
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple and universal headspace GC-FID method for accurate quantitation of volatile amines in pharmaceuticals†
Congchao
You
,
Tien
Ho
,
Victor
Rucker
,
Jerry
Yeh
and
Lin
Wang
 *
*
Analytical Development and Operations, Gilead Sciences, 333 Lakeside Drive, Foster City, CA 94404, USA. E-mail: Lin.Wang@gilead.com
First published on 30th August 2023
Abstract
Volatile amines are reagents commonly used in pharmaceutical manufacturing of intermediates, active pharmaceutical ingredients (APIs), and drug products as participating regents for chemical reactions and optimization of product yield. Due to their compound specific daily allowable intake, residual volatile amines are required by regulatory agencies to be monitored and controlled in pharmaceutical products intended for human consumption. However, the accurate quantification of residual volatile amines in pharmaceutical entities can often be challenging as these analytes may chemically react and/or interact with the sample matrix. Herein, we describe a simple and universal headspace gas chromatography with flame ionization detection (HS-GC-FID) method capable of separating 14 commonly used volatile amines. The chemical activity of the volatile amines with the API matrix were mitigated by using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as an additive to reduce matrix effects in conventional high-boiling diluents. The addition of DBU drastically improved the detectability and method accuracy of the residual volatile amines in an acidic API, namely, Ketoprofen®. Additionally, DBU was employed as a GC deactivation reagent to ensure interfacial adsorption of the analytes to GC components were reduced, thereby improving method precision. Method validation showed acceptable linearity, limit of detection, limit of quantitation, solution stability, precision, and robustness. Separation specificity, evaluated by observing the chromatographic resolution of the volatile amines with one-another and against a set of 23 common residual solvents, were shown to be acceptable for most peak pairs.
1 Introduction
Volatile amines are often used in the manufacturing of pharmaceutical entities, such as process intermediates, active pharmaceutical ingredients (APIs), and drug product intermediates to accelerate reaction kinetics and minimize undesired side reactions, both of which favour increased product yields and maintain product quality.1,2 Global guidelines have been established to regulate daily exposure levels of common volatile amines with respect to the dosage and consumption duration of the API.3–8 To ensure patient safety and product quality while adhering to global regulatory requirements, a robust analytical strategy for controlling volatile amines must be defined prior to pharmaceutical manufacturing. As such, the development of accurate and sensitive analytical methods to enable the control of volatile amines is especially important in the pharmaceutical industry.Chromatographic and electrophoresis separation techniques are often used to analyze volatile amines in various sample types, as they enable selective and sensitive quantitation when applied with appropriate sample preparation strategies.1,9–12 For example, Riekkola et al.13 applied commercial solid phase microextraction (SPME) technology coupled to gas chromatography-mass spectrometry (GC-MS) to analyze volatile amines in wastewater and atmosphere,13 while Anderson et al.14 developed nickel-coordinated polymeric ionic liquids as SPME coatings to improve extraction selectivity and detection sensitivity of these analytes from tap and lake water.14 An orthogonal strategy to reduce the potential activity of amines, as well as improve their detection sensitivity is by chemical derivatization of the analytes.15,16 Coupling derivatization chemistry to chromatographic systems with sensitive MS detection can improve the quantification of amines to trace/ultra-trace levels.17–19
Headspace GC (HS-GC) has also been previously applied for the analysis of volatile amines in various sample matrices, due to its simplicity, ease of operation, and analytical performance in both research and quality control environments. For example, Xie et al.20 reported an automated strategy to quantify aliphatic amines in epoxy hardeners using reaction-based HS-GC.20 Raghuram et al.12 successfully applied HS-GC for determining diethylamine (DEA) and triethylamine (TEA) in two APIs, and improved amine recovery by adding sodium hydroxide to the headspace sample solution.12
Although there have been diverse approaches to quantifying volatile amines, the analysis of these compounds in pharmaceutical entities presents an ongoing challenge for pharmaceutical scientists. As these basic analytes can exhibit high activity and/or reactivity towards the sample matrix or components of the instrumentation, the analytical performance and recovery of volatile amines can be compromised, especially in trace-level analyses.3,12,21
Herein, we describe a simple and universal HS-GC-FID method capable of separating and quantifying 14 volatile amines commonly used in pharmaceutical chemistry workflows. For the first time, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was exploited as an additive in conventional high-boiling sample diluents during sample preparation to address the intrinsic chemical activity/reactivity of volatile amines towards pharmaceutical samples. Three API samples with varying degrees of acidity/basicity, namely, Bictegravir Sodium (BIC; neutral API), Emtricitabine (FTC; basic API), and Ketoprofen® (KET; acidic API), were used as real samples to determine the effects of DBU as a sample deactivation reagent in the recovery of volatile amines. The presence of basic DBU effectively passivated the API sample matrix and significantly improved method sensitivity, accuracy, and precision of the residual volatile amines in the acidic API. Being a high-boiling organic base, DBU was also employed as an instrument deactivation reagent prior to sample injection to ensure the interfacial adsorption of the analytes to the GC inlet were reduced, thereby improving method precision. The analytical performance of this method was validated for all volatile amines in two high-boiling diluent systems. The separation specificity, evaluated by observing the chromatographic resolution of the volatile amines with one-another and against a set of 23 common residual solvents, were shown to be acceptable for most peak pairs. This enables the consolidation of separate residual solvent and volatile amine methods into a single universal method, thereby improving analysis throughput.
2 Experimental
2.1 Chemicals and reagents
All amines and residual solvents were of chromatographic quality or ACS grade. Methylamine (MA) solution (40 wt% in water), dimethylamine (DMA) solution (40 wt% in water), t-butylamine (TBA), butylamine (BA), pyrrolidine (PYR), pyridine (PY), N-methylmorpholine (NMM), tetramethylethylenediamine (TMEDA), N,N-diisopropylethylamine (DIPEA), 2,6-lutidine (26L), N-methylimidazole (NMI), 2,2,6,6-tetramethylpiperidine (TMP), and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were obtained from Sigma-Aldrich, Inc. (St. Louis, MO, USA). Diethylamine (DEA) and triethylamine (TEA) were obtained from ACROS, Carlsbad, CA. Diisopropylamine (DIPA) was obtained from Oakwood Chemical, Estill, SC, USA. N,N-Dimethylacetamide (DMAc) and N-methyl-2-pyrrolidone (NMP) were obtained from Honeywell, Charlotte, NC, USA. Acetone, acetonitrile (ACN), 2-butanol, n-butanol, t-butanol, benzene, dichloromethane (DCM), diisopropyl ether, N,N-dimethylformamide (DMF), 1,4-dioxane, ethanol (EtOH), ethyl acetate (EtOAc), n-heptane, n-hexane, isopropyl acetate (IPAc), isopropyl alcohol (IPA), methanol (MeOH), 2-butanone, methyl ethyl ketone (MEK), methyl isobutyl ketone (MIBK), methyl t-butyl ether (MTBE), 2-methyl tetrahydrofuran, tetrahydrofuran (THF) and toluene were obtained from Sigma-Aldrich, Inc. (St. Louis, MO, USA).Bictegravir Sodium (BIC) and Emtricitabine (FTC) were obtained from Gilead Sciences, Inc., Foster City, CA, USA. Ketoprofen® (KET) was obtained from Sigma-Aldrich, Inc. (St. Louis, MO, USA). The names, structures, and abbreviations of all volatile amines, APIs, as well as DBU are tabulated in Table S1.†
2.2 Instrumentation and software
An Agilent 7890 GC-FID, equipped with a 7697A headspace sampler, was used in all experimental studies. Instrument operation and data collection was carried out using the Empower™ 3 Chromatographic Data System (Waters, Milford, MA, USA). The separation of all volatile amines was achieved using the Restek, Rtx-Volatile Amine (30 m × 0.32 mm, 5.0 μm, part number 18077) column, while the liner used was the Topaz straight inlet liner (2.0 mm id, cat# 23313), both of which were obtained from Restek, Bellefonte, PA. Headspace vials (20 mL) and aluminum crimp caps with PTFE-lined septa were used for sampling. GC and headspace sampler conditions applied in the analysis of the volatile amines are shown in Table S2.† Representative chromatograms for the separation of the 14 volatile amines in 5% (v/v) DBU/DMAc are shown in Fig. S1.†2.3 Preparation of analytical standards
All stock standards and dilutions were prepared in volumetric flasks using grade A glassware. A variety of diluent systems, namely, 0.1%, 5%, or 10% (v/v) DBU in either DMAc or NMP, were evaluated in the preparation of all standards and samples.Amine stock standard solutions were prepared at 2.5 mg mL−1 in both diluent systems (5% (v/v) DBU/DMAc or 5% (v/v) DBU/NMP). The amine working standard solution was prepared at 0.1 mg mL−1 in both diluents. The intermediate standard solution contained approximately 0.05 mg mL−1 of DEA, 0.5 mg mL−1 of DMA, PYR, and NMI, and 0.1 mg mL−1 of TBA, BA, DIPA, TEA, PY, NMM, TMEDA, DIPEA, 26L, and TMP in both diluents.
The limit of quantitation solution (LOQ) contained approximately 0.005 mg mL−1 of DEA, 0.05 mg mL−1 of DMA, PYR, and NMI, and 0.01 mg mL−1 of TBA, BA, DIPA, TEA, PY, NMM, TMEDA, DIPEA, 26L, and TMP in both diluents. This corresponds to 0.005%, 0.05%, and 0.01% w/w of amine relative to a nominal sample concentration of 100 mg mL−1 of API.
The specificity solution was prepared to evaluate chromatographic retention and resolution of the volatile amines with one another, as well as residual organic solvents that are common to the pharmaceutical manufacturing process. Specificity solutions were made by preparing a composite of 23 residual solvents at 0.1 mg mL−1 and 15 amines at 0.1 mg mL−1 in both diluents.
2.4 Preparation of API sample solutions
The accuracy of the method was assessed by the recovery of 14 amines spiked at the LOQ level in a neutral API salt: Bictegravir Sodium (BIC, Gilead Sciences), an API free base: Emtricitabine (FTC, Gilead Sciences), and an API free acid: Ketoprofen® (KET). The role of DBU in mitigating API matrix effects and improving method accuracy was studied by varying the sample diluent system (e.g., 0%, 0.1%, 5%, or 10% v/v of DBU in DMAc). The API matrix accuracy samples spiked with LOQ solution were prepared at 100 mg mL−1 in 1.0 mL of a LOQ solution. The API control samples were prepared at 100 mg mL−1 in the diluent. The recovery, obtained from triplicate sample preparations, was determined by comparing the experimental weight percent of the analyte against the weight percent of its theoretical spiking concentration.3 Results & discussion
The 14 volatile amines selected in this study form part of a comprehensive list of amines that have been used within Gilead in the preparation of synthetic intermediates and APIs for the past two decades. The development and validation of a generic HS-GC-FID method for chromatographic separation and analysis of multiple volatile amines in a single experimental run is vital for a fast and reliable turnaround of analytical results. The proposed method conditions can also serve as a starting point for method development in order to analyze other challenging polar amines not represented here. Development of the key method parameters and validation data are discussed herein.3.1 Mitigating matrix activity and improving method accuracy using DBU as a diluent additive
One of the main challenges in the analysis of residual volatile amines is the chemical activity intrinsic to these analytes. Due to their elevated chemical activity, amines can readily adsorb to GC instrument components during chromatographic separation, which eventually leads to a deterioration of method accuracy. Instrument manufacturers have previously developed technologies, such as amine-specific GC columns and deactivated inlet components, in order to mitigate interfacial adsorption issues.1 However, method accuracy and recovery remain an extraordinary challenge for pharmaceutical scientists trying to analyze amines in a complex API matrix.3,12,21 Since amines can react and/or interact with the API sample itself especially under elevated sampling temperatures of the HS-GC, the recoverable quantities of these analytes can be significantly compromised particularly at the low concentration levels required by regulatory agencies for analysis and/or control.To combat issues regarding the chemical activity of amines in API matrices, we exploited DBU as a diluent additive during sample preparation. DBU is an organic base (pKa = 13.5) with a relatively high boiling point (Tb = 261 °C). As such, DBU can be applied in excess quantities as a competing agent to readily react with the API matrix in place of the volatile amine analytes. This in turn will facilitate the free partitioning of the volatile amines in the headspace, while at the same time minimizing matrix interference from the API sample. The high boiling point of DBU can also be of substantial benefit when exploited for headspace GC analysis. Since the boiling point of DBU is significantly higher than those of the volatile amines, using excess quantities of DBU will not likely interfere with the separation and resolution of the volatile amines.
The recovery results for all volatile amines from BIC, FTC, and Ketoprofen prepared in varying concentrations of DBU in DMAc are compared in Tables S3 and S4† respectively. As shown in Table S3,† the recoveries of amines from the neutral BIC API were generally superior when using pure DMAc as a diluent (recovery range = 90.0% to 115.9%). Increasing the concentration of DBU in the DMAc diluent resulted in poorer recoveries for some amines from the neutral BIC API. For example, the recovery of NMI was significantly reduced from 93.4% to 65.9% when the concentration of DBU was increased to 5% v/v in DMAc. Slightly poorer recoveries were also observed for DMA, BA, and PYR when in DBU was added to the diluent system. In regard to precision, the % RSD obtained for all volatile amines in every diluent system were acceptable and below 10%.
The recovery of volatile amine from the basic FTC API showed varying result when using DMAc as a diluent with and without DBU (Table S4†). For example, as the concentration of DBU in DMAc increased, the recovery of PYR was dramatically reduced from 45.9% to 18.6%. Poorer recoveries were also observed for DMA, BA, and NMI when increasing the concentration of DBU in DMAc. On the other hand, matrix effects were reduced for several volatile amines when DBU was added to the diluent. For example, the recovery of TBA improved from 83.8% (in pure DMAc) to 92.9% (in 10% (v/v) DBU/DMAc). Additionally, matrix effects resulting in the over-recovery of PY, NMM, DIPEA, 26L, and TMP when using pure DMAc as the diluent (e.g., 117.0%, 114.7%, 117.5%, 117.1%, 116.4%, respectively) was reduced when DBU was added to the system (Table S4†). The precision of the recovery study in FTC was acceptable for all diluent systems, with % RSD values below 10%.
As BIC and FTC are neutral and basic compounds, respectively, the addition of another basic component, such as DBU, may impact the chemical activity of volatile amines on a case-by-case basis. Higher levels of DBU may further add to the complexity of the matrix itself, thereby propagating matrix effects and impeding the recovery of the volatile amines in neutral salts such as BIC. Contrarily, the addition of DBU may improve the recovery of some volatile amines from a basic API, such as FTC. Therefore, empirical evaluations should be considered when using this deactivation reagent to mitigate matrix effects in neutral or basic pharmaceutical entities.
In contrast to using DBU as an additive in the analysis of neutral or basic APIs, there is significant advantage in exploiting DBU for analyzing acidic APIs, such as KET. As shown in Table 1, the recovery of most amines was greatly improved when adding DBU to the DMAc diluent. For example, the recovery of DEA improved from 31.0% to 99.4% when using 10% (v/v) DBU/DMAc as a diluent versus pure DMAc. Similarly, the recovery of TBA and TMP improved from 21.2% to 92.9% and 28.4% to 111.4%, respectively, when using 10% (v/v) DBU/DMAc versus pure DMAc. A comparison of chromatograms obtained from spiked KET samples dissolved in DMAc or 5% (v/v) DBU/DMA is shown in Fig. 1. A significant improvement in both sensitivity was observed for several amines, most notably PYR, wherein this amine was barely quantifiable in pure DMAc. When using 5% (v/v) DBU/DMAc as a diluent, the PYR peak was easily integratable, and the recovery improved from 3.7% to 28.5% (Table 1). It is worthy to note that there is no optimal concentration of DBU that can be widely applied for every amine studied. For example, using 5% (v/v) DBU/DMAc can improve the recovery of DMA from 44.6% to 69.5% (versus pure DMAc); however, using 10% (v/v) DBU/DMAc will result in a reduction of the recovery to 45.1%. Analogously, DIPEA was best recovered from KET using 0.1% (v/v) DBU/DMAc compared to every other diluent system. The precision of the recovery study was also greatly improved for most volatile amines when using DBU in the diluent system. The % RSD of all volatile amines ranged from 7.5% to 37.4%, 0.6% to 10.2%, 0.5% to 14.5%, and 0.8% to 2.8% for DMAc vs. 0.1%, 5%, and 10% (v/v) DBU/DMAc, respectively. Specifically, the % RSD of BA significantly improved from 37.4% to 2.1%, 9.3% and 2.0% in pure DMAc vs. 0.1%, 5%, and 10% (v/v) DBU/DMAc, respectively (Table 1).
| Volatile amine | DMAc | 0.1% (v/v) DBU/DMAc | 5% (v/v) DBU/DMAc | 10% (v/v) DBU/DMAc | ||||
|---|---|---|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | |
| DMA | 44.6 | 9.6 | 60.1 | 4.8 | 69.5 | 2.4 | 45.1 | 0.1 |
| TBA | 21.2 | 10.2 | 39.4 | 3.1 | 88.5 | 1.7 | 92.9 | 0.8 |
| DEA | 31.0 | 11.5 | 45.4 | 7.3 | 96.1 | 2.9 | 99.4 | 1.1 |
| BA | 9.7 | 37.4 | 19.2 | 2.1 | 38.4 | 9.3 | 34.8 | 2.0 |
| DIPA | 49.8 | 8.8 | 68.9 | 2.7 | 92.1 | 5.0 | 91.2 | 1.9 |
| PYR | 3.7 | 18.4 | 7.5 | 3.0 | 28.5 | 14.2 | 16.4 | 1.9 |
| TEA | 71.5 | 9.9 | 85.2 | 2.1 | 83.3 | 3.7 | 92.6 | 0.9 |
| PY | 97.6 | 12.6 | 112.8 | 1.0 | 105.0 | 8.3 | 109.7 | 1.6 |
| NMM | 94.0 | 10.6 | 105.9 | 0.6 | 97.4 | 7.4 | 98.6 | 1.3 |
| TMEDA | 58.1 | 9.5 | 74.9 | 4.8 | 110.8 | 0.5 | 96.6 | 1.7 |
| DIPEA | 80.5 | 7.5 | 91.7 | 5.0 | 66.1 | 7.2 | 79.1 | 1.4 |
| 26L | 96.8 | 14.9 | 112.0 | 3.0 | 93.9 | 11.3 | 93.2 | 2.8 |
| NMI | 59.1 | 10.1 | 87.6 | 10.2 | 76.3 | 14.5 | 73.6 | 2.2 |
| TMP | 28.4 | 11.1 | 48.4 | 2.0 | 102.9 | 7.1 | 111.4 | 1.1 |
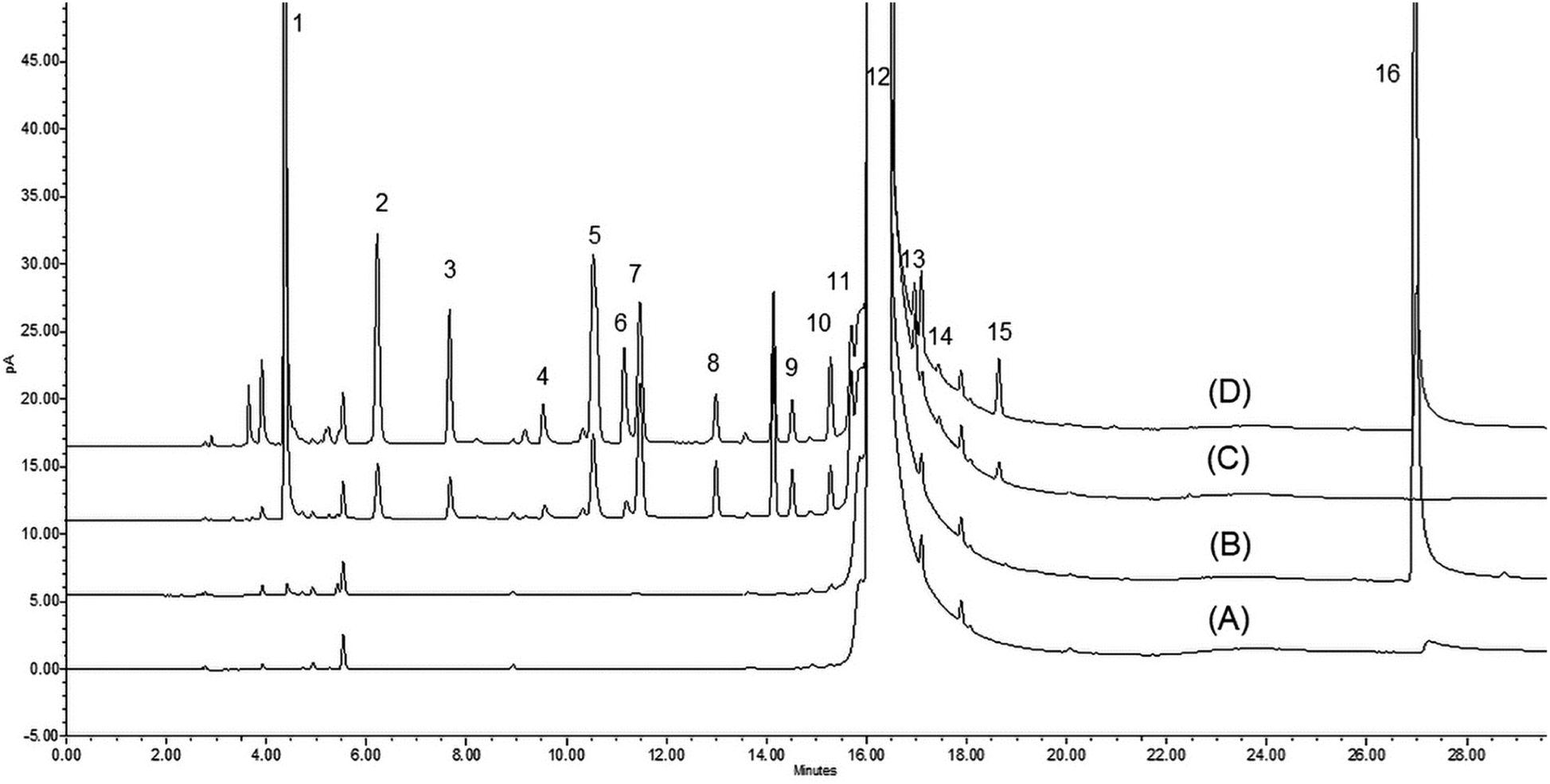 | ||
| Fig. 1 Overlaid chromatograms comparing LOQ-spiked KET samples prepared in DMAc vs. 5% (v/v) DBU/DMAc (A) DMAc, (B) 5% v/v DBU/DMAc, (C) Ketoprofen sample spiked with LOQ in DMAc, (D) Ketoprofen sample spiked with LOQ in 5% (v/v) DBU/DMAc. (1) DMA, (2) TBA, (3) DEA, (4) BA, (5) DIPA, (6) PYR, (7) TEA, (8) PY, (9) NMM, (10) TMEDA, (11) DIPEA, (12) DMAc, (13) 26L, (14) NMI, (15) TMP, (16) DBU. | ||
Per the aforementioned examples using real pharmaceutical APIs, there seems to be “sweet-spot” DBU concentrations unique to the analyte and sample matrix that will require empirical studies to fully optimize method accuracy. Nonetheless, the addition of DBU clearly played a critical role in improving the recovery of volatile amines, especially in an acidic API. As mentioned previously, this benefit is to be expected as DBU can react and/or interact with the acidic API, thereby permitting the volatile amines to freely partition in the GC vial headspace. Based on the sensitivity and recovery results generated for volatile amines from API samples, 5% (v/v) DBU in either DMAc or NMP were selected as the final diluent systems to evaluate analytical performance and method validation.
It is noteworthy that inorganic bases, such as NaOH, can also be added to the diluent system to facilitate the recovery of volatile amines,12 similar to DBU. However, the addition of NaOH should be evaluated and optimized based on empirical studies. Preliminary evaluations of 0.01 N and 0.5 N NaOH were carried out to determine whether the inorganic base can provide similar matrix deactivation results as DBU. However, artifact peaks which coeluted with some of the amines of interest (i.e., DMA and TMEDA), were observed from API sample matrices containing NaOH (Fig. S3†). Therefore, this inorganic additive was not suitable for the scope of this study.
3.2 Improving method precision by system passivation using DBU
In order to mitigate interfacial adsorption of volatile amines to instrument components and maintain chromatographic performance, it is essential to ensure the GC flow path be appropriately passivated. A simple but effective passivation strategy would be to pre-emptively inject a diluent system containing DBU to inhibit active sites present in the GC flow path, specifically targeting the GC inlet, prior to sample injection. To demonstrate the capability of DBU as a deactivation reagent, a series of six injections of the LOQ solution (prepared in 5% (v/v) DBU/DMAc) was performed after three pre-emptive blank injections with either pure DMAc or 5% (v/v) DBU/DMAc.As shown in Table S5,† the precision of the LOQ solution injections was superior when the GC system had been pre-exposed to 5% (v/v) DBU/DMAc with % RSD ranging from 0.5% to 5.9%, compared to pure DMAc (% RSD ranging from 0.9% to 11.2%). The mitigation of interfacial adsorption was clearly represented in the case of PYR Fig. 2. Without passivation using DBU, the peak area of PYR continued to increase throughout the six consecutive injections of the LOQ solution with peak areas ranging from 151 to 174. However, by passivating the GC system prior to injection, the peak areas of PYR remained consistent throughout the six consecutive injections, with peak areas ranging from 165 to 170. Based on the results, DBU may be universally applied as a system deactivation reagent during instrument readiness preparation. It is worthy to emphasize that DBU possesses a relatively high boiling point (Tb = 261 °C); therefore, analyst should ensure complete elution of the reagent during each run by using appropriate GC temperature programs.
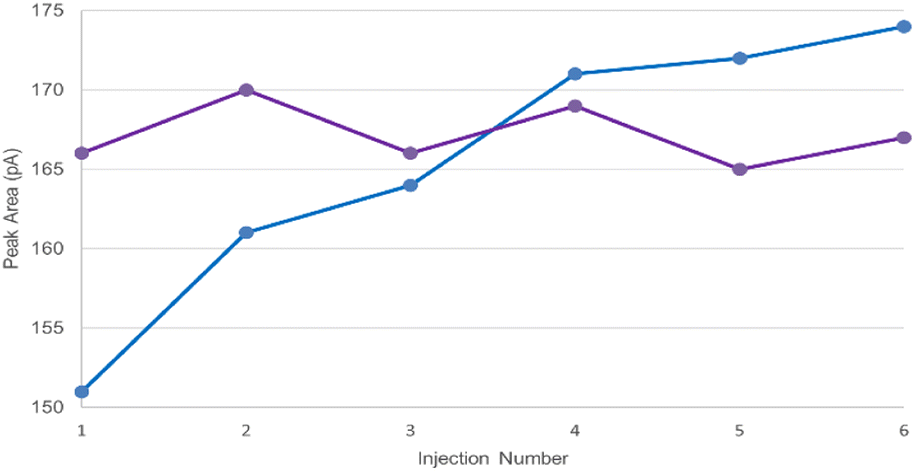 | ||
Fig. 2 Injection-to-injection peak area for pyrrolidine following ( ) system passivation injections using DMAc and ( ) system passivation injections using DMAc and ( ) system passivation injections using 5% (v/v) DBU/DMAc. ) system passivation injections using 5% (v/v) DBU/DMAc. | ||
3.3 Analytical performance and validation
To ensure that the method can be applicable for pharmaceutical analysis, the analytical performance of the method was validated in accordance with ICH Q2 (R2) and conventional quality and compliance practices.22 Method validation was performed following cGMP procedures and analytical performance attributes, namely, specificity (selectivity), sensitivity (LOD and LOQ) and precision, linearity, solution stability, as well as separation robustness were evaluated.The chromatographic specificity was evaluated by preparing a composite solution containing 14 amines and 23 common residual solvents at 0.1 mg mL−1 in 5% (v/v) DBU/DMAc or 5% (v/v) DBU/NMP. The specificity results for the 15 amines combined with 23 common residual solvents are tabulated in Table S6,† and chromatograms for the analytes in 5% (v/v) DBU/DMAc and 5% (v/v) DBU/NMP are shown in the Fig. S4 and S5,† respectively. The volatile amines are well-separated from each other, while a few peak coelutions were observed when the volatile amines are combined with common residual solvents in either diluent systems (i.e., DEA/MTBE, and DIPA/benzene). Nevertheless, most peak pairs were well resolved with one another, and peak shapes remained highly symmetrical for the analytes tested. Based on these results, the proposed method can be applied as a single universal GC method which enables rapid analysis of both volatile amines and target residual solvents in pharmaceutical samples, so long as the peaks of interest are appropriately resolved from each other. It is worthy to note that although DMAc and NMP were applied as high-boiling diluents to study the analytical performance of the proposed method as a proof-of-concept, other high-boiling diluents (i.e., 1,3-dimethyl-2-imidazolinone, N,N-dimethylformamide, etc.8) can also be evaluated during method development. As HS-GC methods have previously been developed separately for either volatile amines or residual solvents, the prospect of combining multiple methods into a single method would undoubtedly enhance operational efficiency and analytical throughput in all phases of pharmaceutical development and testing.
Linearity was determined by analysis of standard solutions prepared through serial dilution covering the expected range of the method, namely, 0.005 mg mL−1 (LOQ), 0.01 mg mL−1, 0.025 mg mL−1, 0.05 mg mL−1, 0.1 mg mL−1, 0.5 mg mL−1, 1.25 mg mL−1, and 2.5 mg mL−1. All linearity standards were prepared in either NMP or DMAc diluent systems containing 5% (v/v) DBU. As shown in Table S7,† the linearity of all amines was achieved in the concentration range studied, up to 3 orders of magnitude, with correlation coefficients (R2) ranging from 0.999 to 1.000 in both diluent systems. The amine 26L was not studied in the NMP diluent system due to the presence of a chromatographic artifact in this diluent. The method precision as represented by the % RSD of the LOQ (n = 6) was acceptable for all amines in both diluent systems and ranged from 0.3% to 3.0% and 0.6% to 3.1% in 5% (v/v) DBU/DMAc and 5% (v/v) DBU/NMP, respectively. Regarding detectability, the LOD for all amines ranged from 0.0025 to 0.025 mg mL−1, which enables the method to achieve limits described in ICH Q3C6 when appropriate sample concentrations are evaluated.
The separation robustness was assessed by deliberately varying method conditions one factor at a time. The GC oven temperature program gradient was altered to ±0.5 °C min−1 from the original method condition. Additionally, the flow rate was adjusted to ±0.1 mL min−1 from the original method condition. No significant impact on the method chromatography was observed and the separation is deemed robust. Regarding standard solution stability (evaluated per solution recovery studies), the volatile amines are stable up to 7 and 8 days in 5% (v/v) DBU/NMP and 5% (v/v) DBU/DMAc, respectively, when stored under room temperatures at working standard concentrations. The standard solution stability is also consistent at the LOQ solution concentration for up to 7 days, while only DMA showed stability for 6 days under both diluent systems at the sensitivity level (Table S8†).
Overall, the validation of analytical performance demonstrated the applicability of the method for pharmaceutical analysis to quantify volatile amines in complex API samples.
4 Conclusions
Driven by the commitment to expedite new products to market and improve patient access, the industry seeks responsible solutions to facilitate lab efficiency, whilst maintaining product quality and minimizing environmental impact. In the spirit of accelerating lab productivity and strictly adhering to regulatory guidance, we established a rapid and universal HS-GC-FID method that enables the quantification and control of volatile amines with improved analytical performance. By exploiting DBU as an additive in the sample diluent, we significantly reduced the chemical activity of volatile amines in an acidic API sample matrix, thereby leading to superior method sensitivity, precision and accuracy. When used as a GC deactivation reagent, DBU was demonstrated to mitigate potential interfacial adsorption of volatile amines to the GC inlet and improved method precision. The validation of the method demonstrated its applicability as a starting point for scientists to analyze volatile amines in potentially reactive pharmaceutical entities. Finally, the separation specificity demonstrated for volatile amines and residual solvents offers the prospect of combining multiple methods into a single universal method, further enabling analytical throughput and reduce waste generation during pharmaceutical development and testing.Author contributions
Congchao You: methodology, formal analysis, writing – original draft preparation; Tien Ho: data curation, writing – reviewing and editing; Victor Rucker: resources, funding acquisition, writing – reviewing and editing; Jerry Yeh: resources, writing – reviewing and editing; Lin Wang: conceptualization, supervision, project administration, writing – reviewing and editing.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
The authors extend their appreciation to the following scientists and colleagues for performing the method validation: Suneetha Kambampati, Yu Lee, Anna Peh, Tara Grove, and Bryan Ding (ADO, Gilead, Foster City, CA, USA), Terence Cheng and Bryan Karolat (GAU, Gilead, Alberta, Canada).References
- G. C. Graffius, B. M. Jocher, D. Zewge, H. M. Halsey, G. Lee, F. Bernardoni, X. Bu, R. Hartman and E. L. Regalado, J. Chromatogr. A, 2017, 1518, 70–77 CrossRef CAS PubMed.
- L. Hong, W. Sun, D. Yang, G. Li and R. Wang, Chem. Rev., 2016, 116, 4006–4123 CrossRef CAS PubMed.
- L. Dai, A. C. Quiroga, K. Zhang, H. B. Runes, D. T. Yazzie, K. Mistry, N. P. Chetwyn and M. W. Dong, LCGC North Am., 2010, 28, 54–66 CAS.
- E. P. Hayes, R. A. Jolly, E. C. Faria, E. L. Barle, J. P. Bercu, L. R. Molnar, B. D. Naumann, M. J. Olson, A. M. Pecquet, R. Sandhu, B. K. Shipp, R. G. Sussman and P. A. Weideman, Regul. Toxicol. Pharmacol., 2016, 79, S39–S47 CrossRef CAS PubMed.
- United States Pharmacopeia, General Chapter, 〈467〉 Residual Solvents, 2023, https://doi.org/10.31003/USPNF_M99226_08_01, accessed 17 Februrary, 2023, DOI:10.31003/uspnf_m99226_08_01.
- ICH Harmonised Guideline, Impurities: Guideline for Residual Solvents, Q3C(R8), https://database.ich.org/sites/default/files/ICH_Q3C-R8_Guideline_Step4_2021_0422_1.pdf, accessed 20 March, 2023 Search PubMed.
- European Pharmacopoeia 11.0, Impurities: Guidelines for Residual Solvents (CHMP/ICH/82260/2006), 5.4. Residual Solvents Search PubMed.
- European Pharmacopoeia 7.0, 2008, 2.4.24. Identification and control of residual solvents Search PubMed.
- P. Neyer, L. Bernasconi, J. A. Fuchs, M. D. Allenspach and C. Steuer, J. Clin. Lab. Anal., 2020, 34, 1–8 CrossRef PubMed.
- I. Ueta, Y. Nakamura, H. Fujikawa, K. Fujimura and Y. Saito, Chromatographia, 2018, 82, 317–323 CrossRef.
- F. N. Ferreira, J. C. Afonso, F. V. M. Pontes, M. Castro Carneiro, A. A. Neto, M. L. B. Tristão and M. I. C. Monteiro, J. Sep. Sci., 2016, 39, 1454–1460 CrossRef CAS PubMed.
- P. Raghuram, I. V. Soma Raju and J. Sriramulu, Chromatographia, 2010, 71, 963–966 CrossRef CAS.
- A. Helin, T. Ronkko, J. Parshintsev, K. Hartonen, B. Schilling, T. Laubli and M. L. Riekkola, J. Chromatogr. A, 2015, 1426, 56–63 CrossRef CAS PubMed.
- K. Yavir, P. Eor, A. Kloskowski and J. L. Anderson, J. Sep. Sci., 2021, 44, 2620–2630 CrossRef CAS PubMed.
- M. Asif Iqbal, J. E. Szulejko and K.-H. Kim, Anal. Methods, 2014, 6, 5697–5707 RSC.
- M. A. Farajzadeh and N. Nouri, Anal. Chim. Acta, 2013, 775, 50–57 CrossRef CAS PubMed.
- L. Balino-Zuazo and A. Barranco, Food Chem., 2016, 196, 1207–1214 CrossRef CAS PubMed.
- S. C. Cunha, M. A. Faria and J. O. Fernandes, J. Agric. Food Chem., 2011, 59, 8742–8753 CrossRef CAS PubMed.
- S. A. Zdravkovic, J. Pharm. Biomed. Anal., 2015, 112, 126–138 CrossRef CAS PubMed.
- W.-Q. Xie, Y.-X. Gong and K.-X. Yu, Anal. Methods, 2017, 9, 2440–2444 RSC.
- M. Shou and H. Qiu, J. Pharm. Anal., 2021, 11, 251–256 CrossRef PubMed.
- ICH Harmonised Guideline, Validation of Analytical Procedures, Q2(R2), https://database.ich.org/sites/default/files/ICH_Q2-R2_Document_Step2_Guideline_2022_0324.pdf, accessed 01 August, 2023 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ay00956d |
| This journal is © The Royal Society of Chemistry 2023 |