
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous determination of 22 fatty acids in total parenteral nutrition (TPN) components by gas chromatography-mass spectrometry (GC-MS)†
Mark Dennis
Chico Retrato
 *ab,
Siyuan
Qiu
a,
Anna
Lundquist
b,
Aida Zuberovic
Muratovic
c,
Farshid Mashayekhy
Rad
a,
S. J. Kumari A.
Ubhayasekera
a and
Jonas
Bergquist
a
*ab,
Siyuan
Qiu
a,
Anna
Lundquist
b,
Aida Zuberovic
Muratovic
c,
Farshid Mashayekhy
Rad
a,
S. J. Kumari A.
Ubhayasekera
a and
Jonas
Bergquist
a
aDepartment of Chemistry – Biomedical Center, Analytical Chemistry and Neurochemistry, Uppsala University, Uppsala, Sweden. E-mail: mark.retrato@kemi.uu.se
bInnovation and Development Department, Fresenius-Kabi, Uppsala, Sweden
cSwedish Food Agency (Livsmedelsverket), Uppsala, Sweden
First published on 8th May 2023
Abstract
Evaluating total parenteral nutrition (TPN) products for quality assurance and quality control is crucial due to the chemical complexity of its components. With the advent of exploring different approaches for analysing TPN components using tandem mass spectrometry techniques, there is still a need for a robust and reproducible method for industrial routine analyses. This study allows simple, simultaneous determination of 22 fatty acids (FAs) commonly found in TPN components using gas chromatography-mass spectrometry (GC-MS). Five different transesterification techniques were applied for the FA standards and the sodium methoxide in methanol-dimethyl carbonate method was selected due to its good methylation efficiency. Fatty acid methyl esters (FAMEs) were separated in gas chromatography using an HP-5MS UI column with helium as the carrier gas. Mass spectrometry was used to fragment and quantify FAMEs using electron ionization (EI) and selected ion monitoring (SIM) mode. The analytical method was evaluated using the guidelines from the US Food and Drug Agency (FDA) and European Medicines Agency (EMA) in compliance with the International Council for Harmonization (ICH) document Q2(R2). Correlation coefficients (R2) of the calibration curves for FAMEs were 0.99, except for C24:1 n-9 and C24:0, both R2 = 0.98. The limits of detection (LOD) and quantification (LOQ) were found to be 1.69 μg mL−1 and 5.14 μg mL−1, respectively. The linear range was from 3.10–179.9 μg mL−1 for most FAMEs, except for C18:1 n-7 (3.96–224.9 μg mL−1) and C18:1 n-9 (6.30–349.57 μg mL−1). The intra-day and inter-day precision coefficients of variance (CV) of the method were less than 11.10% and 11.30%, respectively. Freeze-thaw cycles and ambient temperature measurements were performed for assessing sample stability. The validated method was applied to analyse major TPN components-fish and olive oils, and an unidentified lipid sample. The presented GC-MS method is simple and robust in the identification and quantification of 22 fatty acids simultaneously in the tested TPN components.
1. Introduction
Increased consciousness and interest from the general public regarding nutrient content in food and pharmaceutical products call for a thorough component analysis for quality assurance and quality control. Providing a robust and steadfast analytical method in evaluating these nutritional components is essential for the food and pharmaceutical industries in keeping their product standards and optimum quality for consumer intake. Moreover, establishing an appropriate method for analysing total parenteral nutrition (TPN) products, administered mostly for critically ill patients is crucial due to the chemical complexity of its components, particularly in lipid content.1,2 For instance, analytical techniques in analysing fatty acids (FAs) in food and pharmaceutical products has evolved through the recent years from using gas chromatography-flame ionization detector (GC-FID) methods3–9 to gas chromatography-mass spectrometry (GC-MS) methods10–19 due to the higher sensitivity of the MS detectors,20,21 giving a better estimate of FAs present in the analyte of interest.Fatty acids are widely found in nature and they can occur in different forms-free fatty acids or bound fatty acids. Bound FAs are more abundant than free FAs physiologically since fatty acids can react with other substances to form esters.22,23 For instance, fatty acid molecules are esterified with three hydroxyl groups of glycerol to form glycerides, of which triglycerides are the most common. Triglycerides are non-polar substances that are stored in the adipose tissues in the dehydrated form and are the most abundant energy-productive substances in the body. Triglycerides become triacylglycerols upon hydration which are essential for several metabolic processes, mainly for insulation and protection, and as carriers of fat-soluble compounds. These triacylglycerols in solid and liquid form are collectively referred to as fats and oils, respectively.24
Fatty acids can also be esterified with two hydroxyl groups in glycerol-3-phosphate to form glycerophospholipids or the amino group on sphingosine can be combined with a molecule of fatty acid in the form of an amide bond to form sphingolipids. Phospholipids are amphiphilic molecules, i.e. they have hydrophilic nitrogen- or phosphorus-containing group at one end and a hydrophobic tail consisting of a fatty acid chain at the other.25 With this property, phospholipid molecules have hydrophilic ends close to each other and hydrophobic ends close to each other, and together with other molecules such as proteins, cholesterol, and glycolipids forming the lipid bilayer, the structure of biological cell membranes.26 In addition, studies show that phospholipids are important signalling molecules for disease progressions, and these conditions such as diabetes and some cardiovascular diseases are associated with disorders of phospholipid metabolism.27,28 Animal phospholipids are mostly abundant in egg yolks, animal offal, milk, and animal brain tissues whereas plant phospholipids are commonly found in seeds, grains, and nuts, such as soy, sesame, and peanuts. Fatty acids can also be combined with glucose or other sugars to form glycolipids.29,30
Due to the complex chemistry of FAs, there is a multitude of analytical techniques developed for FA quantification, including several GC-MS methods. These developed methods, however, are either (1) targeting only essential fatty acids;31–33 (2) imposing limitations to some fatty acids of interest;11,13,23,34–36 (3) mostly focused on fatty acid extraction from human samples;13,15,18,23,34,36–38 (4) have complicated sample preparation technique,4,32,39,40 (5) merely evaluating dysregulated fatty acids;23,41,42 and (6) needing analytical method validation.10,12,14,20,43 Thus, there is a need for straightforward GC-MS methods that allow the analysis of FAs as classified by their chain length and degree of unsaturation and are suitable for industrial routine applications, particularly in evaluating the quality of the TPN products.
The study aims to develop a simple GC-MS method for analysing 22 FAs commonly present in TPN components ranging from short-chain FAs (C6:0) to long-chain FAs (C24:0), perform analytical method validation using the guidelines from the US Food and Drug Agency (FDA)44 and European Medicines Agency (EMA)45 in compliance with the International Council for Harmonization (ICH) document Q2(R2) (March 2022),46 and apply this method in different TPN components. This study also focuses on evaluating five (5) fatty acid transesterification techniques converting them to fatty acid methyl esters (FAMEs) and selecting an optimized method for derivatization that would be flexible across sample types.
2. Materials and methods
2.1. Chemicals and standards
Sodium methoxide (5.4 M, 30 wt% solution with methanol), naphthalene (≥99.0% purity), acetyl chloride (≥99.0% purity), potassium hydroxide (≥85.0% purity), and sodium carbonate (≥99.5% purity) were purchased from Merck Life Science AB (Solna, Sweden). Anhydrous sodium sulfate (≥99.9% purity) and sulfuric acid (95–97%) were purchased from VWR (Stockholm, Sweden). All other solvents and chemicals in the highest purity (LC-MS grade) were purchased from Fisher Scientific (Göteborg, Sweden) unless otherwise stated. All the fatty acid methyl esters (FAMEs) and their mixtures (>99.0% purity), free fatty acids (99.0% purity), and all triglycerides (TG) (>99.0% purity) were purchased from Larodan AB (Solna, Sweden). Total parenteral nutrition (TPN) components-fish and olive oils, and the unidentified lipid sample were obtained in collaboration with Fresenius-Kabi (Uppsala, Sweden).2.2. Instrumentation
A single quadrupole mass spectrometer with a gas chromatographic system (Agilent Technologies, Inc., 8860 GC system, CA, USA) was utilized for GC-MS fatty acid quantification. Chromatographic separation of FAMEs was performed through an HP-5MS UI column (30 m × 0.250 mm × 0.25 μm, Agilent Technologies, Inc., CA, USA). The helium (>99.999% purity) at a constant flow rate of 1.0 mL min−1 was used as the carrier gas. The injector temperature was set to 250 °C and the injection volume was set to 1.0 μL. All GC injections were introduced to the instrument using the split mode with a 1:10 split ratio. The initial column temperature was set to 60 °C and held for 1 min, followed by an increase in temperature at a rate of 25 °C min−1 to 240 °C, then the final temperature was held for 13.3 min. The temperatures for the mass transfer line and ion source were set to 220 °C and 230 °C, respectively. The quadrupole temperature was set to 150 °C. FAME standards and samples were ionized in the electron impact ionization (EI) in positive mode at 70 eV. The full scan mode was applied for qualitative analysis from 35–400 amu with a step size of 0.1 m/z, while the selected ion monitoring (SIM) mode was used for the quantitative analysis of FAMEs. The GC-MS profile and spectra of FAME standards and the samples analysed from TPN components were processed using the MassHunter Workstation GC/MS software (Agilent Technologies, Inc., CA, USA).2.3. Derivatization methods for fatty acids
2.4. Method validation
Analytical method validation for the optimized GC-MS procedure for the identification and quantification of 22 FAME standards was performed using the guidelines from the FDA44 and EMA45 in compliance with the ICH Q2(R2) document (Validation of Analytical Procedures. March 2022. EMA/CHMP/ICH/82072/2006).46For the ambient temperature stability measurements, the QC standards stored at −80 °C were taken out from the freezer and kept at room temperature for 30 min prior to analysis. After the first analysis, the QC standards were kept at room temperature for the entire duration of the investigation. The analyses were repeated three times with a time interval of 24 hours between repetitions (24 h, 48 h, 72 h) corresponding to three consecutive days.
3. Results and discussion
3.1. GC-MS method development for FA analysis
Twenty-two (22) standard FAMEs in a broad range-from short-chain FAs (C6:0) to long-chain FAs (C24:0) were analysed using the optimized GC-MS method (Fig. 1). In general, fatty acids are not volatile enough for direct GC analysis due to their highly-polar carboxyl group, where the hydrogen bond formation from the FAs leads to adsorption issues.47 Hence, the conversion of FAs to their fatty acid methyl ester (FAME) counterparts is an essential part of GC analysis in lipid-rich samples to broaden the range of detection. FAME derivatization converts less volatile and thermally-labile substances into compounds that can be analysed in the gaseous state.48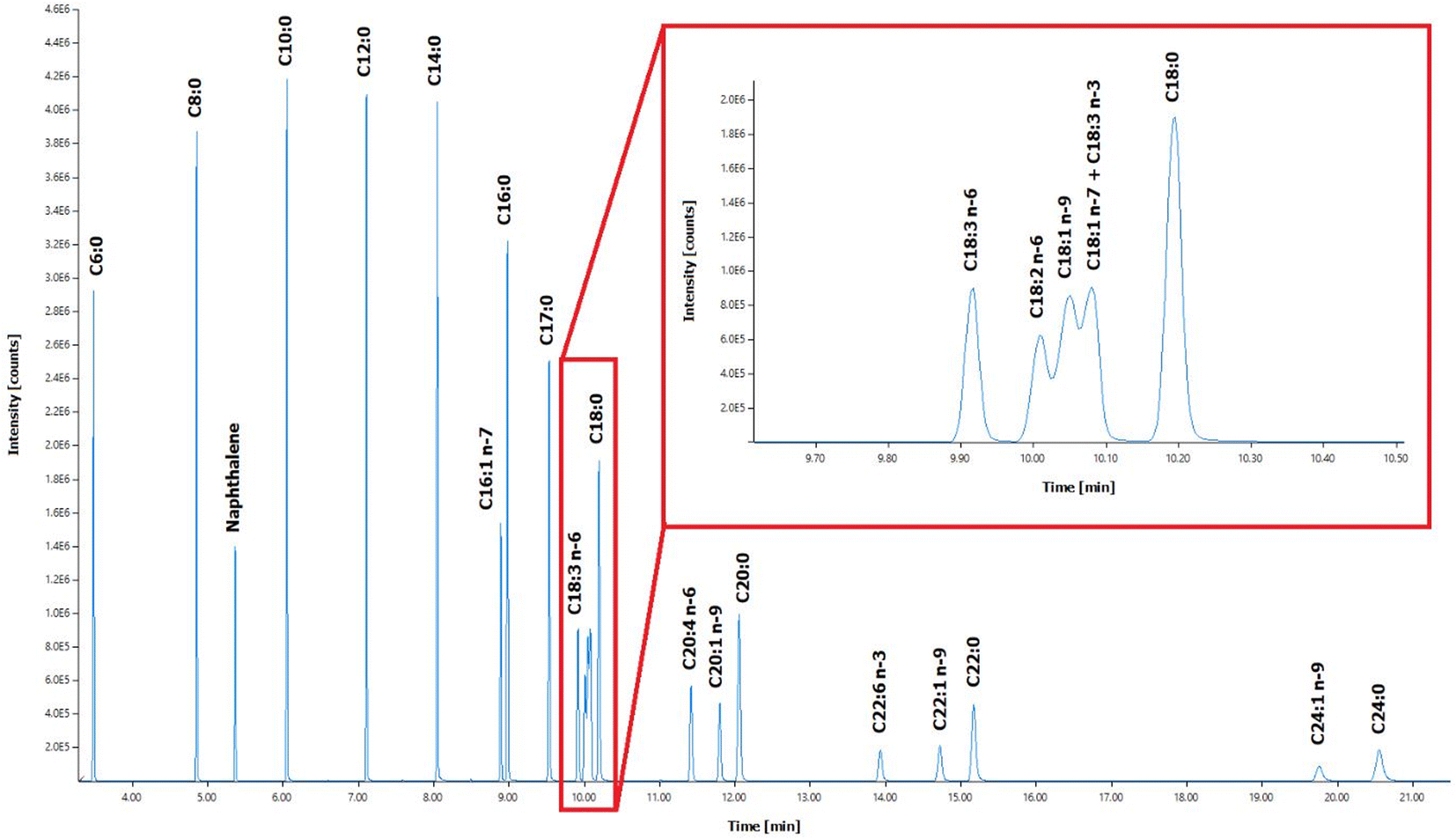 | ||
| Fig. 1 Chromatographic separation profile of 22 FAME standards and internal standard (IS) naphthalene in GC-MS. Inset: Zoomed chromatographic separation profile for C18:3 n-6, C18:2 n-6, C18:1 n-9, C18:1 n-7, C18:3 n-3, and C18:0. | ||
Applying the FAME derivatization step for the analysis of TPN components allows conversion of the high-boiling point esterified FAs and their degradation products, as well as the free FAs contained in the sample itself, into a low-boiling point, easily volatilized FAMEs. In effect, analysing FAMEs instead of underivatized FAs in GC improves analytical separation without tailing effect. In addition, the lack of ionizable functional groups in the structure of the FAs in the gas phase leads to poor sensitivity in mass spectrometry, so modification of the fatty acid structure using chemical derivatization techniques is an effective approach in increasing ionization efficiency in electron ionization (EI) source and improving the mass spectrometric response.49 In this analysis, the peak retention time of FAMEs during the chromatographic separation is related to the length of the carbon chain and the number of unsaturated bonds in the FAs. The FAMEs of isomers are merely different in spatial structure and similar in physicochemical properties, so the peak retention times overlap, representing a challenge in separation. The selected ion monitoring (SIM) mode of mass spectrometry was used to find out the characteristic ions of each fatty acid methyl ester, and the multiple fatty acid methyl esters were collected in segments by time, which improves the separation of most fatty acid methyl esters. The SIM qualitative and quantitative characteristic ions of FAMEs were determined according to their standard mass spectra. The selected ions that can be seen are mainly the base peak ions and the parent ions or ions with higher abundance close to the parent ions (see ESI Table S1†). Different FAMEs have different retention times, and different scan time windows were set for each FAME considering the slight drift of the retention time as well. Hence, different dwell times were selected ensuring sufficient data points per peak.
Due to their very similar physicochemical properties, the separation of the fatty acids C18:2 n-6, C18:1 n-9, C18:3 n-3, and C18:1 n-7 was not optimal using the capillary column (Fig. 1). Optimizing the gradient ramp-up procedure further or using a longer capillary column may improve the separation of these four FAs. By comparing the standard mass spectra of these four FAMEs (see ESI Fig. S1 and S2†), ions 294 and 292 were selected as the characteristic ions of C18:2 n-6 and C18:3 n-3, respectively; ions 222 was selected as the characteristic ion of C18:1 n-9 and C18:1 n-7, which are isomers of each other, could be well separated by SIM or SIE (selected ion extraction) modes and could be quantified separately (see ESI Table S1†). Although C18:1 n-9 and C18:1 n-7 cannot be separated very effectively, oil samples contain only C18:1 n-9, then this FA can be quantified alone using the optimized method. In case when C18:1 n-9 and C18:1 n-7 are both present in the same sample, they are quantified as one peak.
3.2. Validation of the GC-MS method
The 22 fatty acid methyl esters were quantified by the internal standard method and the linear range, linear regression equation, and correlation coefficients were obtained by linear fitting of the concentration of each analyte to the corresponding peak area ratios (Table 1). Good linearity for the 22 FAMEs within the quantification range greater than R2 = 0.9920, except for C24:1 n-9 and C24:0 (with R2 = 0.9840 and R2 = 0.9877, respectively), indicating that the potential actual content of each fatty acid composition could be accurately detected within the concentration range of the standard curve. The method limits of quantification (MLOQ) and instrumental limits of quantification (LOQ) of 22 fatty acids were determined at 10 σ/S and S/N ≥ 10, respectively. From Table 1 and it was observed that the LOD and LOQ of short-chain FAs (C6:0 to C:14) and long-chain FAs (C22:6 n-3 to C24:0) were higher than those of medium-chain (C16:1 n-7 to C18:0) and medium-long-chain (C20:4 n-6 to C20:0) FAs due to their highly-distinct isolation windows. Moreover, the ease of separation in both ends of the spectrum characterized by peak resolution is very much evident from the GC-MS profile of the FAMEs analysed.| Compound | Linear equation | Linear range (μg mL−1) | R 2 | LOD (μg mL−1) | LOQ (μg mL−1) | LOD, S/N ≥ 3 (μg mL−1) | LOQ, S/N ≥ 10 (μg mL−1) |
|---|---|---|---|---|---|---|---|
| C6:0 | y = 0.013x + 0.0521 | 3.14–174.24 | 0.9973 | 5.16 | 15.64 | 0.043 | 0.142 |
| C8:0 | y = 0.0158x + 0.063 | 3.18–176.67 | 0.9965 | 5.96 | 18.07 | 0.033 | 0.111 |
| C10:0 | y = 0.0176x + 0.0558 | 3.20–177.52 | 0.9966 | 5.89 | 17.85 | 0.030 | 0.101 |
| C12:0 | y = 0.0176x + 0.0669 | 3.20–177.52 | 0.9950 | 7.10 | 21.50 | 0.029 | 0.097 |
| C14:0 | y = 0.0166x + 0.0563 | 3.22–178.37 | 0.9957 | 6.66 | 20.17 | 0.031 | 0.103 |
| C16:1 n-7 | y = 0.0078x + 0.0129 | 3.14–174.27 | 0.9978 | 4.61 | 13.98 | 0.065 | 0.217 |
| C16:0 | y = 0.0167x + 0.022 | 3.16–175.30 | 0.9983 | 4.23 | 12.82 | 0.031 | 0.104 |
| C17:0 | y = 0.0152x + 0.3519 | 3.16–179.86 | 0.9975 | 5.04 | 15.27 | 0.035 | 0.117 |
| C18:3 n-6 | y = 0.0066x − 0.0109 | 3.10–172.20 | 0.9997 | 1.69 | 5.14 | 0.080 | 0.267 |
| C18:2 n-6 | y = 0.0003x − 0.001 | 3.18–176.18 | 0.9982 | 4.27 | 12.93 | 0.102 | 0.338 |
| C18:1 n-9 | y = 0.0004x − 0.001 | 6.30–349.57 | 0.9995 | 4.47 | 13.55 | 0.115 | 0.385 |
| C18:3 n-3 | y = 0.0002x − 0.0004 | 3.14–174.43 | 0.9986 | 3.67 | 11.11 | 0.051 | 0.171 |
| C18:1 n-7 | y = 0.0004x + 0.0003 | 3.96–224.93 | 0.9992 | 3.54 | 10.72 | 0.070 | 0.233 |
| C18:0 | y = 0.0151x + 0.0261 | 3.14–174.43 | 0.9982 | 4.16 | 12.60 | 0.036 | 0.121 |
| C20:4 n-6 | y = 0.0063x − 0.0247 | 3.23–179.09 | 0.9989 | 3.38 | 10.23 | 0.082 | 0.273 |
| C20:1 n-9 | y = 0.006x − 0.0075 | 3.13–173.39 | 0.9981 | 4.32 | 13.09 | 0.086 | 0.288 |
| C20:0 | y = 0.0138x − 0.0113 | 3.10–172.20 | 0.9978 | 4.53 | 13.72 | 0.039 | 0.129 |
| C22:6 n-3 | y = 0.004x − 0.0339 | 3.23–179.09 | 0.9924 | 8.90 | 26.97 | 0.132 | 0.440 |
| C22:1 n-9 | y = 0.0055x − 0.024 | 3.14–174.27 | 0.9941 | 7.63 | 23.13 | 0.092 | 0.308 |
| C22:0 | y = 0.0122x − 0.043 | 3.17–175.64 | 0.9943 | 7.54 | 22.84 | 0.043 | 0.143 |
| C24:1 n-9 | y = 0.0045x − 0.0372 | 3.14–174.27 | 0.9840 | 12.59 | 38.14 | 0.117 | 0.392 |
| C24:0 | y = 0.0102x − 0.0671 | 3.10–172.20 | 0.9877 | 10.88 | 32.98 | 0.052 | 0.173 |
Among the 22 FAs analysed in GC-MS, the FAs C18:3 n-6, C20:1 n-9, and C20:0 were selected as the study targets for evaluating the accuracy, repeatability, and reproducibility of the method. These FAME QC standards were analysed at three concentration levels-high (140 μg mL−1), medium (48 μg mL−1), and low (5 μg mL−1). Each concentration level was measured three times in parallel for three consecutive days. The method accuracy was assessed by evaluating the QC standard concentrations measured from the calibration curve, then subtracting their actual concentrations to obtain the error values presented in Table 2. The QC standards were selected at high concentration levels and their determination was repeated 10 times each day for three days to obtain the repeatability and reproducibility of the method. The results were expressed as pooled standard deviations and coefficients of variance (CV) as shown in Table 3. The measured values for method accuracy, repeatability, and reproducibility comply with the acceptance criteria from the ICH Q2(R2) guidelines46 (accuracy within ±15%; precision with a CV of less than ±15%).
| QC samples used | FAME | Concentration, μg mL−1 | Mean concentration, μg mL−1 | % Mean error |
|---|---|---|---|---|
| 5 μg mL−1 (low) | C18:3 n-6 | 4.79 | 5.28 | 7.77 |
| C20:1 n-9 | 4.82 | 5.09 | 7.31 | |
| C20:0 | 4.79 | 4.94 | 6.93 | |
| 48 μg mL−1 (medium) | C18:3 n-6 | 44.00 | 45.11 | 3.52 |
| C20:1 n-9 | 44.30 | 43.76 | 5.05 | |
| C20:0 | 44.00 | 42.69 | 4.55 | |
| 140 μg mL−1 (high) | C18:3 n-6 | 134.96 | 132.10 | 2.99 |
| C20:1 n-9 | 135.90 | 131.78 | 5.02 | |
| C20:0 | 134.96 | 124.85 | 8.27 |
| FAME | Repeatability | Reproducibility | ||||||
|---|---|---|---|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | ||||||
| Intraday spooled, μg mL−1 | Intraday CV, % | Intraday spooled, μg mL−1 | Intraday CV, % | Intraday spooled, μg mL−1 | Intraday CV, % | Interday spooled, μg mL−1 | Interday CV, % | |
| C18:3 n-6 | 5.43 | 4.07 | 4.56 | 3.40 | 8.81 | 6.13 | 7.96 | 5.81 |
| C20:1 n-9 | 5.79 | 4.39 | 12.31 | 9.16 | 9.69 | 6.64 | 11.19 | 8.14 |
| C20:0 | 5.50 | 4.38 | 13.99 | 11.09 | 9.28 | 6.77 | 11.24 | 8.68 |
For the freeze-thaw cycle and ambient temperature stability measurements, the FAs C18:3 n-6, C20:1 n-9, and C20:0 were selected (see ESI Tables S2 and S3†). The stability tests were performed following FDA and EMA guidelines. Results showed that the CVs of the QC standards within 2, 3, and 4 freeze-thaw cycles were at a maximum of 12.6%, which meets the acceptance criteria of the ICH Q2(R2) guidelines for stability testing (CV of ±15%). The CV of the QC standards left at room temperature for 24 h and 48 h, meet the acceptance criteria (CV of ±15%), but some of the QC standards (C20:1 n-9 and C20:0) standing for 72 h at room temperature were above 15% (see ESI Tables S2 and S3†). Therefore, freezing FA standards and samples while using this GC-MS method is important. Degradation compounds from FAs such as n-alkenals and dienals are associated with rancid taste and odor in food products. Oxidation of fatty acids affects the nutritional value of food by decomposition of vitamins, and unsaturated essential fatty acids or can even give rise to toxic compounds.50
Despite the fact that there are numerous GC-MS method development studies that have been validated for the time being, its application in analyzing these pharmaceutical-grade raw materials for producing TPNs for critically-ill patients, is a first of its own. Moreover, the method offers a broad range of fatty acids from C6:0 to C24:0 in 21 min with better resolution as compared to closely related existing methods, where fatty acids from C12:0 to C24:0 were analyzed in 23 min51 In addition, most methods are mainly focused on analysing major contents, such as docosahexaenoic acid (C22:6) for fish oils52 and linoleic acid (C18:2) and oleic acid (C18:1) for olive oils.53
3.3. Selection of the transesterification technique for the TPN components
In this study, three (3) different TPN components as samples with the same weights were methylated using five (5) different derivatization methods as described in the experimental section. These techniques were chosen because they represent different approaches in methyl esterification methods applied to FAs for GC-MS analysis-acid-catalyzed methylation (sulfuric acid method), base-catalyzed methylation (sodium methoxide and potassium hydroxide methods), an acid–base combination (potassium hydroxide-sulfuric acid method), and acetyl chloride method. The same amount of TG 17:0 was added to the samples and the spiked recovery of TG 17:0 was used to characterize the methyl esterification yield.The spiking of the TPN components with TG C17:0 was done because these test samples do not contain TG 17:0 and its fatty acid C17:0. The C17:0 methyl ester generated after derivatization is miscible with the other FAMEs present in the samples. The peak retention time of heptadecanoic acid methyl ester is distinct from the other FAMEs from the developed GC-MS method. Therefore, by adding TG 17:0 to the sample and measuring its esterification rate, various interferences such as matrix effects can be deducted, which is equivalent to the spiked recovery experiment of the pretreatment process. The summary of the FA contents of interest (C16:0, C18:2 n-6, C18:1 n-9, and C18:0) in three different samples per derivatization method was obtained (see ESI Table S4†) as well as the methyl esterification yield of each transesterification method (see ESI Table S5†).
Among the five FAME derivatization techniques performed, the potassium hydroxide method produced the least favourable result for the methylation of all three different TPN components compared to the other four methods. The acetyl chloride, sulfuric acid, and sodium methanol methods have relatively similar methylation efficiencies. The potassium hydroxide-sulfuric acid method was slightly more efficient than these other three methods but the disadvantage was that this method required a two-step reaction and heating, which consumes more chemicals and time. Given that the methyl esterification efficiency of the sodium methoxide method was not the highest among the five methods, it is straightforward and does not require heating and complex setup, hence this method was selected for further derivatization optimization and to utilize with the analysis of TPN components.
3.4. Optimization of the sodium methoxide method
To improve the methylation efficiency using the selected sodium methoxide method, further derivatization experiments in various temperatures and shaking times were performed on unidentified lipid samples. The spiking of TG 17:0 was done to evaluate the esterification rate, and the effects of different reaction temperatures (25, 60, 70, 80 °C) (see ESI Fig. S3a†) and shaking times (1, 5, 10, 15, 20 min) on the methylation of FFAs were investigated (see ESI Fig. S3b†). The temperature changes applied in the derivatization process did not significantly improve the methylation efficiency or the esterification rate, so the sodium methoxide method was performed at room temperature (25 °C) for preparing the TPN components prior to GC-MS analysis. On the other hand, in the evaluation of the optimum shaking time, the methylation yield did not change significantly for the lipid samples used after 15 min, so 15 min was selected as the shaking time for this method.3.5. Evaluating spike recoveries in TPN component matrices
The recoveries of the method were investigated by using three different TPN components as matrices. Each sample was added with triglyceride mixed standard solutions. Three parallel samples were set up for each matrix and each sample was determined three times. The recoveries were obtained after correction for the determination of fatty acid methyl esters using TG C17:0 (see ESI Table S6†). Results revealed that among the three samples, the recoveries ranged from 95.69 to 112.48% for C8:0, 89.62 to 95.20% for C14:0, and 98.40 to 108.94% for C18:0, which met the acceptable recovery range (−50% to +20%) from the ICH Q2(R2) guidelines.463.6. Fatty acid content analysis of TPN components using sodium methoxide derivatization method in GC-MS
Fish and olive oils, and the unidentified lipid samples were evaluated as TPN components for this study. The three samples were prepared according to the optimized sodium methoxide method, three parallel samples were set for each sample, and each sample was determined three times. The results are reported within the 95% confidence interval (see ESI Table S7†). The total ion current chromatograms of the three samples compared with the total ion current chromatograms of the standard FAMEs used are shown in Fig. 2. | ||
| Fig. 2 Comparison of total ion chromatograms (TIC) of different TPN components-fish oil (A), olive oil (B), and unidentified lipid sample (C) with respect to the 22 FAME standards (D) analysed in the developed GC-MS method with an optimized sodium methoxide FAME derivatization technique. | ||
There were six (6) saturated, four (4) monounsaturated, and five (5) polyunsaturated fatty acids detected from the unknown lipid samples, with the highest C18:1 n-9 content of 185.48 ± 18.89 mg g−1, followed by C16:0, C18:2 n-6 and C18:0 with 144.03 ± 16.13, 125.20 ± 12.83 and 90.82 ± 9.19 mg g−1, respectively.
The fatty acid composition of the olive oil samples detected were 6 saturated, 3 monounsaturated, and 2 polyunsaturated fatty acids, respectively, with the highest content of C18:1 n-9 at 921.80 ± 12.36 mg g−1, followed by C18:2 n-6 and C16:0 at 146.27 ± 4.94 and 122.38 ± 1.75 mg g−1, respectively. The major fatty acids reported in a separate study for olive oil are palmitic (C16:0), palmitoleic (C16:1), stearic (C18:0), oleic (C18:1), linoleic (C18:2) and linolenic (C18:3) acids; myristic (C14:0) and eicosanoic acids are present in trace amounts.54 The results of this experiment are in good coherence with the earlier reported fatty acid composition of olive oil.
The fatty acid composition of the fish oil samples detected were 7 saturated, 5 monounsaturated, and 5 polyunsaturated fatty acids, with the highest C22:6 n-3 content of 342.14 ± 23.61 mg g−1, followed by C18:1 n-9, C18:0 and C24:1 n-9 with 87.86 ± 3.44 mg g−1, 30.29 ± 0.83 mg g−1 and 27.39 ± 0.99 mg g−1, respectively. A previous study determined that fish oil is rich in various ω-3 fatty acids such as C20:5 n-3 (EPA), C22:6 n-3 (DHA), C16:3 n-3, and C18:3 n-3. Moreover, fish oil contains various saturated fatty acids C14:0, C16:0, C18:0 and C20:0, monounsaturated fatty acids C16:1 n-7, C18:1 n-9, C18:1 n-7, C20:1 n-9, C22:1, and polyunsaturated fatty acids C18:2 n-6, C18:3 n-6, C20:4 n-6, and C22:5 n-6.55 The fatty acid composition of fish oil determined in this experiment was comparable to the previously reported results, specifically the abundance of ω-3 fatty acids.55 Therefore, the optimized sodium methoxide derivatization and the developed GC-MS method provide a simple and straightforward technique that offers robust quantification of FAs in various carbon chain lengths from C6:0 to C24:0 across TPN components.
4. Conclusions
A simple, rapid, and simultaneous determination of 22 FAs with a wide range of carbon-chain lengths (from C6:0 to C:24:0) using GC-MS was developed with an optimized FAME derivatization method using sodium methoxide. The method was evaluated for linearity, repeatability, reproducibility, freeze-thaw and ambient temperature stability. The validation results were compliant with the threshold ranges specified in the ICH Q2(R2) document on which the FDA and EMA guidelines for analytical method validation are based. The tested TPN components-fish and olive oils, and an unidentified lipid sample were found to have FA levels that were comparable to previous studies, which offers flexibility for the validated method to analyze similar sample types in the future, particularly for more TPN components for pharmaceutical applications. In summary, the simplicity and robustness of this method in analyzing FAs can contribute widely to routine analyses of nutrient content for quality assurance and quality control across food manufacturing and pharmaceutical industries.Author contributions
MDCR, AL, and JB designed the study. MDCR, SQ, FMR and KU performed the experimental work. MDCR and SQ analysed the data. MDCR wrote the manuscript drafts. All authors revised and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Fresenius-Kabi AB is acknowledged for the support.Notes and references
- L. Otero-Millan, N. L. Rivero, A. B. Rodicio, N. G. Beloso, J. L. L. Soto and G. Pineiro-Corrales, Clin. Nutr., 2021, 45, 19–25 Search PubMed.
- T. Y. Ren, L. Cong, Y. Q. Wang, Y. L. Tang, B. Tian, X. Lin, Y. Zhang and X. Tang, Expert Opin. Drug Delivery, 2013, 10, 1533–1549 CrossRef CAS PubMed.
- V. J. dos Santos, A. N. E. Nicacio, R. M. Suzuki, P. B. F. Biondo, L. Maldaner and J. V. Visentainer, J. Iran. Chem. Soc., 2021, 18, 375–383 CrossRef CAS.
- C. S. R. Ferreira, P. D. S. Santos, N. B. M. Sinosaki, C. E. R. Senes, J. V. Visentainer and O. O. Santos, Rev. Virtual Quim., 2020, 12, 1575–1585 CrossRef CAS.
- L. Jarukas, G. Kuraite, J. Baranauskaite, M. Marksa, I. Bezruk and L. Ivanauskas, Appl. Sci., 2021, 11 Search PubMed.
- N. P. Kalogiouri, N. Manousi, I. Mourtzinos, E. Rosenberg and G. A. Zachariadis, Food Anal. Methods, 2022, 15, 761–771 CrossRef.
- A. D. B. Piccioli, C. S. R. Ferreira, P. D. S. dos Santos, C. E. R. Senes, J. V. Visentainer and O. O. Santos, J. Chromatogr. Sci., 2022, 60, 725–731 Search PubMed.
- P. C. Quero-Jimenez, L. A. A. Felipe, M. E. J. Rodriguez, R. M. Ruiz, J. O. P. Garcia, J. B. D. Lopez and O. N. Montenegro, J. Pharm. Pharmacogn. Res., 2021, 9, 208–221 CrossRef CAS PubMed.
- A. A. Rydlewski, J. S. Pizzo, L. P. Manin, M. B. Galuch, P. D. S. Santos, C. Zapiello, O. O. Santos and J. V. Visentainer, Chem. Pap., 2020, 74, 2799–2812 CrossRef CAS.
- K. A. Akinyede, G. D. Hughes, O. E. Ekpo and O. O. Oguntibeju, Plants, 2022, 11(8), 998 CrossRef CAS PubMed.
- M. Q. Chen, Y. D. Zhang, F. G. Wang, N. Zheng and J. Q. Wang, Separations, 2021, 8, 118 CrossRef CAS.
- D. Eisenstecken, J. Stanstrup, P. Robatscher, C. W. Huck and M. Oberhuber, Int. J. Dairy Technol., 2021, 74, 215–224 CrossRef CAS.
- J. Gray, B. C. Guo, R. Bearden and J. Manka, J. Mass Spectrom., 2022, 57(4), e4817 CrossRef CAS PubMed.
- M. Guerrero-Esperanza, K. Wrobel, K. Wrobel and J. J. Ordaz-Ortiz, J. Food Compos. Anal., 2023, 115, 104963 CrossRef CAS.
- V. Lolli, M. Dall'Asta, A. Caligiani, D. Del Rio, M. A. De La Fuente and P. Gomez-Cortes, J. Food Compos. Anal., 2022, 107 Search PubMed.
- A. Savych, R. Basaraba, N. Muzyka and P. Ilashchuk, Pharmacia, 2021, 68, 433–439 CrossRef CAS.
- L. R. Xu, S. H. Wang, A. L. Tian, T. R. Liu, S. Benjakul, G. S. Xiao, X. G. Ying, Y. H. Zhang and L. K. Ma, Food Chem.: X, 2023, 17 Search PubMed.
- H. Yisak, E. E. Yaya, B. S. Chandravanshi and M. Redi-Abshiro, J. Food Compos. Anal., 2022, 107 Search PubMed.
- N. Zhang, T. Y. Chen, S. Ye, S. K. Gao and Y. Y. Dong, Separations, 2022, 9, 314 CrossRef CAS.
- X. Gao, K. X. Chen, M. M. Chi and K. M. Qin, Int. J. Anal. Chem., 2020, 2020 Search PubMed.
- R. A. Aparicio-Ruiz, D. L. G. Garcia-Gonzalez, M. T. Morales, A. L. Lobo-Prieto and I. Romero, Talanta, 2018, 187, 133–141 CrossRef CAS PubMed.
- A. Tres, C. D. Nuchi, N. Magrinya, F. Guardiola, R. Bou and R. Codony, Animal, 2012, 6, 1005–1017 CrossRef CAS PubMed.
- S. J. K. A. Ubhayasekera, J. Staaf, A. Forslund, P. Bergsten and J. Bergquist, Anal. Bioanal. Chem., 2013, 405, 1929–1935 CrossRef CAS PubMed.
- P. C. Calder, JPEN, J. Parenter. Enteral Nutr., 2015, 39, 18s–32s CrossRef PubMed.
- B. L. Peterson and B. S. Cummings, Biomed. Chromatogr., 2006, 20, 227–243 CrossRef CAS PubMed.
- T. Wang and D. Y. Zhou, Curr. Opin. Food Sci., 2017, 16, 15–20 CrossRef.
- E. Cifkova, M. Holcapek, M. Lisa, D. Vrana, J. Gatek and B. Melichar, Anal. Bioanal. Chem., 2015, 407, 991–1002 CrossRef CAS PubMed.
- M. Mapstone, A. K. Cheema, M. S. Fiandaca, X. G. Zhong, T. R. Mhyre, L. H. MacArthur, W. J. Hall, S. G. Fisher, D. R. Peterson, J. M. Haley, M. D. Nazar, S. A. Rich, D. J. Berlau, C. B. Peltz, M. T. Tan, C. H. Kawas and H. J. Federoff, Nat. Med., 2014, 20, 415 CrossRef CAS PubMed.
- K. Abe and Y. Tamai, Neurochem. Res., 1982, 7, 861 Search PubMed.
- H. Zhang, J. Oh, T. S. Jang, B. S. Min and M. Na, Food Chem., 2012, 131, 1097–1103 CrossRef CAS.
- S. Lee, D. K. Lim, S. Y. Baek, D. Seo, J. S. Park, B. M. Kwak, J. Won, J. Lee and B. Kim, J. Anal. Sci. Technol., 2020, 11 CAS.
- H. H. Chiu and C. H. Kuo, J. Food Drug Anal., 2020, 28, 60–73 CrossRef CAS PubMed.
- R. J. Pawlosky, J. R. Hibbeln and N. Salem, J. Lipid Res., 2007, 48, 935–943 CrossRef CAS PubMed.
- H. Wang, Y. Liu, J. Shao, Y. Luo, W. Cai and L. Chen, Anal. Lett., 2020, 53, 2320–2336 CrossRef CAS.
- F. B. Xia, L. S. Guo, P. Cui, Q. Xu, J. D. Huang, H. W. Zhou and W. Shen, J. Pharm. Biomed., 2023, 223 Search PubMed.
- L. X. Yao, E. A. Davidson, M. W. Shaikh, C. B. Forsyth, J. E. Prenni and C. D. Broeckling, Anal. Bioanal. Chem., 2022, 414, 4391–4399 CrossRef CAS PubMed.
- A. Ferracane, I. Aloisi, M. Galletta, M. Zoccali, P. Q. Tranchida, G. Micalizzi and L. Mondello, Anal. Bioanal. Chem., 2022, 414, 8423–8435 CrossRef CAS PubMed.
- H. Kuiper, C. Campbell and H. Vesper, J. Am. Oil Chem. Soc., 2021, 98, 10 Search PubMed.
- M. H. Li, R. R. Zhu, X. X. Song, Z. J. Wang, H. B. Weng and J. Y. Liang, Analyst, 2020, 145, 2692–2700 RSC.
- M. A. Vargas-Munoz, V. Cerda, G. T. Palomino and E. Palacio, Anal. Bioanal. Chem., 2021, 413, 3833–3845 CrossRef CAS PubMed.
- J. W. Ren, E. L. Mozurkewich, A. Sen, A. M. Vahratian, T. G. Ferreri, A. N. Morse and Z. Djuric, Curr. Pharm. Anal., 2013, 9, 331–339 CrossRef CAS PubMed.
- B. W. Kail, D. D. Link and B. D. Morreale, J. Chromatogr. Sci., 2012, 50, 934–939 CAS.
- M. Boga, A. Ertas, M. A. Yilmaz, M. Kizil, B. Ceken, N. Hasimi, T. Y. Ozden, S. Demirci, I. Yener and O. Deveci, Iran. J. Pharm. Res., 2016, 15, 393–405 CAS.
- Q2(R2), Validation of Analytical Procedures, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q2r2-validation-analytical-procedures, accessed January, 2023 − Search PubMed.
- ICH Q2(R2), Validation of Analytical Procedures - Scientific Guideline, https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures-scientific-guideline, accessed January, 2023 Search PubMed.
- ICH Quality Guidelines, https://www.ich.org/page/quality-guidelines, accessed January, 2023.
- E. Tammekivi, S. Vahur, O. Kekisev, I. D. van der Werf, L. Toom, K. Herodes and I. Leito, Anal. Methods, 2019, 11, 3514–3522 RSC.
- A. Topolewska, K. Czarnowska, L. P. Halinski and P. Stepnowski, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 990, 150–157 CrossRef CAS PubMed.
- W. W. Christie, Preparation of ester derivatives of fatty acids for chromatographic analysis, 1993 Search PubMed.
- D. Ansorena and I. Astiasaran, Meat Sci., 2004, 67, 237–244 CrossRef CAS PubMed.
- E. Kish-Trier, E. L. Schwarz, M. Pasquali and T. Yuzyuk, Clin. Mass Spectrom., 2016, 2, 11–17 CrossRef.
- T. Yi, S. M. Li, J. Y. Fan, L. L. Fan, Z. F. Zhang, P. Luo, X. J. Zhang, J. G. Wang, L. Zhu, Z. Z. Zhao and H. B. Chen, Lipids Health Dis., 2014, 13, 190 CrossRef PubMed.
- M. Radzimierska-Kazmierczak, K. Smigielski, M. Sikora, A. Nowak, A. Plucinska, A. Kunicka-Styczynska and K. H. Czarnecka-Chrebelska, Molecules, 2021, 26, 3074 CrossRef CAS PubMed.
- S. M. Wabaidur, A. AlAmmari, A. Aqel, S. A. Al-Tamrah, Z. A. Alothman and A. Y. B. H. Ahmed, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1031, 109–115 CrossRef CAS PubMed.
- F. Sahena, I. S. M. Zaidul, S. Jinap, A. M. Yazid, A. Khatib and N. A. N. Norulaini, Food Chem., 2010, 120, 879–885 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Workflow of GC-MS method development in analysing TPN components. The retention times and selected ion monitoring (SIM) parameters for 22 FAMEs and internal standard (IS) naphthalene in GC-MS. Freeze-thaw stability (−80 °C/room temperature) testing of FAME standards. Ambient stability testing of FAME standards. The four main FA contents in the different TPN component matrices (n = 3) detected by the five derivatization methods (mg g−1). Methyl esterification yield of five derivatization methods in different TPN component matrices (n = 3). Recovery measurements in three study targets & horbar; C8:0, C14:0, and C18:0 in different TPN component matrices (n = 3). Quantification of fatty acids expressed in mg g−1 present in different TPN component matrices (n = 3) using the validated GC-MS method and optimized sodium methoxide FAME derivatization technique. Standard mass spectra and chemical structure of FAMEs. Extraction chromatogram of ions 222, 292 and 294 featuring C18:2 n-6, C18:1 n-9, C18:3 n-3, and C18:1 n-7. Optimization of the sodium methoxide FAME derivatization technique. See DOI: https://doi.org/10.1039/d3ay00407d |
| This journal is © The Royal Society of Chemistry 2023 |