RBBR), was used to calibrate the assay and allowed the kinetics of the keratinase-catalysed reaction to be determined. The assay was also used to investigate substrate pre-treatment, as well as different reaction quenching/work up conditions. Milling and washing of the KA substrate provided the best reproducibility and centrifugation was the most effective method for removing unreacted starting material. This assay was then applied to investigate the reduction of the keratin disulfide bond on keratinase-catalysed degradation. This optimised, improved and robust method will enable identification of keratinases ideally suited for application in the valorisation of the α-keratin found in natural wool fibres.
Introduction
Keratins are the third most abundant natural biopolymer, forming the epidermal structures of mammals, birds, and fish in forms as diverse as feather(s), horn(s), and slime.1 Despite the abundance, coarse wool is radically undervalued. In one year, the UK alone produces 32 million kg of wool, mainly by the food industry where wool is seen as a by-product.2 This volume of waste is problematic because environmental keratin degradation carried out by natural microorganisms is a slow process. However, keratin could be a valuable source of organic materials such as biopolymers and amino acid building blocks. This sustainable valorisation requires developments in keratin processing and accelerated degradation in order to utilize disgarded fibres.Keratin fibres such as those found in wool and feathers have a higher-order structure. Structurally, keratins are tightly packed insoluble proteins with high cysteine content (10–17% in hair and wool).3 The keratins oxidised cysteine residues form intermolecular disulfide bonds giving the fibre its strength. The keratin itself displays two forms: α-keratin (the predominant form in mammals i.e. wool, hair) and β-keratin (the major form found in reptiles and birds i.e. feathers).1 α-keratin forms helices, whereas, the tougher β-keratin forms more dense β-sheets.1 The mammalian keratin fibre is then coated in cuticle scales protecting the fibre from degradation.1,4
Conventionally, keratin can be hydrolysed by high pH or superheated water treatment (140–170 °C), requiring a large energy input.5 Keratinases are a biocatalytic alternative, which are a subclass of proteases (EC3.4)6 produced by bacteria and fungi that naturally breakdown keratin, as shown in Scheme 1.
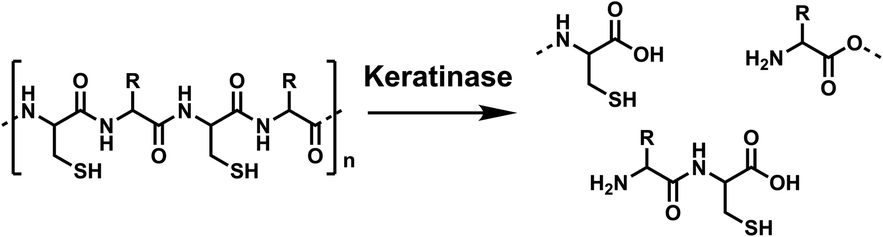 | ||
| Scheme 1 Hydrolysis of the keratin polymer by keratinases, from poly-amide chains to soluble amino acids and peptides. Created in ChemDraw. | ||
Due to their range of potential industrial applications (detergents, leathers and pharmaceuticals) keratinases have attracted a significant amount of interest.7 A search of the National Center for Biotechnology Information (NCBI) database with ‘keratinase’ returns over 9000 proteins (released between 1995 and 2023).8 The search lists >100 characterised, sequenced keratinases, with tens of thousands of patent applications referring to the application of keratinases. Despite widespread interest and application, there is motivation to find novel keratinases or optimise known keratinases to aid in developing the biotechnology sector.8 A major issue is the many reports describing enzymes with keratinase activity where they use different assay techniques (e.g. substrates, unit of activity, work up) which make studies difficult to compare. Despite reviewing the field we were only able to find comparable data of 18 keratinases, we extracted their kinetic parameters and used these to compile a comparison with Proteinase K (PK); a commonly used broad-spectrum serine protease with proven keratinase activity (Fig. 1, Table S1†). The broad protease, chymotrypsin, displayed the poorest activity, whereas the enzyme from Bacillus sp. MTS was particularly active. Despite the comparison with PK, the extracellular keratinases, isolated from B. sp. MTS, could be a cocktail – indicated by the 3 bands on their SDS-PAGE gel. However, this analysis highlighted the requirement for a rigorous, reproducible, standardised keratinase assay. This in-depth review of the published data provided guidance with respect to the initial conditions to begin our assay optimisation (e.g. enzyme concentration, temperature and buffers, Table S2†).
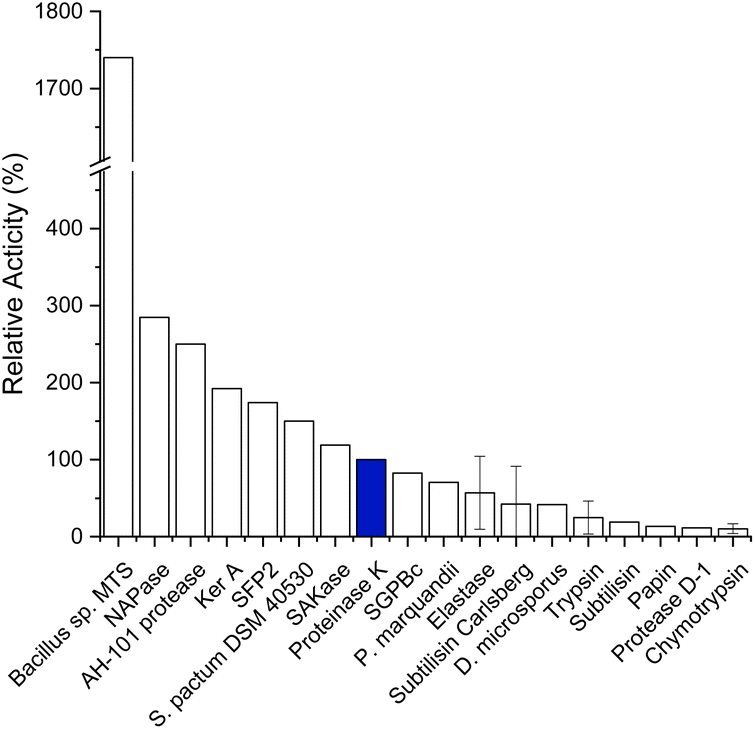 | ||
| Fig. 1 Enzymatic activity of keratinases relative to the activity of Proteinase K (PK, blue). Relative activities are calculated from published data, standard deviation given where multiple publications have been reported. Reported data and references found in Table S1.† Created in Origin. | ||
Keratinase activity can be assayed using the substrate keratin azure (KA), a commercially available, blue-dyed wool composed of α-keratin. This relies on the chemical and spectroscopic properties of an anthraquinone derivative, Remazol Brilliant Blue R dye (RBBR, 1, also known as reactive blue 19, Fig. 2) which is measured at 595 nm. The dye is covalently bound to the keratin fibre9 and as the keratinase breaks down the insoluble wool, it releases smaller soluble peptides and amino acids with the dye still bound. Keratinase activity is therefore proportionate to the increase in absorbance of the reaction solution at 595 nm.
 | ||
| Fig. 2 (A) Structure of the anthraquinone derived Remazol Brilliant Blue R dye (RBBR, 1). (B) Image of the keratin azure (KA) substrate. Created in ChemDraw. | ||
The earliest reported KA assay was sourced to a 1977 paper, where Roach et al. used the method to determine the keratinase activity of a Lactobacilli strain with a KA substrate from Calbiochem.10 Since then, similar KA assays have been used, but in the absence of KA standardised procedures, it has been difficult to compare and contrast published data. For example, Sigma-Aldrich, the vendor of KA, have a protocol for how to carry out this assay but, it lacks experimental details which preclude the application of a reliable method.11 Recently, Gonzalo et al. used KA in a comparison with various keratinous materials and proposed the keratinase activity be calibrated against the commercially available PK.12 This solution also offers the prospect of a universal positive control which would facilitate comparisons between publications.
This paper reports our efforts to develop a standard keratinase activity assay using a combination of KA, PK and another established keratinase, Subtilisin Carlsberg (SC). This goal was achieved firstly by calibrating the assay using the commercial RBBR dye (ε595 = 8586 M−1 cm−1, pH 8.0) and then subsequently quantifying the amount of soluble dyed peptide released by the action of the enzyme on the KA. The commercial PK biocatalyst was used to optimise the method in terms of KA pre-treatment (milled, autoclaved and/or washed), assay conditions (mixing technique) and work up (filtration, acid treatment). The method allowed for a comparative study of the activity of PK with SC. The optimised assay was also used to explore the impact of the disulfide-bond reducing agent (tris(2-carboxyethyl)phosphine, TCEP, Scheme 2) on keratinase activity. The final assay method described here using the commercial KA substrate is suitable for the screening of keratinase activity.
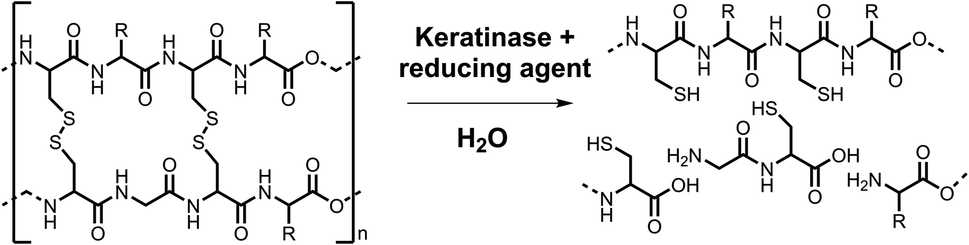 | ||
| Scheme 2 The reduction of disulfide bonds with the hydrolysis of cross-linked keratin by keratinases and reducing agents. From interlinked polypeptide chains to soluble amino acids and peptides with reduced cysteine residues. Created in ChemDraw. | ||
Experimental
Materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 930 Da, PDB code: 1IC6)13 and Subtilisin Carlsberg (272614 Da, PDB code: 3UNX)14 were purchased from Sigma-Aldrich as lyophilised powders, stored at −20 °C, and solubilised in buffer on the day of use to 50 μM.
930 Da, PDB code: 1IC6)13 and Subtilisin Carlsberg (272614 Da, PDB code: 3UNX)14 were purchased from Sigma-Aldrich as lyophilised powders, stored at −20 °C, and solubilised in buffer on the day of use to 50 μM.
Instrumentation
Methods
Calibration curve
A RBBR stock 1 mM was made with buffer (PK: 50 mM Tris–HCl, pH 9.0, SC: 50 mM Tris–HCl, pH 8.0). In triplicate, serial dilutions were performed with buffer to make a range of concentrations between 500.0 and 7.8 μM. 200 μL was pipette into a 96-well plate and measure the absorbance using the plate reader method described above and in ESI (Table A†). Using the data a line of best fit was created in order to get the equation of the line.Work up optimisation
The KA assay was performed with milled KA wool (5.0 mg, 1.0 % w/v) which was washed three times with deionised water (1 mL). The reaction was performed as described in the standard assay. The samples were then treated using the various work up procedures: iced (30 min), centrifuged (13 kRPM, 10 min), filtered (17 mm 0.45 μm PTFE filter) or the addition of TCA (10% w/v, 3:1 TCA:reaction sample, 30 min incubation the centrifuge 10 min, 13 kRPM). Each process was completed in isolation and in every combination of two treatments. Transferring 200 μL to a 96-well plate and measure the absorbance at 595 nm as per the plate reader method above and in the SM (Table A†).
Effect of addition of reducing agent
Results and discussion
Measuring the spectroscopic properties of the RBBR dye and calibrating the KA assay
We began by measuring a UV-vis spectrum of the RBBR dye and observed a broad absorbance between 450 and 700 nm with the expected maximum at 595 nm (Fig. 3). We also made a comparison with another blue dye, Coomassie brilliant blue R, which can be seen to not align with the RBBR Dye standard, Fig. S7.† | ||
| Fig. 3 UV-vis spectra comparing the absorbance of Proteinase K (PK), hydrolysed KA (H-KA) and RBBR dye (150 μM), showing the close similarities between the dye and KA and the common peak at 595 nm. The hydrolysed KA was prepared by adding PK (50 μM) to buffer (Tris–HCl buffer, 50 mM, pH 8.0) containing 0.5% w/v KA, the reaction Eppendorfs were shaken for 3 h at 50 °C on a thermoshaker. Created in Origin. | ||
We first optimised the enzyme assay conditions (buffer/pH, temperature and enzyme concentration) for a commercial source of PK and Subtilisin Carlsberg (Sigma-Aldrich) on a semi-optimised KA assay method as to ensure the proteinases were preforming at their optimal activity (Fig. S1–S3†). Next, we used PK with the KA substrate to compare the spectroscopic properties of the released dyed peptide by the action of the enzyme. After incubation of the reaction mixture containing 50 μM PK and 0.5% w/v KA at 50 °C for 3 h, the reaction was stopped by filtration through a PTFE filter to remove unreacted fibres, Fig. 4A. The UV-vis analysis of the resulting solution showed that it displayed a similar spectrum to the RBBR dye standard over the Coomassie dye (Fig. 3 and S7†).
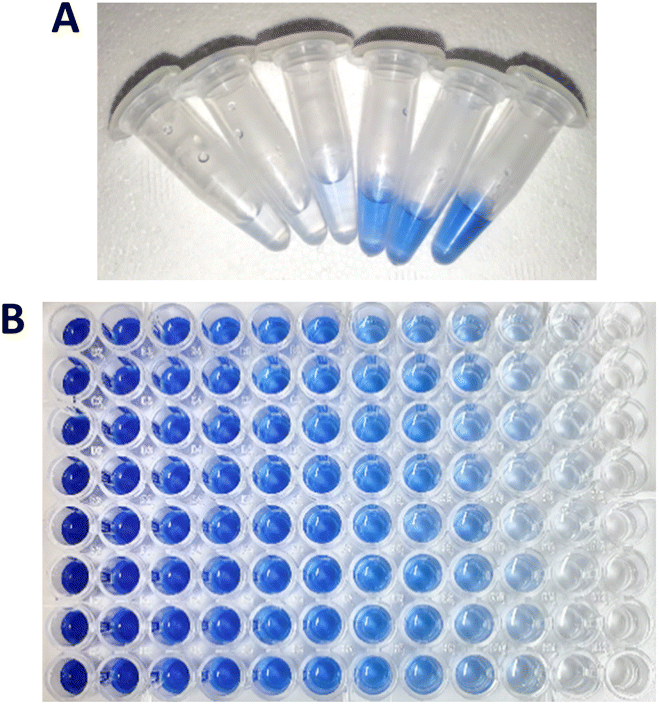 | ||
| Fig. 4 (A) KA reaction solutions after reaction with keratinases, showing the increase in soluble dyed peptides, which can be analysed via UV-vis using a wavelength of 595 nm. The reaction: PK (50 μM) added to buffer (Tris–HCl buffer, 50 mM, pH 8.0) containing 0.5% w/v KA, the reaction was then shaken for 3 h at 50 °C on a thermoshaker. (B) Calibration plate of RBBR standard samples 0–900 μM. | ||
Having confirmed the KA dye was RBBR the assay was calibrated using the RBBR dye standard, Fig. 4B. Calibration curves for the absorbance of aqueous RBBR at 595 nm (ESI, Fig. S8 and S9†) in buffer were linear in the region between 3.9 and 125 μM (r value of 0.999). Using the Beer–Lambert law (ESI†), the molecular absorbance coefficient (ε595) was calculated as 8586.2 M−1 cm−1 ±75.3 (at pH 8.0) and 7545.9 M−1 cm−1 ±127.7 (at pH 9.0), which is comparable to the literature value of 8266 M−1 cm−1 (pH 3.5).15 The standard curves can be used to report results for the KA assay in SI units – either in terms of total dyed peptide released (μM) as an end-point assay or as the rate of released dyed peptide over the time-course of the assay (μM h−1). We used (μM h−1) as the unit for rate since it has the advantage of facilitating comparison between assays of keratinases under different reactions conditions (time, pH and temperature).
Having carried out the reactions in convenient Eppendorf tubes, we then transferred the filtrate to a plate reader to allow multiple high throughput measurements at 595 nm, similar to Fig. 4B. Initially there was a large error which we attribute to the solid nature and the variable dyeing of the KA substrate, which contributes to its inherent heterogeneous composition. We found that changing the volume of wool per sample helped (0.5 to 1.0% w/v) but also the mixing (Fig. S4 and S5†). A thermoshaker was initially used, however, this only gives horizontal mixing. With a change to a blood spinner with top over mixing, improved our errors giving confidence in the results achieved (Fig. S5†). This change enabled us to accurately determine the units of activity of the PK with KA as 73.78 μM h−1 ± 6.85 (Fig. 5, PK, unprocessed). We also used this method to determine the keratinase activity of SC (14.60 μM h−1 ± 10.24) and revealed that the PK is 5 times more active on the whole, unprocessed KA substrate (Fig. 5, SC, unprocessed). The high errors with SC were consistent across initial investigations, which saw improvement with assay optimisation.
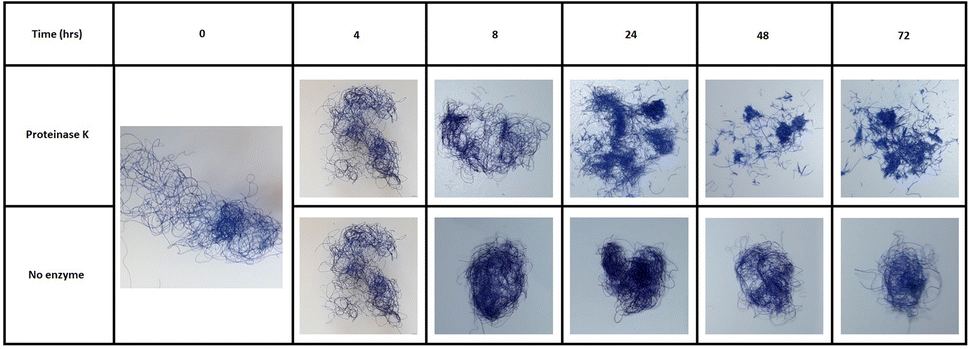 | ||
| Fig. 5 The visible degradation of blue dyed wool to short chopped fibres by a keratinase over 72 hours. The reaction: PK (50 μM) added to buffer (Tris–HCl buffer, 50 mM, pH 8.0) containing 0.5% w/v KA (5.0 mg), the reaction was then shaken for 0–72 h at 50 °C on a thermoshaker. | ||
It was also interesting to observe the easily visible structural changes of the KA fibres when incubated with the PK biocatalysts during an extended 72 h time course (Fig. 5). This effect was also visibly seen after 3 h under microscope (Fig. S6†).
Pre-treatment
The still heterogeneous substrate from the initial analysis prompted us to further improve the assay. Another factor that affects the comparability of assay results is what preparation, if any, is done to the KA substrate before the assay. Martinez et al. and Okoroma et al. have highlighted that treatments such as autoclaving, milling, and solvent washing (EtOH or dH2O), can denature the keratin structure, solubilise keratin, or remove protective lipid layers, potentially increasing susceptibility to proteolytic attack.16,17 While some publications use the unprocessed KA as supplied, as implied by Sigma-Aldrich,11,18 others apply one or more pre-treatments before performing the assay, for example grinding,11,12,19 chopping,20,21 sterilisation,21,22 and washing.23 To our knowledge, there is no published study about the effect that such processing of the substrate has on the KA assay.We performed the assay using KA that was unprocessed (UP), and KA that was processed in three ways as followed; [1] washed once with deionised water (UP-W1), or washed three times (UP-W3). [2] Autoclaved (AC), autoclaved then washed once (AC-W1), washed three times (AC-W3). [3] Milled with a ball mill (M), milled and washed once (M-W1) and finally milled then washed three times (M-W3) (Fig. 6).
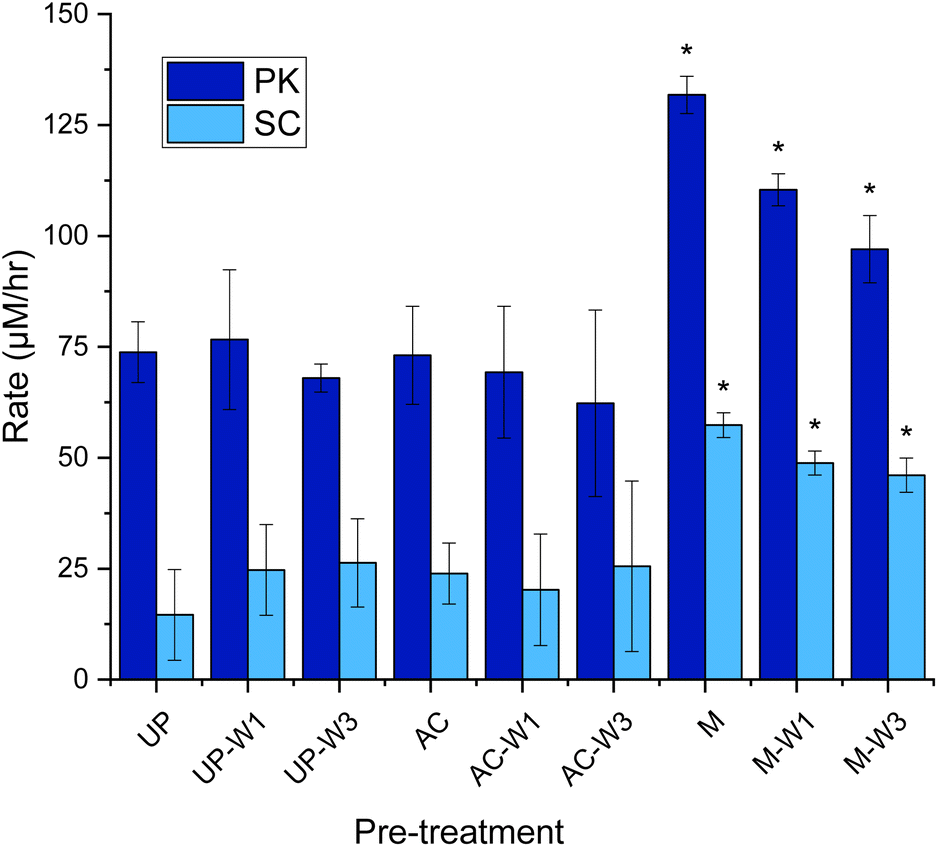 | ||
| Fig. 6 Rate of dyed peptide released during the keratin azure assay with various pre-treated substrates. Unprocessed (UP), autoclaved KA (AC, 130 °C for 15 min) and milled KA (M, 30 Hz s−1 for 3 min), with and without being washed with distilled water (1 mL) once (UP-W1, AC-W1, M-W1), or three times (UP-W3, AC-W3, M-W3). The wash was discarded. To 1.0% w/v of treated KA: buffer (Tris–HCl buffer, 50 mM, pH 9.0/8.0) and PK/SC (50 μM) were added. After being mixed in an Eppendorf for 3 h at 60 °C in an incubator, samples were filtered (17 mm 0.45 μm PTFE filter). 200 μL was transferred to a 96-well plate and was measured using a plate reader at 595 nm. See Fig. S11† for endpoint graph. When compared to UP, p < 0.05*. Created in Origin. | ||
With PK, it was found that neither washing nor autoclaving pre-treatments of KA influence the dye release into solutions when compared with the UP-KA substrate (Fig. 6). In contrast these pre-treatments caused larger errors with the SC biocatalyst. The largest, significant gain in keratinase activity was observed when the KA substrate was ball milled (Fig. 6). Compared with the UP substrate, this milled and washed substrate gave 1.3 and 3.2 fold increase in dyed peptide released with PK and SC, respectively. A small amount of dye is leeched out without enzyme treatment witch is increased in the M sample without any washing (observed in the controls, see Fig. S12†). The errors were also reduced and we suggest that this milling step has resulted in homogenising the KA sample and making it easier to handle (Fig. S10†). Although the UP KA is still a useable substrate in the keratinase assay, we suggest that simple ball milling, combined with three washes (M-W3), reduces the background and improves the detection of keratinase activity.
Post-treatment
The third source of variation that occurs when performing the KA assay is the processing/work up of the assay solution. The goal here is to stop enzyme activity, remove unreacted substrate and retain the soluble dyed peptides for quantification on a plate reader. Recent studies have used combinations of filtration (F),12,18,24,25 ice quenching (I),18,23–25 centrifugation (C),11,12,18,21–25 and tricholoroacetic acid precipitation (TCA)12,19,20,26 to stop the reaction. There appears to be no published analysis of the practical use and optimal benefits of these methods. Gonzalo et al. did show that treating the samples with TCA (2–10%) made their KA assay with PK 20-fold less sensitive, as keratin-derived peptides with dye attached precipitate at the low pH.12 Therefore, they suggest a process using only filtration, but offer no evidence comparing this with other processing methods.Hence, it made sense to investigate the effect of the processing techniques on the optimal KA assays described earlier for PK and SC. Taking forward the results found in Fig. 6, each sample started with M-W3 KA as the substrate and was then incubated for 3 hours and then processed 11 different ways using the previously described processing techniques (F, I, C and TCA), individually and in combination, as shown in Fig. 7. These show that the post-treatment technique had a great effect on the measured keratinase activity.
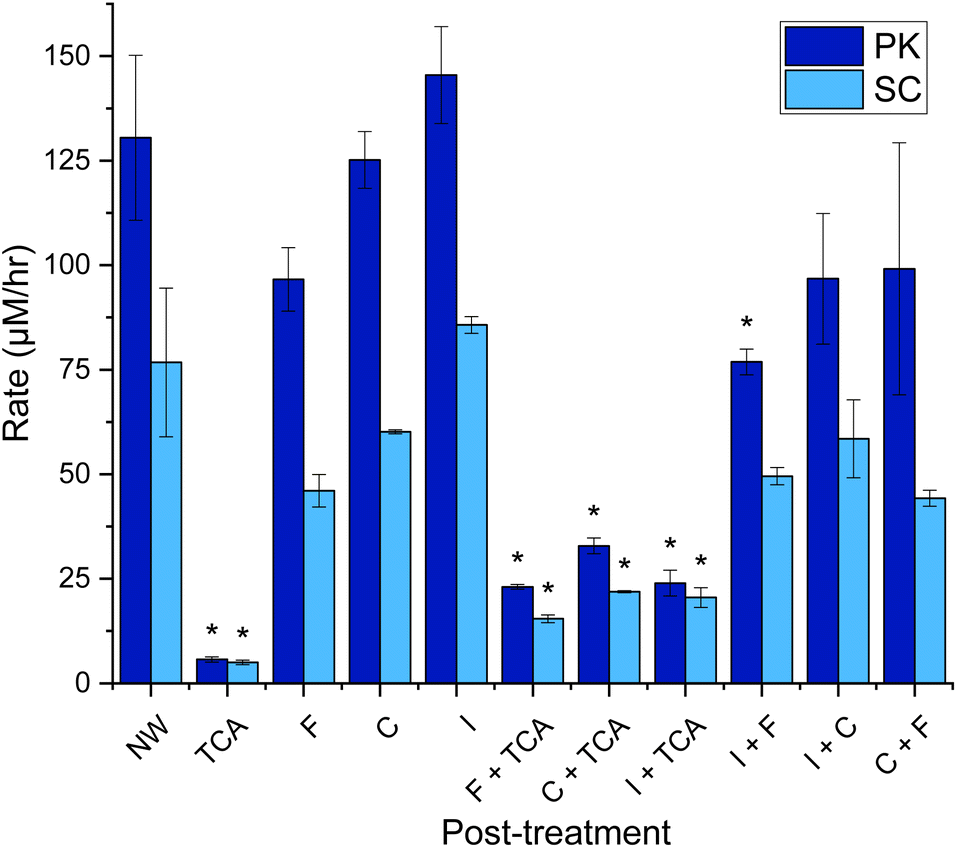 | ||
| Fig. 7 Rate of dyed peptide released in keratin azure assay post-treatment using 11 different techniques Milled KA (30 Hz s−1 for 3 min) washed with distilled water (1 mL) three times. The wash was discarded. To the KA (1.0% w/v) buffer (Tris–HCl buffer, 50 mM, pH 9.0/8.0) and PK/SC (50 μM) were added. After being mixed in an Eppendorf for 3 h at 60 °C in an incubator, samples were iced (30 min), centrifuged (13 kRPM, 10 min), filtered (17 mm 0.45 μm PTFE filter) and/or treated with TCA (10% w/v, 3:1 TCA:reaction sample, 30 min incubation the centrifuge 10 min, 13 kRPM). 200 μL was transferred to a 96-well plate and was measured using a plate reader at 595 nm. See Fig. S13† for endpoint graph. When compared to NW, p < 0.05*. Created in Origin. | ||
Our data also agrees with the observation by Gonzalo et al.,12 which shows that TCA quenching reduces the signal by 82–87% in KA assay solutions using both PK and SC enzymes (Fig. 7). This loss is accompanied by the appearance of a blue precipitate, clearly derived from the substrate and any RBBR-bound peptide products. This large loss of signal means that results from non-TCA quenched assays should not be compared with results from a TCA quenched assay. Both F and C can be used to remove unreacted KA substrate, stopping the reaction. Filtration does appear to reduce the signal (a blue residue was observed on the filter retentate), due to removing larger KA fragments. Similarly, centrifugation does remove the largest/intact fragments (a small blue pellet is observed), but a higher concentration of soluble peptide retained in the sample with a ∼30% increase from F to C (for both PK & SC). A final way of terminating the reaction is to lower the temperature by placing the assays on ice, where the activities of both PK and SC are minimal. This process resulted in small increases (within error) in the amount of dye observed compared with the no workup (NW) samples attributed to the opaque sample post-iced incubation. Combinations of these processing treatments failed to improve the measurements with TCA treatment remaining the poorest. Therefore, we conclude that the simplest and most economical (to avoid filter waste) way to work up the KA assay is to centrifuge the sample before decanting into a cuvette or well plate, as appropriate.
Reducing agents
Since the α-keratin core structure contains high concentrations of intramolecular disulfide bonds, we used the KA assay to investigate the effects of chemical reduction on the activity of both PK and SC. Previous studies had reported using standard, reversible reagents such as dithiothreitol (DTT) and/or 2 -mercaptoethanol (β-ME) with varying outcomes.27–30 For example, a recombinant keratinase (Ker) was isolated from a metagenomics screen that displayed >90% sequence identity to the enzymes from Bacillus and Brevibacillus bacteria. The characteristics of this serine protease were tested with a range of keratin substrates (none were dyed) and it was found that both DTT and β-ME reduced the activity by ∼80%.27 In contrast, the keratinase isolated directly from Brevibacillus parabrevis CGMCC 10798 retained >99% activity in the presence of β-ME.28Only a handful of keratinases have been tested with reducing agents added to the reaction mix (rather than pre-incubated with the keratinase or substrate), most show an increase in activity.12,31–34 To our knowledge no studies have investigated the effect the reducing agent may have on the keratin vs. keratinase.
Here, instead of using the reversible DTT and β-ME, we employed TCEP since it reduces disulfide bond(s) rapidly and irreversibly. As PK contains disulfide bonds (SC does not) we incorporated these considerations into our experimental design to investigate TCEPs affect on the activity of the keratinases. The experiments are as described below and illustrated in Fig. S15:†
(1) Enzyme incubation (EI). To analyse the effect on the keratinase. Reducing agent incubated with keratinase for 30 min and then added to KA.
(2) Wool incubation (WI). To analyse the effect of reducing the KA. The TCEP reducing agent incubated with KA and washed away before assaying.
(3) In reaction (IR). To investigate any cumulative effect on the enzyme and KA substrate. A reducing agent is added directly to the KA assay.
Our results show that using the TCEP reagent (up to 1 mM) to chemically reduce the disulfide bonds of keratin had a relatively small impact of the keratinase activity of both PK and SC on the KA substrate (Fig. 8). For PK, the only statistically significant change was for the IR incubation where we observed a 15.7% decrease in rate (Fig. 8A). Surprisingly, PK activity was not significantly affected by the incubation with TCEP in the EI incubation, despite having two disulfide bonds.13 Protocols provided by vendors recommend using DTT (5 mM) to enhance the protease activity of PK suggesting that disulfide bonds are not essential for catalysis.35
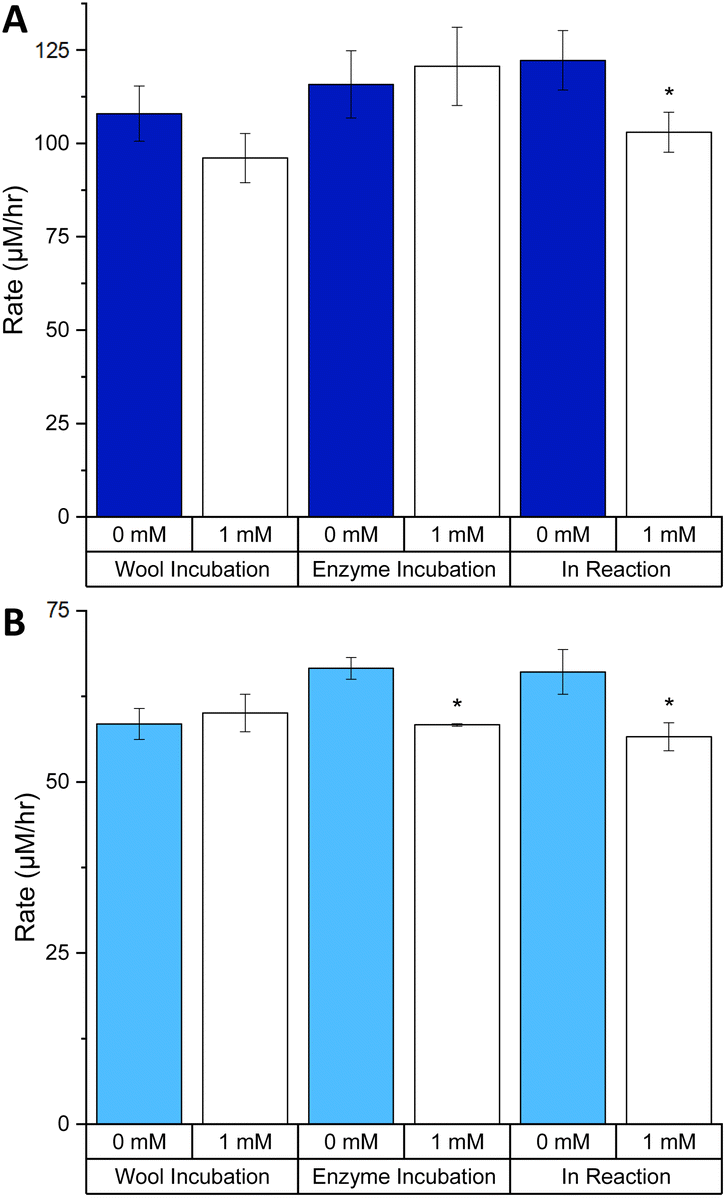 | ||
| Fig. 8 The effects of TCEP on the rate of keratinase activity with different incubations: (A) PK and (B) SC. Milled KA (30 Hz s−1 for 3 min) washed with distilled water (1 mL) three times. The wash was discarded. TCEP was added to wool only (TCEP: 50 μL, 10 mM), enzyme only (TCEP: 50 μL, 10 mM, enzyme 50 μL, 50 μM, incubated for 30 min at 25 °C with 400 μL of buffer) or in the reaction (TCEP: 50 μL, 10 mM) as illustrated in Fig. S15.† In the reactions were also KA (1.0% w/v) buffer (Tris–HCl buffer, 50 mM, pH 9.0/8.0) and PK/SC (50 μM). After being mixed in an Eppendorf for 3 h at 60 °C in an incubator, samples were centrifuged (13 kRPM, 10 min). 200 μL was transferred to a 96-well plate and was measured using a plate reader at 595 nm. See Fig. S16† for endpoint graph. When comparing 1 mM to 0 mM for each experiment, p < 0.05*. Created in Origin. | ||
A similar analysis of SC showed that enzyme activity displayed modest, but significant, change in both the EI and IR incubations, a decrease of 12.4 and 14.3%, respectively (Fig. 8B). In contrast to PK, the SC enzyme contains no disulfide bond so any observed difference must solely be due to the effect on the KA substrate. The exact details of the impact of TCEP on KA cannot be elucidated without more information of the exact dyeing process of keratin with RBBR. Overall, it is apparent that there is no gain in activity for both PK and SC with the addition of 1 mM TCEP.
Despite our results using a chemical reducing reagent, previous studies have suggested that disulfde bond reductases (DSBRs) work together with keratinases to degrade wool in a natural setting. For example, one study,32 isolated two types of extracellular enzymes (a keratinase, named protease D-1 and a DSBR) from the keratin-degrading bacterium Stenotrophomonas sp. strain D-1. The DSBR enhanced the keratinase activity of both PK and SC on human hair keratin. Our robust KA assay could be used to screen for novel reductases that enhance the activity of various keratinases.
Conclusions
Our goal at the start of this investigation was to optimise an assay that could be used to screen enzymes that display keratinase activity. We began with a thorough investigation of the literature and found multiple assays which varied with respect to substrate, common methodology and analysis. In most cases there was a lack of calibration using a robust standard. To that end, we chose a commercially-available keratin dyed with a RBBR azure dye (KA) as our standard substrate. We had found KA to have been used previously, but these assays had not been calibrated with the RBBR dye standard using the appropriate molecular absorbance coefficients (ε595). Using this data, we determined a limit of detection (LOD) = 3.9 μM, a limit of quantification LOQ = 7.8 μM and linearity between 3.9 and 125 μM (see SM for calculations). We then optimised the assay using KA and well known enzymes with keratinase activity (PK and SC).Two major procedures, which had the biggest impacts on the quality of our data (e.g. sensitivity), was the pre-treatment of the KA substrate and work up of the assay sample. Our observations suggest washing the KA 3 times with water after ball-milling to produce a consistent, homogeneous keratinase substrate. Previous methods, with 1% v/v TCA treatment to precipitate/remove enzyme and unreacted KA substrate led to the loss of RBBR dye signal. Our method shows that a substantial gain was achieved by simple centrifugation of the assay sample after keratinase treatment. We additionally created an experimental procedure for the investigation of reducing agents (here TCEP) which can be used in future to delineate the impact on the observed activity. This can be used interchangeably for different enzymes and substrates (i.e. ‘raw’ wool or feather).
Taken together, we now have produced a standard operating procedure (SOP) that can be easily followed to quantify keratinase activity (Fig. 9 and ESI†). This method is highly reproducible and allows for both retrospective and future comparisons between studies. Our current efforts are focused on using this KA SOP to screen for novel α-keratin keratinases and to identify other factors (such as DSBRs) that will improve the wool-degrading activity of these industrially useful enzymes.
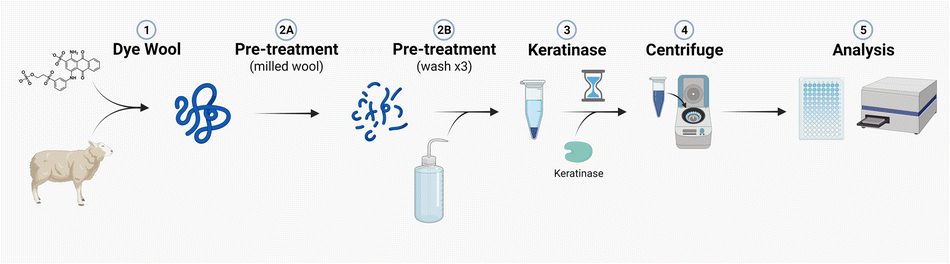 | ||
| Fig. 9 Illustration of the optimised keratin azure assay. Created with BioRender. | ||
Author contributions
Rhona M. Cowan; data collection and analysis, method development, formal analysis, investigation, project administration, manuscript writing and editing. Eleanor Birch; data collection and analysis, method development, manuscript writing and review. Grace Nisbet; data collection and analysis. Chimaeze Onyeiwu; conceptualization. Clare Campbell; conceptualization and sample preparation. Ian Archer; conceptualization and project management. Dominic J. Campopiano; conceptualization, data analysis, manuscript writing and reviewing.Conflicts of interest
Prickly Thistle Ltd and Johnstons of Elgin are commercial businesses but have no conflicts to declare.Acknowledgements
We would like to thank IBioIC and the School of Chemistry, University of Edinburgh for funding (PhD studentship to RMC) and IBioIC for the feasibility grant to allow this work to be completed. We thank Simon Cotton from Macnaughton Holdings for initial discussions of this project.Notes and references
- J. McKittrick, P. Y. Chen, S. G. Bodde, W. Yang, E. E. Novitskaya and M. A. Meyers, JOM, 2012, 64, 449–468 CrossRef.
- UKRI Innovate UK, Wool Innovation Action Plan to Support a Circular and Sustainable Future, https://iuk.ktn-uk.org/wp-content/uploads/2023/05/Wool-Innovation-Action-Plan.pdf, (accessed June 2023) Search PubMed.
- Z. Peng, J. Zhang, G. Du and J. Chen, ACS Sustain. Chem. Eng., 2019, 7, 9727–9736 CrossRef CAS.
- C. R. Robbins, Chemical and Physical Behaviour of Human Hair, Springer, Berlin, 5th edn, 2012, ch. 1, pp. 1–104 Search PubMed.
- P. Bhavsar, M. Zoccola, A. Patrucco, A. Montarsolo, G. Rovero and C. Tonin, Text. Res. J., 2017, 87, 1696–1705 CrossRef CAS.
- BRENDA Enzyme Classifications, https://www.brenda-enzymes.org/ecexplorer.php?browser=1%26f[nodes]=%26f[action]=open%26f[change]=170#170, (accessed October 2023) Search PubMed.
- B. Vidmar and M. Vodovnik, Food Technol. Biotechnol., 2018, 56, 312–328 CAS.
- NCBI Keratinase Search, https://www.ncbi.nlm.nih.gov/search/all/?term=keratinase, (accessed June 2023) Search PubMed.
- R. M. Christie, Colour Chemistry, The Royal Society of Chemistry, UK, 2001, 1st edn, ch. 8, pp. 135–147 Search PubMed.
- S. Roach, D. C. Savage and G. W. Tannock, Appl. Environ. Microbiol., 1977, 33, 1197–1203 CrossRef CAS PubMed.
- Sigma Aldrich Protocol for a Keratin Azure Assay, https://www.sigmaaldrich.com/deepweb/assets/sigmaaldrich/marketing/global/documents/254/658/keratin_azure_substrate.pdf, (accessed June 2023) Search PubMed.
- M. Gonzalo, R. Espersen, W. A. Al-Soud, F. Cristiano Falco, P. Hägglund, S. J. Sørensen, B. Svensson and S. Jacquiod, Microb. Biotechnol., 2020, 13, 984–996 CrossRef CAS PubMed.
- C. Betzel, S. Gourinath, P. Kumar, P. Kaur, M. Perbandt, S. Eschenburg and T. P. Singh, Biochem, 2001, 40, 3080–3088 CrossRef CAS PubMed.
- S. J. Fisher, M. P. Blakeley, M. Cianci, S. McSweeney and J. R. Helliwell, Acta Crystallogr., 2012, 68, 800–809 CrossRef CAS PubMed.
- P. R. Moreira, E. Almeida-Vara, G. Sena-Martins, I. Polónia, F. X. Malcata and J. C. Duarte, J. Biotechnol., 2001, 89, 107–111 CrossRef CAS PubMed.
- E. A. Okoroma, H. Garelick, O. O. Abiola and D. Purchase, Int. Biodeterior. Biodegrad., 2012, 74, 54–60 CrossRef CAS.
- J. P. De Oliveira Martinez, G. Cai, M. Nachtschatt, L. Navone, Z. Zhang, K. Robins and R. Speight, Catalysts, 2020, 10, 184 CrossRef.
- S. Hamiche, S. Mechri, L. Khelouia, R. Annane, M. El Hattab, A. Badis and B. Jaouadi, Int. J. Biol. Macromol., 2019, 122, 758–769 CrossRef CAS PubMed.
- F. Akram, I. U. Haq and Z. Jabbar, Int. J. Biol. Macromol., 2020, 164, 371–383 CrossRef CAS PubMed.
- L.-X. Lv, M.-H. Sim, Y.-D. Li, J. Min, W.-H. Feng, W.-J. Guan and Y.-Q. Li, Process Biochem., 2010, 45, 1236–1244 CrossRef CAS.
- J. D. Allpress, G. Mountain and P. C. Gowland, Lett. Appl. Microbiol., 2002, 34, 337–342 CrossRef CAS PubMed.
- H. Hänel, J. Kalisch, M. Keil, W. C. Marsch and M. Buslau, Med. Microbiol. Immunol., 1991, 180, 45–51 CrossRef PubMed.
- X. Liang, Y. Bian, X.-F. Tang, G. Xiao and B. Tang, Appl. Microbiol. Biotechnol., 2010, 87, 999–1006 CrossRef CAS PubMed.
- N. E. Nnolim, L. Mpaka, A. I. Okoh and U. U. Nwodo, Microorganisms, 2020, 8, 1304 CrossRef CAS PubMed.
- M. Ben Elhoul, N. Zaraî Jaouadi, H. Rekik, M. Omrane Benmrad, S. Mechri, E. Moujehed, S. Kourdali, M. El Hattab, A. Badis, S. Bejar and B. Jaouadi, Int. J. Biol. Macromol., 2016, 92, 299–315 CrossRef CAS PubMed.
- Y. Jagadeesan, S. Meenakshisundaram, V. Saravanan and A. Balaiah, Int. J. Biol. Macromol., 2020, 163, 135–146 CrossRef CAS PubMed.
- C. Su, J.-S. Gong, R.-X. Zhang, L.-Y. Tao, W.-F. Dou, D.-D. Zhang, H. Li, Z.-M. Lu, Z.-H. Xu and J.-S. Shi, Int. J. Biol. Macromol., 2017, 95, 404–411 CrossRef CAS PubMed.
- R.-X. Zhang, J.-S. Gong, C. Su, D.-D. Zhang, H. Tian, W.-F. Dou, H. Li, J.-S. Shi and Z.-H. Xu, Int. J. Biol. Macromol., 2016, 93, 843–851 CrossRef CAS PubMed.
- M. A. Hassan, T. H. Taha, G. M. Hamad, M. Hashem, S. Alamri and Y. S. Mostafa, Int. J. Biol. Macromol., 2020, 153, 561–572 CrossRef CAS PubMed.
- M. O. Babalola, A. O. Ayodeji, O. S. Bamidele and J. O. Ajele, Biotechnol. Lett., 2020, 42, 2673–2683 CrossRef CAS PubMed.
- B. Böckle, B. Galunsky and R. Müller, Appl. Environ. Microbiol., 1995, 61, 3705–3710 CrossRef PubMed.
- S. Yamamura, Y. Morita, Q. Hasan, K. Yokoyama and E. Tamiya, Biochem. Biophys. Res. Commun., 2002, 294, 1138–1143 CrossRef CAS PubMed.
- R. Sharma and R. Gupta, Bioresour. Technol., 2012, 120, 314–317 CrossRef CAS PubMed.
- S. Parinayawanich, D. Sittipol, Y. U. S. Ajingi, S. Rodpan, K. Pattanapanyasat and N. Jongruja, Biocatal. Agric. Biotechnol., 2021, 36, 102146 CrossRef CAS.
- Novagen Protocol for Proteinase K, https://www.sigmaaldrich.com/deepweb/assets/sigmaaldrich/marketing/global/documents/428/170/TB271.pdf, (accessed Aug 2023) Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ay01433a |
| ‡ These authors contributed equally. |
| § Current address: Ingenza Ltd, Roslin Innovation Centre, Charnock Bradley Building, Easter Bush Campus, Bush Farm Road, Roslin EH25 9RG. |
| ¶ Current address: Bioconnect Ireland Knockaconny, Monaghan, H18 KV40, Ireland. |
| This journal is © The Royal Society of Chemistry 2023 |