
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimisation of plutonium separations using TEVA cartridges and ICP-MS/MS analysis for applicability to large-scale studies in tropical soils†
Sophia M.
Dowell
 ab,
Thomas S.
Barlow
a,
Simon R.
Chenery
a,
Olivier S.
Humphrey
a,
Job
Isaboke
c,
William H.
Blake
b,
Odipo
Osano
c and
Michael J.
Watts
*a
ab,
Thomas S.
Barlow
a,
Simon R.
Chenery
a,
Olivier S.
Humphrey
a,
Job
Isaboke
c,
William H.
Blake
b,
Odipo
Osano
c and
Michael J.
Watts
*a
aInorganic Geochemistry, Centre for Environmental Geochemistry, British Geological Survey, Nottingham, NG12 5GG, UK. E-mail: mwatts@bgs.ac.uk
bSchool of Geography, Earth and Environmental Sciences, University of Plymouth, Plymouth, Devon PL4 8AA, UK
cSchool of Environmental Sciences, University of Eldoret, Eldoret, Kenya
First published on 16th August 2023
Abstract
The analysis of plutonium (Pu) in soil samples can inform the understanding of soil erosion processes globally. However, there are specific challenges associated for analysis in tropical soils and so an optimal analytical methodology ensuring best sensitivity is critical. This method aimed to demonstrate the feasibility of sample preparation and analysis of Pu isotopes in African soils, considering the environmental and cost implications applicable to low-resource laboratories. The separation procedure builds upon previous work using TEVA columns, further demonstrating their usefulness for the reduction of uranium (U) interference in ICP-MS analysis with enhanced selectivity for Pu. Here several steps were optimised to enhance Pu recovery, reducing method blank concentration, and improving the separation efficiency through the determination of the elution profiles of U and Pu. The elimination of the complexing agent in the eluent, increased the spike recovery by improving matrix tolerance of the plasma, and simplified the separation procedure, improving throughput by 20%. The subsequent method was validated through the analysis of Certified Reference Material IAEA-384, where high accuracy and improved precision of measurement were demonstrated (measured value 114 ± 12 versus certified value 108 ± 13 Bq kg−1). Optimisation of the column separation, along with the analysis of the samples using O2 gas in ICP-MS/MS mode to mass shift Pu isotopes away from interfering molecular U ions provided a simple, robust, and cost-effective method with low achievable method detection limits of 0.18 pg kg−1 239+240Pu, applicable to the detection of ultra-trace fallout Pu in African soils.
1. Introduction
Plutonium (Pu) originates in the environment primarily as a consequence of nuclear weapons testing, 520 atmospheric tests were conducted worldwide between 1945 and 1980.1,2 However, only 10% of these experiments were conducted in the southern hemisphere, resulting in significantly less fallout in the tropics than in the mid-latitudes of the northern hemisphere. This makes the analysis of ultra-trace Pu isotopes in tropical soils challenging.3,4 Due to their long retention time and minimal spatial variability, Pu isotopes have been recently utilised as an alternative fallout radionuclide tracer for determining soil erosion rates. Due to the much longer half-lives of 239Pu and 240Pu (24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 110 and 6561 years, respectively), approximately 99% of the original activity remains in soils, allowing for their stable and long-term use as a tracer compared to 137Cs, which has a half-life of only 30 years.5,6 In addition, more than six times as many atoms of 239+240Pu were initially dispersed compared to 137Cs, despite the latter's significantly higher activity in the environment. This combination of long half-life and higher atom content makes mass spectrometry techniques better suited to Pu isotopes, whereas radiometric decay counting techniques are more appropriate for the higher specific activity 137Cs. Consequently, recent developments in mass spectrometry techniques have the potential to increase the sensitivity of quantification of Pu isotopes and subsequently the availability of analytical methods applicable to tropical soils.7,8 This raises the potential of using Pu as a soil erosion tracer in the tropics, where the risk of soil degradation is increasing due to extreme weather patterns.3
110 and 6561 years, respectively), approximately 99% of the original activity remains in soils, allowing for their stable and long-term use as a tracer compared to 137Cs, which has a half-life of only 30 years.5,6 In addition, more than six times as many atoms of 239+240Pu were initially dispersed compared to 137Cs, despite the latter's significantly higher activity in the environment. This combination of long half-life and higher atom content makes mass spectrometry techniques better suited to Pu isotopes, whereas radiometric decay counting techniques are more appropriate for the higher specific activity 137Cs. Consequently, recent developments in mass spectrometry techniques have the potential to increase the sensitivity of quantification of Pu isotopes and subsequently the availability of analytical methods applicable to tropical soils.7,8 This raises the potential of using Pu as a soil erosion tracer in the tropics, where the risk of soil degradation is increasing due to extreme weather patterns.3
Radiometric and mass spectroscopy techniques, such as alpha spectrometry, accelerator mass spectrometry (AMS), and inductively coupled plasma mass spectrometry (ICP-MS), can be used to analyse Pu in a variety of samples.7–10 In recent years, ICP-MS has gained popularity due to its low detection limits, short analytical time, high sample throughput, relatively simple operation, and lower instrument cost.5,11–14 Despite the benefits of ICP-MS, the technique is severely constrained by polyatomic interferences, most notably uranium hydrides (UH+). In addition, 238U tailing interferes with the detection of mass-to-charge ratio (m/z) 239 due to its concentration being several orders of magnitude greater than that of 239Pu in soils.15 This has placed a requirement for a high level of enrichment and elemental separation prior to analysis for the accurate determination of trace Pu isotopes.7 The Pu isotopes can be separated from the interfering isotopes in the matrix and pre-concentrated to assure maximum sensitivity, using techniques such as coprecipitation, ion exchange chromatography, and extraction chromatography.16–19 Utilising the selective TEVA column resin (Eichrom Technologies) to effectively remove U and pre-concentrate Pu is common for determination by ICP-MS.16,20 Nygren et al. (2003)19 reported that TEVA resin exhibited the maximum separation yield of Pu from soils and sediments compared to other methods. This finding is corroborated by the scientific literature and can be attributed to both the relatively low U content of the resin and the large variation in acid dependency of k′ which is greater than 3 orders of magnitude between U and Pu with nitric acid concentration between 2 and 4 M.21 Therefore, this paper concentrates on improving the analysis of ultra-trace Pu isotopes in east African soils by further optimising the chemistry of TEVA columns for the latest generations of mass spectrometric instrumentation, applicable to analysis of large-scale studies in tropical soils.
As a result of recent developments in reaction cell technology, tandem ICP-MS (ICP-MS/MS), also known as triple quadrupole ICP-MS (ICP-QQQ-MS), provides an alternative method of analysis to determine Pu. This method is increasingly utilised for the determination of Pu isotopes due to its enhanced abundance sensitivity, which effectively eliminates the interference of the 238U peak tailing on the measurement of 239Pu and 240Pu.7,8,14,22 In addition, the main interference of 238UH+ on 239Pu can be eliminated by utilising different reaction gases in the collision-reaction cell.4,7,8,10,15,23 The quadrupole mass filter positioned in front of the collision-reaction cell permits the pre-selection of species, thereby enhancing the reaction efficiency of the collision cell and prohibiting the formation of secondary polyatomic interference. In addition, the use of a second quadrupole reduces peak tailing, resulting in enhanced mass resolution. Gases, including ammonia (NH3), carbon dioxide (CO2), and oxygen (O2), have been proposed for the elimination of UH+ interferences by ICP-MS/MS.4,7,15 The most frequently employed of these gases is NH3, which effectively mass shifts the 238U1H interference away from 239Pu via the preferential reaction of U with the NH3 gas. Using this method, detection limits of 0.16 fg g−1 for 239Pu have been achieved; however, despite the ability of NH3 to react with the U interference, the use of NH3 gas poses several safety concerns, making its use undesirable.8,24 Using O2, the lowest detection limits of 0.06 fg g−1 for 239Pu have been reported; this can be attributed to the formation of PuO2+, which effectively mass shifts the Pu isotopes to m/z 271 to avoid interference from 238UH+ at m/z 239.4 The m/z ratio of PuO2+ exceeds the mass range of many early ICP-MS/MS instruments; however, user demands for analysing heavy elements in mass shift modes has led instrument manufacturers developing instruments with an extended m/z range up to 275.
The aim of this study was to accurately determine fallout Pu activity concentrations in tropical soils for the subsequent determination of soil erosion rates with an improved separation and analysis method for ultra-trace Pu determination. The two objectives to achieve this aim are: (1) adapting and optimising a separation method using TEVA cartridges for the removal of matrix interferences with pre-concentration of ultra-trace Pu isotopes to reduce waste and increase throughput; and (2) establishing a robust analytical method for the determination of ultra-trace level Pu isotopes with sufficient sensitivity for African soil samples using oxygen as a reaction gas for ICP-MS/MS.
2. Materials and methods
2.1 Reagents and materials
All reagents used were of analytical grade. Water used throughout had a resistivity of 18.2 MΩ at 25 °C and was obtained from a Milli-Q gradient system (Millipore, MA, USA). Extra-pure nitric acid (70%) was obtained from Thermo Fisher Scientific. Ultra-pure HCl was obtained from ROMIL. A solution of 242Pu (2.1 × 10−3 kBq/14.2 ng) of unknown origin was created from a stock solution (0.21 kBq) using a 100-fold dilution with a solution of 5% HNO3 and 2.5% HCl in water. This 242Pu solution was then used to spike soil samples prior to digestion. Samples were filtered using 0.45 μm hydrophilic PTFE syringe filters (Thermo Fisher Scientific, UK). Oxidation of Pu species was achieved using >97% purity NaNO2 (Sigma-Aldrich). The elution of Pu from the columns used a solution of 0.05 M ammonium oxalate, which was created by dissolving di-ammonium oxalate monohydrate (Supelco, Sigma-Aldrich) in water. Quality control was achieved using certified reference material obtained from the International Atomic Energy Agency (IAEA). The certified reference material IAEA-384 (radionuclides in Fangataufa Lagoon sediment with a certified value of 108 ± 13 Bq kg−1 239+240Pu) was selected, as this reference soil has been widely used within the literature for the determination of Pu isotopes and no suitable soil CRM was available. To account for differences in the sediment CRM and the analysed soils, an in-house reference material was created using soils collected in the UK. Column separations of Pu isotopes were performed using TEVA pre-packed columns (2 ml, 100–150 μm) from Eichrom Technologies. Elemental standard Ir (Spex CertiPrep) was used as an internal standard. Calibration of Pu concentrations via ICP-MS used a U elemental standard solution (Fisher chemical). Silicate sand, which was subjected to the same dissolution procedure as the CRM and samples, was used as the method blank.2.2 Sample collection
As part of this study, two bulk reference soils were collected from farmland within the UK and Zambia. The UK soil was collected from a cattle farm (Hoveringham, Nottinghamshire, England) during August 2020. Soil was collected from 3 locations across the farm, down to a depth of 15 cm. The soil was then dried in an oven overnight, disaggregated, sieved to <2 mm and finally milled ≤53 μm using a planetary ball mill (Retsch GmbH, Germany), ready for dissolution. This UK reference soil was then homogenised and used throughout the method development as both a quality control sample and to optimise the separation method. The Zambian soil was created from the combination of agricultural soils collected from Kitwe, Zambia, by Hamilton et al. (2020).25 The analysed soils were prepared in the same way as the UK reference soil and were used to verify the usefulness of the final method for use on African soils, where Pu levels are much lower than in the Northern Hemisphere.To validate the method for the determination of soil redistribution rates, a soil core was collected within the Oroba valley, Nandi County, Kenya (Fig. 1). Within the valley, a site with no overall soil re-distribution, representative of a reference site, was collected. Here, a core was taken to a depth of 30 cm and broken down into 10 sections to determine the Pu inventory within the site. To collect the sample, a pit was dug, and a bulk density tin with a diameter of 5 cm and a height of 3 cm, was inserted into the pit wall. The sample was then taken from the tin and dried in an overnight oven, weighed, disaggregated, and then sieved to <2 mm. The resulting soil was then weighed again to determine the soil density, before finally being milled to ≤53 μm using a planetary ball mill before dissolution. This sample was then analysed to determine the Pu inventory at the site and to demonstrate the methods usability at depth where levels of Pu are significantly lower.
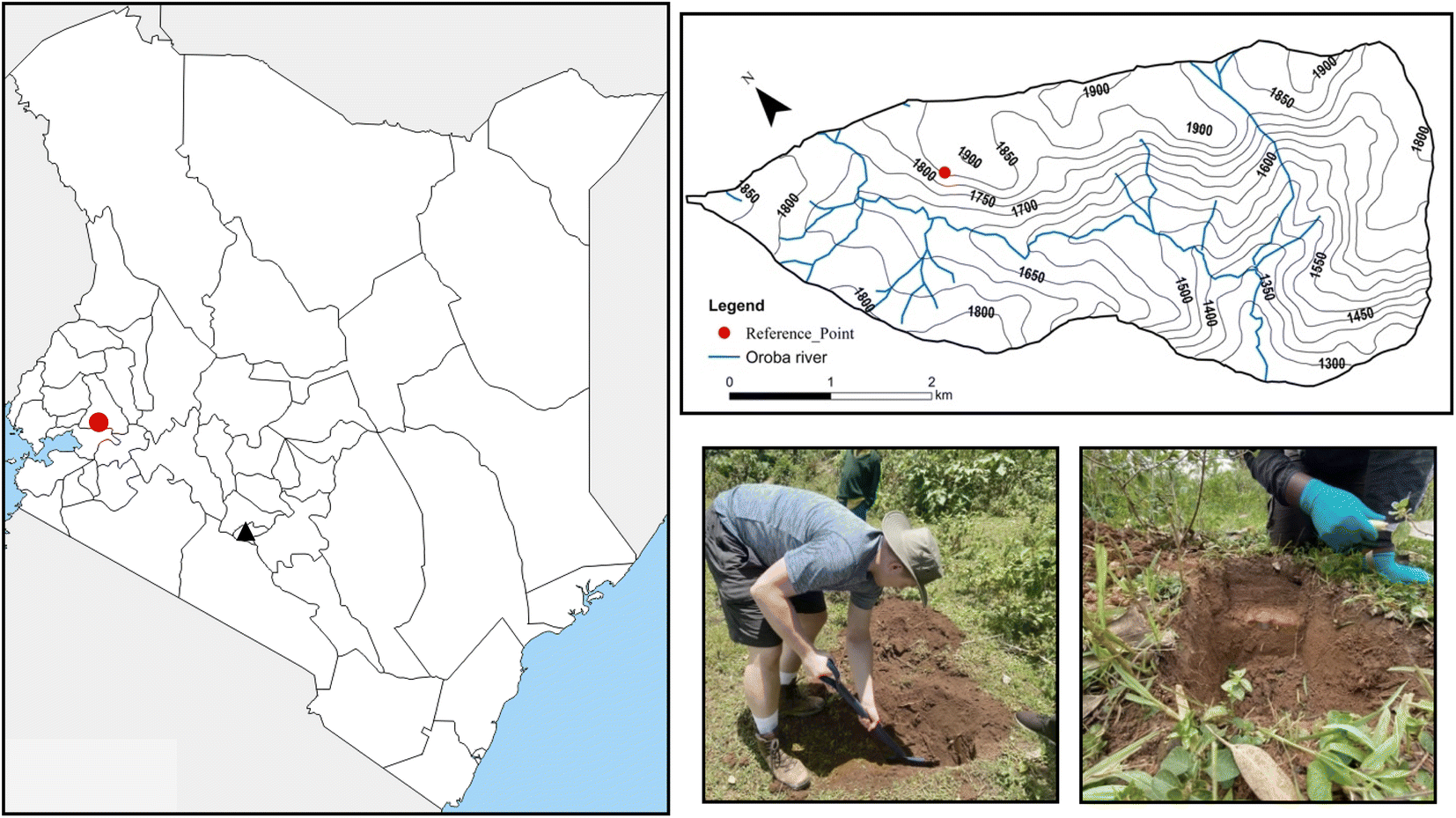 | ||
| Fig. 1 Sample location of soil core within the Oroba valley, Nandi County, Kenya, and sampling technique. | ||
2.3 Sample preparation
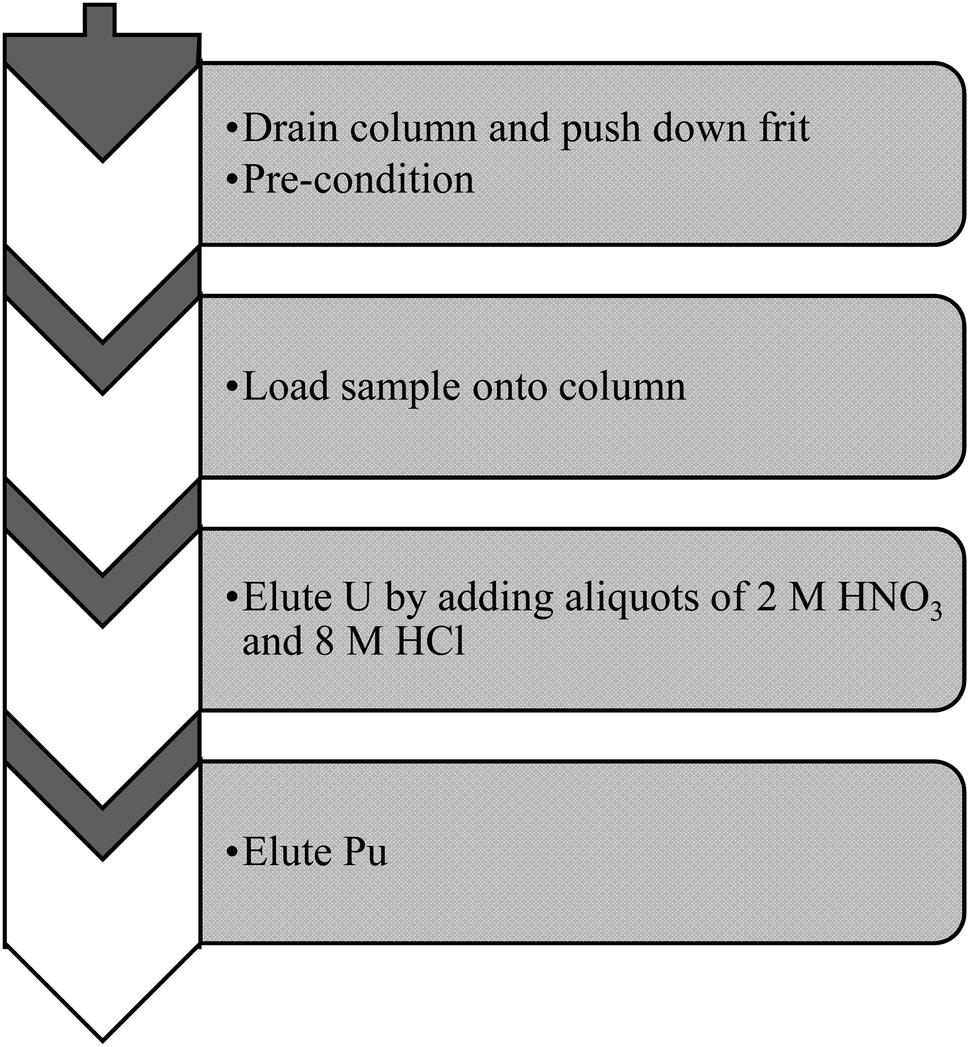 | ||
| Fig. 2 Outline of column separation employed for the preparation of Pu isotopes in soil using a TEVA resin. | ||
2.4 Instrumentation and setup
An ICP-MS/MS (Agilent 8900, Agilent Technologies, Japan) was used for the measurement of Pu isotopes in the soil samples. This instrument was equipped with a collision reaction cell (CRC) between two quadrupole mass filters. For the measurement of plutonium isotopes, O2 was used in the CRC to mass shift both Pu and U to PuO2+ and UO2+ ions for measurement, as demonstrated in Zhang et al. (2021).4 The operating gas modes and default internal standards for each isotope can be seen in Table 1. This mass shifting has the advantage of removing the major interference of UH+ on Pu isotope measurements. The ICP-MS/MS was equipped with an Agilent IaS micro-autosampler and a Cetac Aridus 2 desolvating nebuliser (Teledyne CETAC Technologies, Omaha, USA). This combination required only 1 ml to be used for analysis, ensuring the maximum sensitivity of Pu in the sample through large pre-concentration factors for a given sample size. The instrument was auto-tuned using the Agilent Masshunter software using a 1 μg kg−1 tune solution (SPEX CertiPrep #CL-TUNE-1) for general performance. Agilent typically enables autotuning up to mass 260, and therefore, autotuning on U+ at mass 270 is not possible. The instrument was then manually tuned on 270U+ specifically for Pu and U in oxygen mode. The key tuning parameters were O2 flow rate, Q2 bias, energy discrimination, OctP bias, wait time offset, cell focus and axial acceleration. Optimised parameters used are shown in ESI Table 1.† The instrument was calibrated for sensitivity using a 238U standard as no certified Pu standard was available. It was assumed that the sensitivity of 238U was the same as 239Pu allowing for isotopic abundance (ESI Table 2†).| Element | Isotope | Gas mode | Reaction product | Internal standard |
|---|---|---|---|---|
| U | 238 → 270 | O2 | 238UO2+ | 193Ir |
| Pu | 239 → 271 | O2 | 239PuO2+ | 193Ir |
| Pu | 240 → 272 | O2 | 240PuO2+ | 193Ir |
| Pu | 242 → 274 | O2 | 242PuO2+ | 193Ir |
2.5 Quality control
A variety of laboratory control samples were used to track instrumental performance within and between instrument analyses. These included a certified reference soil obtained from the IAEA, specifically IAEA-384 (Radionuclides in Fangataufa Lagoon sediment), with a certified value of 108 ± 13 Bq kg−1 239+240Pu, which was digested in duplicate within each dissolution batch and then analysed. Background signals were identified by analysis of laboratory control samples (water with 5% HNO3 and 2.5% HCl) and method blanks (20 g silicate sand, which was subjected to the same dissolution procedure as the CRM and samples). Detector performance was monitored using a 1 μg kg−1 tune solution (SPEX CertiPrep #CL-TUNE-1). Internal standard (193Ir) was analysed to correct for instrumental drift and any matrix suppression effects. Soils were analysed in triplicate within each dissolution batch to verify intra-batch analytical measurements for precision.3. Results and discussion
3.1 Soil dissolution
Fig. 3 shows the relative standard deviation (RSD) of 239+240Pu measurements according to different sample masses (g). To ensure the environmental impact (primarily the volume of acid required) of the method was accounted for, the minimum mass of sample with the lowest variability in measurement was selected, and the optimum mass for a UK sample was determined to be 20 g. The 239+240Pu concentration within the UK reference soil was found to be 38.86 ± 1.93 pg kg−1. Due to the much lower fallout in the tropics compared to the mid-latitudes of the northern hemisphere, Pu activity in Europe is on average two-to-three times greater than that in Africa.2 As a result, when working with African soils, it was decided that 50 g would be the preferred mass whilst retaining optimum sensitivity of Pu measurement. Previous studies utilising ICP-MS for the determination of Pu in soils in the northern hemisphere have used masses between 1 and 10 g,6,27–31 however, the results suggest that the error in measurements at these lower masses justifies the use of a greater mass for separation. The use of 50 g as the soil mass for dissolution is supported elsewhere in the literature, e.g. Wilken et al. (2021)3 used 50 g of soil for the determination of Pu in African soils.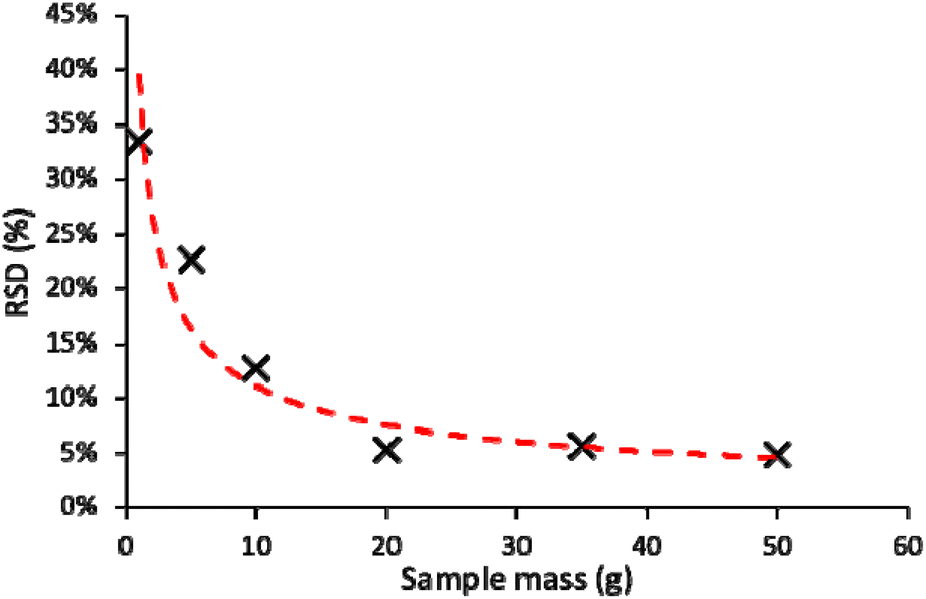 | ||
| Fig. 3 Relative standard deviation (n = 3) in measurement of 239+240Pu isotope concentrations by ICP-MS/MS according to different sample mass of UK soil used for dissolution. | ||
Fig. 4 shows the recovery of the 242Pu spike as a function of the volume of HNO3 per gram of soil. Each sample was digested in triplicate, with the error bars representing two times the standard deviation of the measurements. The spike recovery of 239+240Pu, representative of the whole separation procedure, peaked at 2 ml with the addition of more digest acid, resulting in no significant increase in Pu extraction as the graph plateaus. This is likely as a result of both the decrease in viscosity of the digest solution passing through the column and the increased efficiency of dissolution. The maximum spike recoveries exceed 100% as a result of the overall uncertainty. This compliments with the method suggested by Ketterer et al. (2004),26 which was widely reported in the literature. The benefits of the complete extraction of Pu isotopes from the soil using 2 ml of acid outweigh the environmental and cost implications of using a larger volume.
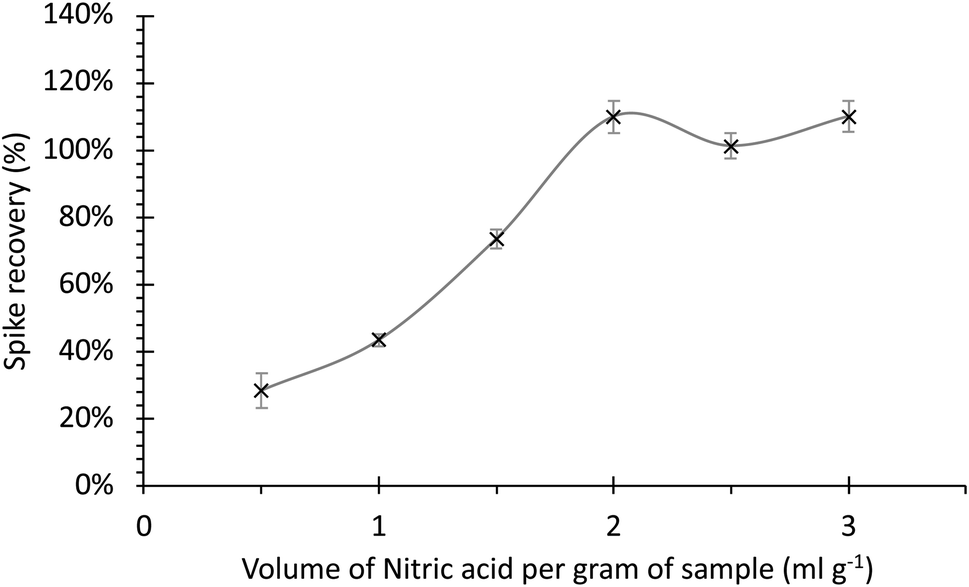 | ||
| Fig. 4 242Pu spike recovery dependant on the volume of nitric acid used for the dissolution of soil samples. | ||
The final step of the dissolution was to centrifuge the mixture to separate the digest from the residual solid. In the initial tests, it was found that spike recoveries were low (30–50%), and so the washing of the solid pellet after centrifugation was investigated. After the initial centrifugation step, the supernatant was transferred into a PTFE beaker, leaving behind a solid pellet. This was then re-distributed using an equivalent volume of DI water to the volume of acid used in the digestion. The centrifuge tube was then shaken vigorously until the solid pellet was fully redistributed into the water. The mixture was then centrifuged again at 3000 rpm min−1 for 15 minutes and the new supernatant was subsequently added to the PTFE beaker containing the digest to adjust the concentration of HNO3 to 8 M. The results showed that the spike recovery was increased by 30% with the addition of a washing step, suggesting that some of the Pu remained within residual acid retained between pores of the centrifugated soil.
3.2 Optimisation of column separation
The Pu in the samples was separated from the sample matrix using a TEVA column. The separation involved preconditioning of the column, loading of the sample, elution of matrix elements, including U isotopes, and finally the elution of Pu isotopes. As part of the method development, the elution of U isotopes and the elution of Pu isotopes were investigated. Due to the major interference caused by UH+ on Pu isotopes during ICP-MS analysis, it was vital to elute and remove the greatest amount of the U isotopes present in the dissolution prior to analysis. The elution of U isotopes was achieved by loading the column with 2 M HNO3 and discarding the eluent. The tetravalent Pu provided maximum uptake in the region of 2–4 M HNO3, while the hexavalent U was eluted, Pu was retained on the resin during this step.32 After loading the sample onto the column, 2 M HNO3 was added to the column 5 ml at a time up to 80 ml. Each of the eluents for every addition of 5 ml was collected and subsequently analysed by ICP-MS/MS for 238U to determine at what volume the maximum U had been eluted from the resin.Fig. 5 shows the percentage U eluted between 0 and 80 ml 2 M HNO3. The greatest proportion of U was eluted within the first 10 ml of acid (98.5%); however, an additional 1.0% was removed up to 60 ml of acid, thereafter minimal U was eluted. For this reason, a volume of 60 ml acid was used to elute the U from the column to optimise the analysis due to the reduction in UH+ formation in the plasma. Portes et al. (2018)27 and Wilken et al. (2021)3 used 5 ml per 30 mg of TEVA resin as the rinse volume of 2 M HNO3 which would be equivalent to approximately 35 ml for the TEVA column used. Although 99% of the U had been eluted when using a volume of 30 ml, the benefit of adding an additional 30 ml of rinse acid had the benefit of reducing the overall U in the final sample for analysis, which is particularly important when working with ultra-trace Pu isotopes in African soils.
 | ||
| Fig. 5 Elution profile of 238U isotope from TEVA resin using eluent 2 M nitric acid. | ||
The elution of Pu isotopes from the TEVA resin has commonly used 0.05 M ammonium oxalate in the literature, as described in Ketterer et al. (2004).26 However, more recently, Metzger et al. (2019)16 found that Pu can alternatively be removed from the TEVA resin using a low acid concentration. The Idaho National Engineering and Environmental Laboratory (INEEL) method uses 0.5 M HCl as the eluent of Pu, and so for this study, the use of both 0.05 M ammonium oxalate and 0.5 M HCl was investigated to optimise the elution of Pu from TEVA columns. The UK reference sample was prepared using the dissolution method detailed and loaded onto the column. After acid rinsing of the column, each sample was eluted using the different eluent matrices in triplicate. The oxalate samples were evaporated to dryness on a hot plate, followed by an addition of concentrated nitric acid and heating to decompose hydroxylamine in the samples, which would otherwise detriment the ICP-MS plasma efficiency. They were then reconstituted into 1 ml of the analysis matrix (5% HNO3 and 2.5% HCl) and subsequently diluted to ×5 with the addition of the internal standard for analysis. The HCl samples did not require removal of the matrix and were simply diluted using a solution of 5% HNO3 and the internal standard. The samples were then analysed by ICP-MS/MS to determine the 242Pu spike recovery. The spike recovery for the ammonium oxalate was 61 ± 11%, compared to the HCl spike recovery of 87 ± 17%. These findings agree with those of Metzger et al. (2019)16 and show that dilute HCl is a more suitable eluent for the determination of Pu than the commonly used ammonium oxalate. The presented method shows comparable spike recoveries with other methods utilising ICP-MS/MS analysis where reported values range between 70 and 90%.15,23,33 Using HCl not only improved spike recovery, but it also eliminated the need for extra steps in the process of removing oxalate from the sample. This increased throughput by 20% and reduced the environment burden through the removal of the complexing agent.
The product sheet for TEVA resin indicated that the retention of U isotopes in dilute HCl is also very low and as a result any remaining U on the column after the rinsing steps is likely to elute alongside the Pu isotopes. In total 0.7 μg kg−1 238U was eluted in the collection step, contributing to 0.5% of the total 238U in the sample. In comparison 0.2 μg kg−1 238U was eluted when using ammonium oxalate as the eluent. With the capabilities of ICP-MS/MS to remove the 238UH+ interference through mass shifting of the Pu isotopes the increased throughput of the method when using dilute HCl outweighs the additional U isotopes being eluted into the sample.
To ensure that the maximum Pu was eluted from the column within the smallest volume to avoid dilution, the volume of eluent acid was investigated. The dilute HCl was added stepwise in 0.5 ml increments to the column and collected to determine the elution profile (Fig. 6). To avoid dilution of the Pu isotopes prior to analysis, the eluent was collected between 1 ml and 4 ml where the maximum Pu was eluted (99%).
 | ||
| Fig. 6 Elution profile of 239+240Pu isotopes using eluent 0.5 M HCl. | ||
3.3 Method performance
Blank samples were prepared in conjunction with the analysed soil samples, these consisted of finely milled silicate sand and enabled the determination of U and Pu originating from the whole dissolution and separation processes. While Pu does not occur naturally in the environment, naturally occurring U can be found throughout the environment, including in labware as well as within reagents. The use of clean acid-leached vials and ultra-pure reagents was employed to minimise the U background detected. In addition, analysis blanks containing the analysis matrix of 5% HNO3 and 2.5% HCl were analysed as part of the analytical procedure. The amounts of U and Pu in the column blanks and ICP-MS measurement only blanks (analytical blanks) were estimated based on count rates and the sensitivity of the day. The analytical blanks yielded an average of 0.009 ± 0.014 μg kg−1 238U and 0.108 ± 0.105 pg kg−1 239+240Pu (n = 32) with limits of detection of 0.021 μg kg−1 and 0.15 pg kg−1 respectively. The method blanks yielded an average (±2σ) concentration of 0.012 ± 0.008 μg kg−1 238U and 0.066 ± 0.121 pg kg−1 239+240Pu (n = 32) with limits of detection of 0.012 μg kg−1 and 0.18 pg kg−1 respectively. These detection limits were comparable with previous studies that used O2 as the reaction gas, such as Zhang et al. (2021)4 who reported detection limits of 0.06 pg kg−1. The detection limits using this method are improved compared to methods using NH3 as the reaction gas (0.16–0.55 pg kg−1) owing to the formation of PuO2+ in the reaction cell, effectively mass shifting the Pu isotopes to m/z 271 away from interfering 238UH+ at m/z 239.8,10,23Validation of the optimised separation method was conducted using the analysis of 239+240Pu in CRM samples IAEA-384 over five separate analytical runs (n = 36). In addition, the UK reference soil was analysed as an indicator of precision within an analytical sequence as well as to monitor performance between different analytical batches. Table 2 reports the measured values for both the CRM sample and the UK reference site (±2σ). The measured value for IAEA-384 indicates both good accuracy and similar precision to the certified value, which was determined by a combination of radiometric (alpha and gamma spectroscopy) and mass spectroscopy techniques. Using a similar method with detection by Thermo X Series II quadrupole ICP-MS (Bremen, Germany), Wilken et al. (2021)3 measured the IAEA-384 reference value to be 102 ± 20 Bq kg−1 over 11 measurements. The method presented shows improved precision through the optimisation of column separation and the use of O2 in a collision cell ICP-MS/MS to reduce the error in measurements.
| Certified value (Bq kg−1) | Measured value (Bq kg−1) | |
|---|---|---|
| IAEA 384 (Fangataufa Lagoon sediment) (n = 36) | 108 ± 13 | 114 ± 12 |
| UK reference sample (n = 27) | — | 0.14 ± 0.02 |
Overall spike recoveries using the method in Table 3 ranged between 67 and 100% with an average of 86% (n = 32). This is comparable with other reported results of spike recovery using ICP-MS/MS in the literature, which range from 70 – 90%.15,23,33 This, along with the low detection limits, accuracy of measurement, and high precision supports the application of this separation and analytical method for the detection of Pu isotopes in soils.
| Step | Description | Optimum volume (ml) | Column volumes | Reagent |
|---|---|---|---|---|
| 1 | Precondition | 4 | 2 | 2 M HNO3 |
| 2 | Load sample | 200 | 100 | — |
| 3 | Wash TEVA | 60 | 30 | 2 M HNO3 |
| 4 | Wash TEVA | 20 | 10 | 8 M HCl |
| 5 | Load column with eluent | 1 | 0.5 | 0.5 M HCl |
| 6 | Elute Pu isotopes | 3 | 1.5 | 0.5 M HCl |
| 7 | Wash TEVA | 2 | 1 | 0.5 M HCl |
| 8 | Wash TEVA | 4 | 2 | 2 M HNO3 |
| 9 | Wash TEVA | 4 | 2 | 0.01 M HNO3 |
| 10 | Store TEVA column | 3 | 1.5 | 0.01 M HNO3 |
3.4 Application for the determination of Pu isotopes in African soil samples
The method reported in Table 3 was then tested on the African soil, which was collected in Zambia by Hamilton et al. (2020).25 The overall 239+240Pu in the topsoil samples was determined to be 29.59 ± 0.97 pg kg−1 (equivalent to 0.09 Bq kg−1) with an average spike recovery of 81% (n = 3). This value is approximately 1.5× smaller than the 239+240Pu concentration found within the UK reference soil and supports the use of 50 g as the sample mass for African soils to achieve maximum sensitivity down the soil profile. The measurement of soil erosion rates using Pu has increases significantly in the literature, and with improvements in the separation and measurement of Pu in soils, the method has the potential to improve understanding of processes influencing erosion and inform mitigation strategies globally.5,14 Wilken et al. (2021)3 demonstrated the usefulness of Pu as an alternative soil erosion tracer in East Africa, informing soil degradation patterns and highlighting the need for additional studies into erosion rates in tropical soils. This study reported measured mean values at sloped cropland sites in the DR Congo and Uganda of a similar magnitude, with reported values between 0.012 and 0.046 Bq kg−1. Through the detection of ultra-trace Pu in African soils as a tracer of soil erosion, the data collected can be used to reinforce sustainable soil conservation measures and aid in the validation of prediction models, allowing for a better understanding of the factors influencing accelerated erosion.To validate the methods usability for studies into soil redistribution rates, Pu inventory was determined at a reference site in the Oroba valley, Nandi County, Kenya. The greatest concentration of 239+240Pu was found at the depth of 7.5 cm (32.30 pg kg−1/0.11 Bq kg−1), whereafter concentrations exponentially decreased (Fig. 7). The usability of the method for the detection of Pu isotopes in African soils is demonstrated by the ability to detect Pu at depths greater than 30 cm with concentrations being an order of magnitude greater than the methods detection limit. The total inventory for the reference site between 0 and 30 cm was determined as 16.86 Bq m−2 and the depth profile was consistent with examples in the literature globally.5,26,30,34,35 The total inventory for the site is consistent with the estimated global fallout reported by Hardy et al. (1973)2 and Kelley et al. (1999)36 for Kenya of 19.2 Bq m−2 and the average latitudinal distribution (0–10° S) of 11.1 ± 7.4 Bq m−2. In addition, the value is in line with the inventories reported by Wilken et al. (2021),3 where although the inventories in this study were lower, they agree with the differences in annual precipitation (960 and 1400 mm per year for Kenya and the White Nile-Congo rift respectively).37
 | ||
| Fig. 7 Depth profile of 239+240Pu inventory at reference site. | ||
4. Conclusion
A modified, robust analytical sequence for the pre-concentration and separation of ultra-trace Pu isotopes in African soils provided increased sensitivity using ICP-MS/MS with O2 as a reaction gas to remove interferences. This improved the determination of fallout Pu activity concentrations in the southern hemisphere where Pu signals are relatively low compared to the northern hemisphere. Accuracy was improved through the elimination of the ammonium oxalate matrix in the eluent, with recoveries improved from 61 to 87%. Removal of the oxalate-sample matrix resulted in more stable plasma conditions, an increase in column separation throughput by 20%, and reduction in reagent consumption/disposal. This method reduced the acid requirement for separations by 80% compared to consensus literature reports, whilst maintaining maximum sensitivity and spike recovery. Additionally, O2 use as a reaction gas provided low method blank measurements by ICP-MS/MS of 0.06 pg kg−1 239+240Pu. The CRM sediment, IAEA-384, evidenced accuracy, with improved precision of measurement of: (±2σ) of 114 ± 12 Bq kg−1 239+240Pu. The total inventory (16.86 Bq m−2) and depth profile at a reference site in Kenya is in strong agreement with the literature which reinforces this methods usefulness in the determination of soil redistribution rates in tropical soils.This method presents a simple, cost-effective, robust sequence with reduced laboratory waste disposal, which is vital to ensure the separation method is applicable to low-resource laboratories. Analysis via ICP-MS/MS with O2 as a reaction gas offers a robust technique with high throughput compared to traditional techniques such as gamma spectroscopy, and therefore lends itself well to field and survey-scale soil erosion assessment. This outcome, along with the low detection limits that are comparable to alternative mass spectrometric methods, makes the method applicable to the detection of ultra-trace fallout Pu in African soils. Due to increasing concern regarding accelerated soil erosion and its impact on sustainable intensification of agriculture in developing countries, this work provides advancements in the detection of 239+240Pu which has proven to be a robust tracer for soil erosion. Furthermore, the optimised analytical method is a powerful tool to drive mitigation strategies through the analysis of ultra-trace Pu in African soils, ultimately improving the determination of soil erosion rates in tropical soils to better inform mitigation strategies.
Author contributions
SMD undertook fieldwork, method development, analyses and writing, TSB analyses, SRC analysis and validation, OSH fieldwork and conceptualisation, JB fieldwork and analyses, WHB conceptualisation and supervision, OO fieldwork, conceptualisation, validation, MJW conceptualisation, funding acquisition, administration, validation, writing and supervision. All authors contributed to the review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is published with the permission of the Executive Director, British Geological Survey. This work has originated from research conducted with the financial support of the following funders: BGS-NERC grant NE/R000069/1 entitled ‘Geoscience for Sustainable Futures’ and BGS Centre for Environmental Geochemistry programmes for financial support, the NERC National Capability International Geoscience programme entitled ‘Geoscience to tackle global environmental challenges’ (NE/X006255/1). In addition, The Royal Society international collaboration awards 2019 grant ICA/R1/191077 entitled ‘Dynamics of environmental geochemistry and health in a lake wide basin’, Natural Environment Research Councils ARIES Doctoral Training Partnership (grant number NE/S007334/1) and the British Geological Survey University Funding Initiative (GA/19S/017). Additional support provided from the British Academy Early Career Researchers Writing Skills Workshop (WW21100104).References
- P. Thakur, H. Khaing and S. Salminen-Paatero, Plutonium in the atmosphere: A global perspective, J. Environ. Radioact., 2017, 175–176, 39–51, DOI:10.1016/j.jenvrad.2017.04.008.
- E. P. Hardy, P. W. Krey and H. L. Volchok, Global inventory and distribution of fallout plutonium, Nature, 1973, 241, 444–445, DOI:10.1038/241444a0.
- F. Wilken, P. Fiener, M. Ketterer, K. Meusburger, D. I. Muhindo, K. van Oost and S. Doetterl, Assessing soil erosion of forest and cropland sites in wet tropical Africa using 239+240Pu fallout radionuclides, SOIL, 2021, 7, 399–414, DOI:10.5194/soil-7-399-2021.
- W. Zhang, J. Lin, S. Fang, C. Li, X. Yi, X. Hou, N. Chen, H. Zhang, Y. Xu, H. Dang, W. Wang and J. Xu, Determination of ultra-trace level plutonium isotopes in soil samples by triple-quadrupole inductively coupled plasma-mass spectrometry with mass-shift mode combined with UTEVA chromatographic separation, Talanta, 2021, 234, 122652, DOI:10.1016/j.talanta.2021.122652.
- C. Alewell, A. Pitois, K. Meusburger, M. Ketterer and L. Mabit, 239+240Pu from ‘contaminant’ to soil erosion tracer: Where do we stand?, Earth-Sci. Rev., 2017, 172, 107–123, DOI:10.1016/j.earscirev.2017.07.009.
- K. Meusburger, P. Porto, L. Mabit, C. La Spada, L. Arata and C. Alewell, Excess Lead-210 and Plutonium-239+240: Two suitable radiogenic soil erosion tracers for mountain grassland sites, Environ. Res., 2018, 160, 195–202, DOI:10.1016/j.envres.2017.09.020.
- X. Hou, W. Zhang and Y. Wang, Determination of Femtogram-Level Plutonium Isotopes in Environmental and Forensic Samples with High-Level Uranium Using Chemical Separation and ICP-MS/MS Measurement, Anal. Chem., 2019, 91, 11553–11561, DOI:10.1021/acs.analchem.9b01347.
- Y. Xu, C. Li, H. Yu, F. Fang, X. Hou, C. Zhang, X. Li and S. Xing, Rapid determination of plutonium isotopes in small samples using single anion exchange separation and ICP-MS/MS measurement in NH3–He mode for sediment dating, Talanta, 2022, 240, 123152, DOI:10.1016/j.talanta.2021.123152.
- C. Shin, H. Choi, H. Kwon, H. Jo, H. Kim, H. Yoon, D. Kim and G. Kang, Determination of plutonium isotopes (238,239,240Pu) and strontium (90Sr) in seafood using alpha spectrometry and liquid scintillation spectrometry, J. Environ. Radioact., 2017, 177, 151–157, DOI:10.1016/j.jenvrad.2017.06.025.
- S. Xing, M. Luo, N. Yuan, D. Liu, Y. Yang, X. Dai, W. Zhang and N. Chen, Accurate determination of plutonium in soil by tandem quadrupole icp-ms with different sample preparation methods, At. Spectrosc., 2021, 42, 62–70, DOI:10.46770/AS.2021.011.
- J. Qiao, X. Hou, M. Miró and P. Roos, Determination of plutonium isotopes in waters and environmental solids: A review, Anal. Chim. Acta, 2009, 652, 66–84, DOI:10.1016/j.aca.2009.03.010.
- L. Cao, W. Bu, J. Zheng, S. Pan, Z. Wang and S. Uchida, Plutonium determination in seawater by inductively coupled plasma mass spectrometry: A review, Talanta, 2016, 151, 30–41, DOI:10.1016/j.talanta.2016.01.010.
- C. S. Kim, C. K. Kim, P. Martin and U. Sansone, Determination of Pu isotope concentrations and isotope ratio by inductively coupled plasma mass spectrometry: a review of analytical methodology, J. Anal. At. Spectrom., 2007, 22, 827–841, 10.1039/B617568F.
- S. M. Dowell, O. S. Humphrey, W. H. Blake, O. Osano, S. Chenery and M. J. Watts, Ultra-Trace Analysis of Fallout Plutonium Isotopes in Soil: Emerging Trends and Future Perspectives, Chem. Afr., 2023 DOI:10.1007/s42250-023-00659-7.
- S. Xing, W. Zhang, J. Qiao and X. Hou, Determination of ultra-low level plutonium isotopes (239Pu, 240Pu) in environmental samples with high uranium, Talanta, 2018, 187, 357–364, DOI:10.1016/j.talanta.2018.05.051.
- S. C. Metzger, K. T. Rogers, D. A. Bostock, E. H. McBay, B. W. Ticknor, B. T. Manard and C. R. Hexel, Optimization of uranium and plutonium separations using TEVA and UTEVA cartridges for MC-ICP-MS analysis of environmental swipe samples, Talanta, 2019, 198, 257–262, DOI:10.1016/j.talanta.2019.02.034.
- Z. Varga, G. Surányi, N. Vajda and Z. Stefánka, Determination of plutonium and americium in environmental samples by inductively coupled plasma sector field mass spectrometry and alpha spectrometry, Microchem. J., 2007, 85, 39–45, DOI:10.1016/j.microc.2006.02.006.
- A. Puzas, P. Genys, V. Remeikis and R. Druteikienė, Challenges in preparing soil samples and performing a reliable plutonium isotopic analysis by ICP-MS, J. Radioanal. Nucl. Chem., 2014, 303, 751–759, DOI:10.1007/s10967-014-3411-8.
- U. Nygren, I. Rodushkin, C. Nilsson and D. C. Baxter, Separation of plutonium from soil and sediment prior to determination by inductively coupled plasma mass spectrometry, J. Anal. At. Spectrom., 2003, 18, 1426–1434, 10.1039/B306357G.
- B. Liu, K. Shi, G. Ye, Z. Guo and W. Wu, Method development for plutonium analysis in environmental water samples using TEVA microextraction chromatography separation and low background liquid scintillation counter measurement, Microchem. J., 2016, 124, 824–830, DOI:10.1016/J.MICROC.2015.10.007.
- E. P. Horwitz, M. L. Dietz, R. Chiarizia, H. Diamond, S. L. Maxwell and M. R. Nelson, Separation and preconcentration of actinides by extraction chromatography using a supported liquid anion exchanger: application to the characterization of high-level nuclear waste solutions, Anal. Chim. Acta, 1995, 310, 63–78, DOI:10.1016/0003-2670(95)00144-O.
- L. Cao, J. Zheng, H. Tsukada, S. Pan, Z. Wang, K. Tagami and S. Uchida, Simultaneous determination of radiocesium (135Cs, 137Cs) and plutonium (239Pu, 240Pu) isotopes in river suspended particles by ICP-MS/MS and SF-ICP-MS, Talanta, 2016, 159, 55–63, DOI:10.1016/j.talanta.2016.06.008.
- W. Bu, M. Gu, X. Ding, Y. Ni, X. Shao, X. Liu, C. Yang and S. Hu, Exploring the ability of triple quadrupole inductively coupled plasma mass spectrometry for the determination of Pu isotopes in environmental samples, J. Anal. At. Spectrom., 2021, 36, 2330–2337, 10.1039/D1JA00288K.
- L. Y. D. Tiong and S. Tan, In situ determination of 238Pu in the presence of uranium by triple quadrupole ICP-MS (ICP-QQQ-MS), J. Radioanal. Nucl. Chem., 2019, 399–406, DOI:10.1007/S10967-019-06695-3/TABLES/3.
- E. M. Hamilton, R. M. Lark, S. D. Young, E. H. Bailey, G. M. Sakala, K. K. Maseka and M. J. Watts, Reconnaissance sampling and determination of hexavalent chromium in potentially-contaminated agricultural soils in Copperbelt Province, Zambia, Chemosphere, 2020, 247, 125984, DOI:10.1016/J.CHEMOSPHERE.2020.125984.
- M. E. Ketterer, K. M. Hafer, V. J. Jones and P. G. Appleby, Rapid dating of recent sediments in Loch Ness: inductively coupled plasma mass spectrometric measurements of global fallout plutonium, Sci. Total Environ., 2004, 322, 221–229, DOI:10.1016/J.SCITOTENV.2003.09.016.
- R. Portes, D. Dahms, D. Brandova, G. Raab, M. Christl, P. Kuhn, M. Ketterer and M. Egli, Evolution of soil erosion rates in alpine soils of the Central Rocky Mountains using fallout Pu and δ13C, Earth Planet. Sci. Lett., 2018, 496, 257–269, DOI:10.1016/j.epsl.2018.06.002.
- F. Calitri, M. Sommer, K. Norton, A. Temme, D. Brandova, R. Portes, M. Christl, M. Ketterer and M. Egli, Tracing the temporal evolution of soil redistribution rates in an agricultural landscape using 239+240Pu and 10Be, Earth Surf. Processes Landforms, 2019, 44, 1783–1798, DOI:10.1002/esp.4612.
- G. Raab, F. Scarciglia, K. Norton, D. Dahms, D. Brandova, R. Portes, M. Christl, M. Ketterer, A. Ruppli and M. Egli, Denudation variability of the Sila Massif upland (Italy) from decades to millennia using 10Be and 239+240Pu, L. Degrad. Dev, 2018, 29, 3736–3752, DOI:10.1002/ldr.3120.
- C. Alewell, K. Meusburger, G. Juretzko, L. Mabit and M. E. Ketterer, Suitability of 239+240Pu and 137Cs as tracers for soil erosion assessment in mountain grasslands, Chemosphere, 2014, 103, 274–280, DOI:10.1016/J.CHEMOSPHERE.2013.12.016.
- Y. Muramatsu, W. Rühm, S. Yoshida, K. Tagami, S. Uchida and E. Wirth, Concentrations of 239Pu and 240Pu and Their Isotopic Ratios Determined by ICP-MS in Soils Collected from the Chernobyl 30-km Zone, Environ. Sci. Technol., 2000, 34, 2913–2917, DOI:10.1021/ES0008968.
- E. P. Horwitz, M. L. Dietz, R. Chiarizia, H. Diamond, S. L. Maxwell and M. R. Nelson, Separation and preconcentration of actinides by extraction chromatography using a supported liquid anion exchanger: application to the characterization of high-level nuclear waste solutions, Anal. Chim. Acta, 1995, 310, 63–78, DOI:10.1016/0003-2670(95)00144-O.
- X. Hou, Radioanalysis of ultra-low level radionuclides for environmental tracer studies and decommissioning of nuclear facilities, J. Radioanal. Nucl. Chem., 2019, 322, 1217–1245, DOI:10.1007/s10967-019-06908-9.
- R. Lal, S. G. Tims, L. K. Fifield, R. J. Wasson and D. Howe, Applicability of 239Pu as a tracer for soil erosion in the wet-dry tropics of northern Australia, Nucl. Instrum. Methods Phys. Res., 2013, 294, 577–583, DOI:10.1016/j.nimb.2012.07.041.
- Y. Xu, J. Qiao, X. Hou and S. Pan, Plutonium in Soils from Northeast China and Its Potential Application for Evaluation of Soil Erosion, Sci. Rep., 2013, 3, 1–8, DOI:10.1038/srep03506.
- J. M. Kelley, L. A. Bond and T. M. Beasley, Global distribution of Pu isotopes and 237Np, Sci. Total Environ., 1999, 237–238, 483–500, DOI:10.1016/S0048-9697(99)00160-6.
- S. E. Fick and R. J. Hijmans, WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas, Int. J. Climatol., 2017, 37, 4302–4315, DOI:10.1002/joc.5086.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ay01030a |
| This journal is © The Royal Society of Chemistry 2023 |