
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnified analysis method for total and inorganic As determination in foodstuffs by hydride generation high-resolution continuum source quartz tube atomic absorption spectrometry†
Lucia
Chirita
 ab,
Eniko
Covaci
ab,
Michaela
Ponta
ab and
Tiberiu
Frentiu
*ab
ab,
Eniko
Covaci
ab,
Michaela
Ponta
ab and
Tiberiu
Frentiu
*ab
aBabes-Bolyai University, Faculty of Chemistry and Chemical Engineering, Arany Janos 11, 400028 Cluj-Napoca, Romania. E-mail: tiberiu.frentiu@ubbcluj.ro
bBabes-Bolyai University, Research Center for Advanced Analysis, Instrumentation and Chemometrics, Arany Janos 11, 400028 Cluj-Napoca, Romania
First published on 8th March 2023
Abstract
A unified analytical method applicable to common foodstuff matrices was developed and characterized for total and inorganic arsenic determination by hydride generation high-resolution continuum source quartz tube atomic absorption spectrometry, which was established based on different sample preparation procedures. This new method was found to be interference-free and cost-effective in terms of reagents consumption for sample preparation and derivatization to arsine for the inorganic arsenic fraction. Microwave-assisted digestion in HNO3–H2O2 for total arsenic and extraction in 0.28 mol L−1 HNO3 by mechanical stirring in a water bath or ultrasound-assisted extraction in 0.01 mol L−1 HCl without separation of inorganic As, all coupled with arsine generation in 0.01 mol L−1 HCl medium with 0.6% NaBH4 in 0.01% NaOH in the presence of 0.2% L-cysteine was found to be suitable for all matrices. The results were statistically compared by applying Tukey's and Dunnett's multiple comparison methods (p > 0.05). The use of external calibration with As(III) standards and standard addition method for quantification showed the lack of non-spectral interferences from the multimineral matrices, resulting in a reliable method for total/inorganic As determination in various foodstuffs. The limits of detection for total/inorganic As using peak height measurement were 0.0044 ± 0.0005/0.0022 ± 0.0003 mg kg−1 (n = 25 days). The overall recovery for total/inorganic As in the certified reference materials was in the range of 98% ± 22%, and 99% ± 24% (k = 2). The extraction of inorganic As in 0.01 mol L−1 HCl and 0.28 mol L−1 HNO3 provided the recovery of 106% ± 25% and 100% ± 25% (k = 2), which was better than in 10 mol L−1 HCl. The precision of measurements in real samples of fish muscle, meat and organs, rice and rice-based baby foods with contents of 0.052–5.29 mg kg−1 total As and 0.005–0.063 mg kg−1 inorganic As was 9.8–18.8% and 8.7–32.0%, respectively, which was calculated based on the combined uncertainty.
Introduction
The quantification and monitoring of total As and its species in food, biological systems and the environment are recognized as a problem globally because some species, especially inorganic ones, exhibit high toxicity and are associated with minor disorders and even skin, lung, liver and kidney cancers.1 Therefore, health-related studies and risk assessment involving As species are of great interest to chemists and scientists in medicine. Arsenic is bioaccumulated in seafood, which is considered the main source of organic species in the human body.2 The total As (tAs) and inorganic As (iAs) fraction are regulated in seafoods, food of animal origin, cereals, sweets, cocoa and chocolate, milk, fruit juice, and food supplements for children and babies in many countries; however, there is no international consensus on their maximum levels (MLs).3,4 In Europe, the ML of iAs is regulated in the range of 0.1–0.3 mg kg−1 in different varieties of rice and rice-based infant formula.5 However, considering the high toxicity and human exposure risk, the European Food Safety Authority (EFSA) and European Commission (EC) recommend the regulation of iAs in food groups and consensus in terms of exposure to As via food. Nevertheless, this is challenging, and thus at present, a provisional tolerable weekly intake (PTWI) of 3 μg kg−1 body weight is proposed, above which the incidence of lung, skin and bladder cancer increases.6,7 Very recently, the EFSA assessed the chronic dietary exposure to iAs in the European population based on data collected across Europe between 2013 and 2018 in terms of the consumption of drinking water and foodstuffs.8 Based on these studies, the EFSA recommends the revision and amendment of Commission Regulation (EC) 1881/2006 with MLs for iAs in other foods, which thus far exist only for rice.9 Accordingly, there is constant interest in the development of sample preparation strategies and spectrometric and chromatographic methods for the determination and speciation of As in biota and abiotic samples.4,10–13 Liquid and direct solid sampling without chemical vapor generation are used for quantitation and speciation of As by inductively coupled plasma mass spectrometry (ICP-MS)14–16 and graphite furnace atomic absorption spectrometry (GFAAS).17–21 Frequently, sample preparation involves preconcentration of As species by liquid–liquid microextraction, microextraction on a solid phase using nanoparticles as sorbent materials coupled or not with derivatization to chemical vapor to increase and achieve the required analytical sensitivity.16,18–23 Undoubtedly, chemical or electrochemical hydride generation (HG)24 with or without arsine preconcentration coupled with atomic absorption spectrometry (HG-AAS),25–28 atomic fluorescence spectrometry (HG-AFS),23,29,30 and inductively coupled plasma optical emission spectrometry/mass spectrometry (HG-ICP-OES/HG-ICP-MS)31–34 has proven to be the most effective and sensitive approaches to determine As in various matrices. The methods based on HG-AAS are easier to implement and have become the most accessible for official control laboratories.25 The hyphenated techniques, such as high-performance liquid chromatography with post-column hydride generation coupled with atomic fluorescence spectrometry (HPLC-HG-AFS),35 inductively coupled plasma optical emission spectrometry with post column HG (HPLC-HG-ICP-OES)36 and inductively coupled plasma mass spectrometry with or without post column HG (HPLC-ICP-MS/HPLC-HG-ICP-MS)13,23,25,33,37–42 provide high sensitivity and selectivity for As speciation. However, the hyphenated techniques are more difficult to implement in routine laboratory settings because of their high cost and the need for highly skilled analysts.Non-conventional technology based on high-resolution continuum source atomic absorption spectrometry (HR-CS-AAS) has demonstrated advantages in terms of sequential multi-elemental analysis and advanced strategies for the correction of spectral interferences.43–46 This new instrumental concept is easier to operate than the conventional techniques based on line source low-resolution atomic absorption spectrometry, resulting in a better signal-to-noise ratio, by recording a more stable and reproducible spectrum, the possibility of signal integration using variable line width, the avoidance of self-absorption, etc. Thus, analytical procedures were developed for the determination of chemical vapor generation elements, such as As, using either direct liquid or solid sampling without derivatization (HR-CS-GFAAS)47,48 or hydride generation (HG-HR-CS-GFAAS) with on line and in situ preconcentration by solid phase microextraction with magnetic nanoparticles and graphite furnace, respectively.49–51 Most approaches reported in the literature for the determination of tAs and iAs use readily available instrumentation and fit-of-purpose methods, aiming to achieve the required analytical performance for a specific sample matrix, rather than providing extended use. However, collaborative trials were organized by the EC for the development and validation of methods based on HG-AAS with line sources or hyphenated techniques such as HPLC-ICP-MS for iAs determination in several foodstuffs. Nevertheless, to date, hydride generation high-resolution continuum source quartz tube atomic absorption spectrometry (HG-HR-CS-QTAAS) has not been the subject of a validation study for As determination in food.13,25,52–55
In this context, this study aimed to develop a unified, robust, reliable, and interference-free method using HG-HR-CS-QTAAS commercial instrumentation for the determination of tAs and iAs at least for the more common foodstuffs. This method does not require separation of the iAs species from the matrix and has the advantage of very low HCl consumption in both the pre-reducing step of As(V) to As(III) with L-cysteine and HG. The HG-HR-CS-QTAAS instrumentation has not been investigated to date for the determination of tAs and iAs under the working conditions presented in this study. For an easy transfer of this method to other laboratories, only commercially available instrumentation and accessible sample processing devices were used. Also, we found that L-cysteine is an efficient reagent for reducing the liquid-phase interferences from transition metal ions and prereduction and derivatization to arsine, resulting in an enhancement in the limit of detection (LOD), accuracy and precision. The working parameters influencing the analytical sensitivity and operation of the HG-HR-CS-QTAAS analytical assembly were optimized using As(III) standard solutions, and then a validation study was conducted by analysing certified reference materials (CRMs) and testing real samples. The analytical potential and versatility of the HG-HR-CS-QTAAS method were demonstrated for a wide variety of foodstuffs including fish muscle, meat and organs of terrestrial animals, mushrooms, rice and baby food. Some aspects related to sample preparation that need special attention were highlighted.
Materials and methods
Instrumentation
The experiments were performed on the HG-HR-CS-QTAAS system provided in Fig. 1, which consisted of a ContrAA 300 spectrometer, an HS55 batch mode hydride generation system and an electric oven equipped with a quartz tube for arsine atomization in an Ar atmosphere (Analytik Jena AG, Jena, Germany). The instrumentation and operation procedure of HG-HR-CS-QTAAS are presented in detail in ESI (Section 1).† The operating conditions of the hydride generator and the composition of the atmosphere in the atomizer proved to be crucial for the efficient atomization of arsine and sensitivity of the method. The radical theory of hydride atomization confirmed that the free hydrogen atoms formed in the atomizer and inlet T-arm are responsible for the atomization of arsine species by collisions. The stability of the absorbance is ensured by the presence of free hydrogen atoms.24,56–58 The free hydrogen atoms are formed in the reaction between the hydrogen resulting from NaBH4 and HCl during HG and traces of oxygen purged from the liquid sample, or additionally introduced in the atomizer. However, it has been reported that the efficient atomization of arsine is possible in an Ar atmosphere with traces of hydrogen and oxygen (O2/H2 ratio < 0.1) without extra flow.24,56–58 The extra introduction of hydrogen and oxygen generated additional water vapor in the QTA, mostly around the temperature of 900 °C, which is similar to that necessary for arsine atomization. In addition, the water vapor resulted from the liquid droplets entrained in the atomizer from the gas–liquid separator quenched the chain of H˙-forming reactions, and thus the atomization of hydrides.24,57 Furthermore, the pseudo-continuum absorption of H2O vapor is a potentially serious spectral interference on the As 193.696 nm line, altering the LOD of methods based on QTA.51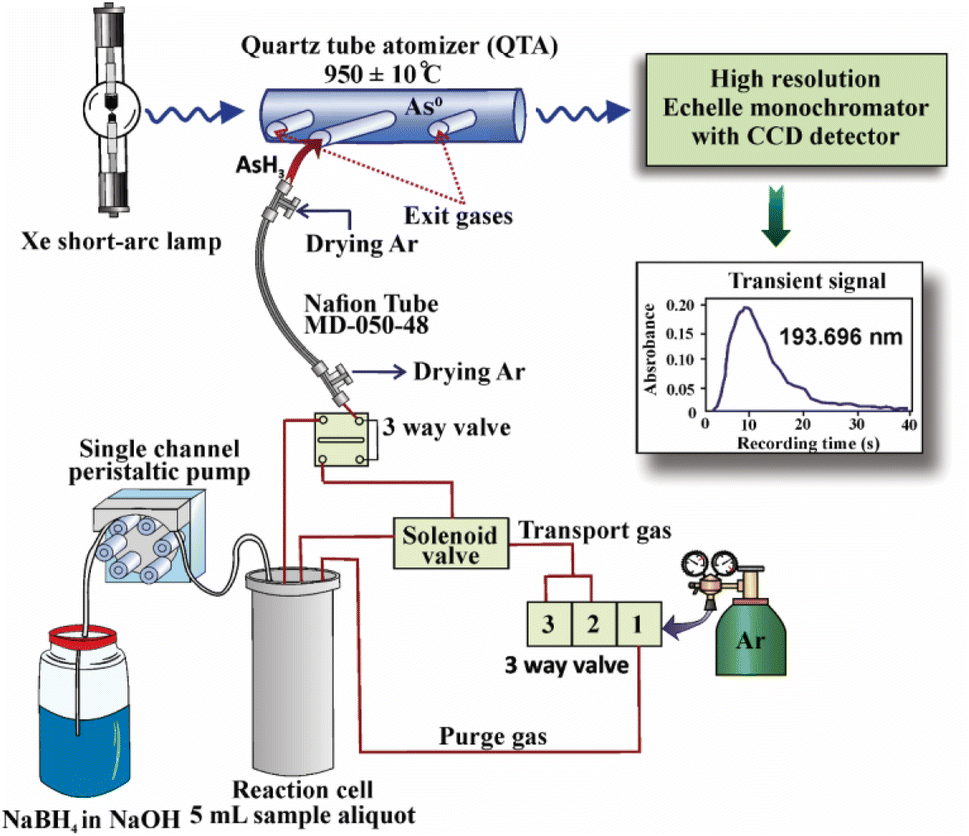 | ||
| Fig. 1 Scheme of the HG-HR-CS-QTAAS system. | ||
During the optimization of the HG-HR-CS-QTAAS method no extra hydrogen and oxygen flows were used. The reaction cell-quartz atomizer assembly was purged with 6 L h−1 Ar for 60 s after sample introduction in the cell and before adding the NaBH4 solution.
Also, the Ar–arsine stream containing traces of H2 and O2 was dried through Perma Pure MD-050-48 Nafion membrane tubing, Chromoservis (Praha, Czech Republic), which removed more than 85% of the water vapor (Target Dew Point of at least −8 °C).
A ContrAA 300 spectrometer equipped with an air-acetylene flame was used for the multielement analysis of the sample matrices. A Berghof MWS3+ system (Berghof, Germany) was used for the microwave-assisted digestion of the samples for tAs determination.
Reagents and solutions
Hydrochloric acid 30% (m/m) Suprapur for As determination (≤0.001 mg L−1 As), nitric acid Ultrapur (60%), hydrobromic acid Suprapur 47% (m/m), hydrogen peroxide 30% (m/m) for analysis, L-cysteine for biochemistry (>99.5%), NaBH4 for analysis (<0.001% As), NaOH (>99%), arsenic(V) ICP standard (1000 mg L−1 in 2–3% HNO3), hydrazine sulfate (≥99%, As < 0.0001%), KI Suprapur (≥99.995%), ascorbic acid (>99%), and dried toluene (max. 0.005% H2O) were purchased from Merck (Darmstadt, Germany). Silicone Antifoam 30% in H2O emulsion from Sigma Aldrich (Taufkirchen, Germany) was used to prevent foaming in the HG stage, especially in the case of sample preparation for iAs determination without separation. Ultrapure water (18 MΩ cm) prepared with a Milli-Q water purification system Millipore (Bedford, USA) was used throughout. The solutions needed in the procedure using L-cysteine proposed in this study were prepared, as well as solutions necessary for the procedure developed in the interlaboratory studies organized by the EC for the determination of tAs and iAs. This protocol involves separation by liquid–liquid extraction in a chloroform–HCl solution system, derivatization to arsine and determination by HG-AAS with a line source.52,53Solutions of 10 mol L−1 HCl and 0.28 mol L−1 HNO3 (2% v/v) as extraction reagents of iAs from food samples were prepared. A stock solution of 0.01 mol L−1 HCl (pH = 2.00 ± 0.01, adjusted by potentiometric titration) was prepared as extraction reagent, arsine generation medium in the presence of L-cysteine, and preparation of arsenic standards and CRMs/test samples. A stock solution of 10% (m/v) L-cysteine in 0.01 mol L−1 HCl (pH = 2.00 ± 0.01) was prepared as a prereductant of As(V) to As(III). Arsenic(III) calibration standards (0–10 μg L−1; n = 8) were prepared by mixing appropriate aliquot volumes of 100 μg L−1 As(V) with 1 mL 10% L-cysteine (pH = 2.00 ± 0.01) and heating on a water bath at 90 °C ± 5 °C for 10 min for the prereduction of As(V) to As(III). A final dilution to 50 mL was performed with HCl solution (pH = 2.00 ± 0.01). A solution of 0.6% (m/v) NaBH4 stabilized in 0.01% (m/v) NaOH in the presence of 0.05% (v/v) antifoam agent was prepared and used as the derivatization reagent to arsine in the approach based on L-cysteine in 0.01 mol L−1 HCl. A solution of 0.2% L-cysteine in HCl (pH = 2.00 ± 0.01) was used as the blank.
A solution containing 5% (m/v) KI and 5% (m/v) ascorbic acid and a solution of 15 mol L−1 hydrazine sulfate were prepared as prereductants of As(V) to As(III).52,53 Arsenic(III) calibration standards (0–10 μg L−1; n = 8) were also prepared by pre-reduction with 10 mL of 5% KI and 5% ascorbic acid in the presence of 25 mL of 37% HCl for 30 min at room temperature, and subsequently dilution to 50 mL with ultrapure water. The protocol was developed in the interlaboratory study for the determination of iAs after extraction in chloroform–1 mol L−1 HCl.53 An appropriate solution containing 1% (m/v) KI and 1% (m/v) ascorbic acid in 50% (v/v) HCl was used as the blank. A solution of 1% (m/v) NaBH4 in 0.33% (m/v) NaOH was used for arsine generation by the procedure described in the literature.52,53
Cross contamination was avoided by washing the reaction cell of the hydride generator with ultrapure water between samples. The PTFE digestion vessels were decontaminated using 5 mL 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 HNO3 and running the same thermal program as for sample digestion for tAs determination. All glassware and sample storage vessels were cleaned by keeping filled with 5% (v/v) HNO3 for 24 h, and then rinsing with ultrapure water. The QTA and end windows were periodically thoroughly cleaned by immersion in chromic acid cleaning solution, followed by rinsing with ultrapure water.
:1 HNO3 and running the same thermal program as for sample digestion for tAs determination. All glassware and sample storage vessels were cleaned by keeping filled with 5% (v/v) HNO3 for 24 h, and then rinsing with ultrapure water. The QTA and end windows were periodically thoroughly cleaned by immersion in chromic acid cleaning solution, followed by rinsing with ultrapure water.
Certified reference materials and test samples
The accuracy of the HG-HR-CS-QTAAS method was checked by analyzing several CRMs or standard reference materials (SRMs) available for tAs and iAs, matching as closely as possible the matrix of the test samples. Also, iAs was quantified in CRMs in which this fraction was not certified and the results were compared to the values found by other authors using various methods for sample preparation and analysis.3,4,13,25,33,52,53,59 The following CRMs were analyzed for the internal validation of the analytical procedure: ERM-BB422 Fish Muscle, BCR-627 Tuna Fish Tissue, ERM-CE278k Mussel Tissue, ERM-BC211 Rice, from the Institute for Reference Materials and Measurements – IRMM (Geel, Belgium), SRM 2976 Mussel Tissue from National Institute of Standards and Technology (Gaithersburg, USA), CS-M-3 Mushroom Powder from Institute of Nuclear Chemistry and Technology (Warsaw, Poland), Tort-2 Lobster Hepatopancreas Reference Material for Trace Metals from National Research Council Canada (Ottawa, Ontario Canada), and IAEA-359 Cabbage from International Atomic Energy Agency (Vienna, Austria).The versatility of the HG-HR-CS-QTAAS method for tAs and iAs determination was demonstrated by analyzing samples of fish muscle (4), pork and chicken meat and liver (4), rice and infant food preparations with rice, meat and vegetables (6).
Sample preparation for tAs determination
The preliminary sample preparation and microwave-assisted sample digestion procedure for tAs determination were similar to that used for Hg determination in foodstuffs.60 Briefly, 0.2–0.5 g CRM or test food sample was digested in a mixture of 9 mL 65% HNO3 and 3 mL 30% H2O2, and the resulting solutions were made up to 25 mL with ultrapure water. Prereduction of As(V) to As(III) in an aliquot volume of 5–20 mL was achieved with 0.5 mL 10% L-cysteine in HCl (pH = 2.00 ± 0.01) by heating in a water bath at 90 °C ± 5 °C for 10 min. After cooling, the solutions were diluted to 25 mL with HCl (pH = 2.00 ± 0.01). Finally, the pH was carefully adjusted by potentiometric titration using either HCl solution or 10% NaOH. The final concentration of L-cysteine in the sample was 0.2% (m/v). Serial dilutions were carried out with HCl (pH = 2.00 ± 0.01) when necessary to enable the final As concentration to fit the calibration range. The tAs concentration was determined with the external calibration method using 5 mL sample mixed in the reaction cell with 3.5 mL of 0.6% (m/v) NaBH4 stabilized in 0.01% (m/v) NaOH.Sample preparation for iAs determination
Several preparation procedures were tested for iAs determination in foodstuffs, as schematically illustrated in the ESI (Section 2, Fig. S1†).| Parameter | Setting |
|---|---|
| Temperature | 90 °C ± 5 °C |
| Heating time | 10 min |
| L-Cysteine concentration | 0.2% (m/v) |
| HCl concentration | 0.01 mol L−1 (pH = 2.00 ± 0.01) |
| Parameter | Setting |
|---|---|
| Analytical wavelength | As 193.696 nm |
| Signal measurement | Peak height or peak area |
| Number of pixels associated to As line | 5 (CP ± 2) |
| Temperature of the quartz atomizer | 950 °C ± 10 °C |
| Ar flow rate | 6 L h−1 |
| Spectrum recording time for peak height/peak area measurement | 20/40 s |
| Purging time of reaction cell with Ar before NaBH4 addition | 60 s |
| NaBH4 concentration | 0.6% (m/v) in 0.01% (m/v) NaOH and 0.05% (v/v) antifoam agent |
| Volume of NaBH4 solution/pumping time | 2–8 mL/5–18 s (optimum 3.5 mL/8 s) |
| HCl concentration in sample | 0.01 mol L−1 (pH = 2.00 ± 0.01) |
| Sample volume | 5 mL |
| Calibration | External using As(III) standard solutions and standard addition method |
| Concentration of As(III) in standards | 0; 0.1; 0.2; 0.5; 1; 2; 5; 10 μg L−1 in HCl (pH = 2.00 ± 0.01) and 0.2% (m/v) L-cysteine |
| Number of repeated measurements of standards and samples | 5 |
In all cases, the determination of iAs was performed using 5 aliquots of 5 mL sample, each mixed with 3.5 mL NaBH4 solution in the reaction cell of the hydride generator. A blank correction was made using an appropriate solution according to the sample preparation procedures.
Method validation
The HG-HR-CS-QTAAS method was validated for the determination of tAs and iAs in terms of LOD, accuracy and precision, inter-day reproducibility and non-spectral matrix effects. The instrumental LOD was evaluated according to the 3σ criterion, LOD = 3sb/m, where m is the slope of the calibration curve and sb is the standard deviation of the background signal (n = 11) of appropriate blank solutions. The LODs for tAs and iAs in foodstuffs were calculated according to the sample preparation procedures. The inter-day reproducibility was assessed based on the parameters of the calibration curve and the LODs achieved over one month. The accuracy of the method was evaluated by recovery and the corresponding extended uncertainty in the laboratory (Ulab = k × uc for k = 2, where uc is the combined standard uncertainty) of tAs and iAs in CRMs with certified values for both fractions. The combined uncertainty was calculated according to the uncertainty propagation law taking into account the stages for sample preparation and analysis (sample weighing, calibration standards and sample preparation, calibration curve fitting, aliquot analysis and uncertainty from the CRM certificate). Furthermore, the CRMs with no certified value for iAs were also analyzed, and the accuracy was checked by comparison with the results reported by other authors who used similar or different methods.4,13 The bias was checked based on the difference between the certified and found mean values in CRMs (Δm) for each of the five sample preparation procedures and both calibration methods. There is no bias when Δm < U and Δm < Ulab, where U and Ulab are the extended uncertainty provided in the certificate and that obtained in the laboratory, respectively. The analysis of the aliquots and the fitting of the calibration curve had the greatest influence in the combined uncertainty for the tAs and iAs determination methods. Tukey's method was used for the multiple comparison of the methods considering the found means and their variances (p < 0.05).65 Also, Dunnett's method was used for multiple comparison of the found means through the used methods and the certified mean, which was considered as the control (p < 0.05).66 In the case of the test samples, the significant differences (p < 0.05) between the results obtained for the sample preparation procedures and the two calibration modes were also checked using Tukey's method. Means significantly different from each other or from the certified mean (p < 0.05) were not considered in the calculation of the pooled mean and recovery and their expanded uncertainties. Compliance with AOAC guidelines was also considered.67 Data analysis was made using the Statistica 12.0 software. The presence or absence of non-spectral effects coming from the mineral matrix was tested by comparing the recoveries obtained using the external calibration and standard addition method, respectively. In all the samples, the concentrations of the elements attributed to mineral matrix were determined using HR-CS-FAAS in acetylene-air flame.Results and discussion
Optimization of the arsine generation from As(III) species and operation of the HG-HR-CS-QTAAS instrument
The optimization of arsine generation was performed in terms of concentration of HCl solution as the reaction medium, L-cysteine as the reductant, NaBH4 solution stabilized in NaOH as the derivatization reagent, content of antifoam agent in NaBH4 and sample-to-NaBH4 solution volumes ratio. The optimization criterion was the highest absorption signal of As 193.696 nm line as the peak height and integrated absorbance. Optimization was done on 5 mL aliquots of solution containing 2 μg L−1 As(III), a flow rate of 6 L h−1 Ar purge flow and a temperature of 950 °C ± 10 °C in a quartz atomizer. These experimental conditions provided efficient arsine generation, purging and atomization. The contents of hydrogen and oxygen in the Ar–arsine flow were not determined, considering that the traces of hydrogen generated by the decomposition of NaBH4 and the O2 purged from the liquid sample, after mixing with NaBH4, were sufficient.The optimal conditions for the pre-reduction of As(V) to As(III) and arsine generation are summarized in Table 1, while the operating conditions of the HG-HR-CS-QTAAS analytical system are presented in Table 2. Under these conditions, the HG-HR-CS-QTAAS method showed good capability for As quantification without the need for preconcentration and versatility in terms of matrix type. The optimization study revealed that some variables related to arsine generation from As(III) were critical for the sensitivity. More details on the influence of the experimental parameters on the As absorption are provided in the ESI (Section 3, Fig. S2–S7†).
Although the analytical procedure requires rigorous control of some variables related to arsine generation (pH/HCl concentration, L-cysteine concentration and NaOH concentration in NaBH4 solution), it exhibits the advantage of a substantial reduction in HCl consumption, which is a very expensive reagent. Thus, the procedure based on L-cysteine is very attractive both from analytical and economic perspective.
The absorbance measurement in peak height mode was preferable, given that the maximum signal appeared within only 10–15 s compared to 40 s in the case of integrated absorbance, which shortened the analysis time. Besides, the LOD was not better when using the peak area measurement because both the sensitivity and signal fluctuation increased, and thus the 3sb/m ratio remained almost constant.
Figures of merit of the HG-HR-CS-QTAAS method
The parameters of the calibration curve and LOD (3σ criterion) obtained in an inter-day reproducibility study from peak height measurements of absorbance under the optimal conditions for arsine generation (HCl solution pH = 2.00 ± 0.01, 0.2% L-cysteine, and 5 mL sample aliquot) are presented in the ESI (Section 4, Table S1†). The LOD for As in solution was assessed using the standard deviation of the blank signal (n = 11) and the slope of the calibration curve. In foodstuffs, the LODs for tAs and iAs were calculated based on the sample preparation protocol. An example of the calibration curve and LOD obtained according to the 3σ criterion is presented in the ESI (Section 4, Fig. S8†), while the blank signals (0.2% L-cysteine solution in 0.01 mol L−1 HCl) are presented in Table S2.† The data in Table S1† indicate the good linearity of the calibration curve and good long-term reproducibility at the 95% confidence level for 25 days (intercept 0.0018 ± 0.0007 a.u.; slope 0.0183 ± 0.0006 L μg−1 and R2 = 0.9986 ± 0.0005). Thus, the relative standard deviations (RSDs) of the slope and LOD were better than 8% and 18%, respectively.The LOD for tAs/iAs in solution in the HG-HR-CS-QTAAS method was 0.070 ± 0.005 μg L−1, which was about 145 times lower than the maximum admitted concentration in drinking water (10 μg L−1 As). Under the same measurement conditions, LOD for tAs in foodstuffs (fish muscle, chicken and pork meat, organs, mushrooms, rice and baby food) was 0.0044 ± 0.0005 mg kg−1 As (0.5 g digested sample made up to 25 mL and dilution of 20 mL aliquot digest to 25 mL).
In different countries, the content of As is regulated in the range of 1–3.5 mg kg−1 tAs and 0.1–0.5 mg kg−1 iAs, respectively, in foods of marine origin (fish, molluscs, seaweed, and crustacea), organs and pork muscle, chicken and turkey, cereals (rice and grains), fresh vegetables (mushrooms), fruit juices, chocolate and chocolate products.4 Only in few foods both iAs and tAs are regulated, while in most cases either tAs or iAs is standardized, which can cause confusion. Unfortunately, the values differ substantially globally and there is no consensus concerning As regulation. However, iAs, which is much more toxic, is controlled in rice intended for the production of food for infants and young children to be in the range of 0.1–0.3 mg kg−1 iAs.5 In this context, certain judgments can be made regarding the applicability of the unified method based on HG-HR-CS-QTAAS for the determination of iAs and tAs in food. For the method proposed by us, the LOD for iAs in food samples was 0.0022 ± 0.0003 mg kg−1 (1 g extracted sample, made up to 25 mL and dilution of 20 mL aliquot solution to 25 mL), up to 46-times lower than the admitted values in rice and rice-based products for infants, while the LOD for tAs (0.0044 ± 0.0005 mg kg−1) was at least 23-times lower than the admitted values in foods. The iAs fraction has gained increasing interest in recent years not only in rice and baby food, but also in other foods of vegetable and animal origin, such as mushrooms, fish muscle and organelles, known as important sources of As exposure to the humans.4
Increasing the volume of the analyzed sample from 5 to 10 mL led to a 2-fold improvement in sensitivity and LOD. However, we chose to employ a 5 mL sample aliquot to reduce the consumption of reagents and amount of waste.
A comparison of the LODs for As in HG-HR-CS-QTAAS and other methods is available in the ESI (Section 5, Table S3†). According to the data in Table S3,† the HG-HR-CS-QTAAS without arsine preconcentration provided better LODs than GFAAS, HG-AAS with a line source, ICP-MS, and methods currently used for the determination of As. Compared with the conventional line-source AAS, HR-CS-AAS enables visualization of the spectral environment in the vicinity of the analytical line and facilitates the control of spectral interferences and improved background correction. The examination of the spectral range in the vicinity of the As 193.696 nm line (±0.1 nm) enabled the effective removal of the interferences coming from oxygen and water vapor contained in the QTA. Thereby, it was possible to achieve very good LODs for both iAs and tAs by HG-HR-CS-QFAAS without preconcentration. This method has proven to be versatile for the analysis of a broad variety of complex matrices, which is the case for foods. Alternatively, the LODs were poorer than that obtained by HG and different detection methods coupled with cryotrapping, liquid–liquid and solid-phase microextraction preconcentration coupled with HG, or direct solid sample analysis by HR-CS-GFAAS with minimal sample processing.
Validation of the HG-HR-CS-QTAAS method for tAs determination in CRMs
The data for the accuracy of the HG-HR-CS-QTAAS method for tAs determination in CRMs by external calibration are presented in Table 3. In all the analyzed samples, the difference between the certified and found values was lower than the extended standard uncertainties for the certified and found values (k = 2). Specifically, the determination of tAs by HG-HR-CS-QTAAS is not affected by systematic errors. The pooled recovery was in the range of 98% ± 22% (k = 2), complying with the AOAC guide related to the determination of As. The minimum acceptance criterion for recovery is 60–115% and 80–110% for As in the range of ≤0.01–0.1 and 0.1–10 mg kg−1 As.67| CRM | Certified value ± Ua (mg kg−1) | Found value ± Ulabb (mg kg−1) | Recovery ± Ulabb (%) |
|---|---|---|---|
| a U – is the expanded uncertainty (k = 2; 95% confidence level). b U lab – is the expanded uncertainty in laboratory (k = 2, n = 5 parallel measurements and 95% confidence level). | |||
| ERM-BC211 rice | 0.260 ± 0.013 | 0.262 ± 0.029 | 101 ± 11 |
| ERM-CE278k Mussel Tissue | 6.7 ± 0.4 | 6.7 ± 0.8 | 100 ± 12 |
| BCR 627 Tuna Fish Tissue | 4.8 ± 0.3 | 4.7 ± 0.7 | 98 ± 15 |
| SRM 2976 Mussel Tissue | 13.3 ± 1.8 | 12.1 ± 2.6 | 91 ± 21 |
| IAEA-359 Cabbage | 0.10 ± 0.04 | 0.10 ± 0.05 | 100 ± 50 |
| Tort-2 Lobster Hepatopancreas | 21.6 ± 1.8 | 20.6 ± 3.1 | 95 ± 15 |
| CSM-3 Mushroom Powder | 0.651 ± 0.026 | 0.662 ± 0.076 | 102 ± 11 |
| ERM-BB422 Fish muscle | 12.7 ± 0.7 | 12.0 ± 1.5 | 94 ± 13 |
| Pooled recovery ± CIb (%) | 98 ± 22 | ||
The robustness of the HG-HR-CS-QTAAS method for tAs determination in terms of non-spectral interference caused by the mineral matrix was assessed by analyzing the same samples diluted 2–1000 times according to the case. No significant differences (95% confidence level) were found between the results obtained for different dilution factors. Consequently, the optimal conditions for AsH3 generation established for standards containing only As(III) were found to be suitable for tAs determination by HG-HR-CS-QTAAS regardless of the food type, which considerably simplified the analytical procedure.
The composition of the mineral matrix causing no non-spectral interferences in the determination of tAs in CRMs and real samples is presented in ESI (Section 6, Table S4†). The lack of matrix effects in the liquid phase from transition metal ions (e.g., Fe, Cr, Mn, Co, Ni, and Zn) was due to their complexation with L-cysteine, which decreased their oxidation capability and reactivity towards NaBH4.
Validation of the HG-HR-CS-QTAAS method for iAs determination in CRMs
The results obtained for the accuracy of the HG-HR-CS-QTAAS method for the determination of iAs in foods with different matrices after extraction in various reagents with or without the separation of iAs by double liquid–liquid extraction in toluene are presented in Table 4. We compared our results with the certified value for iAs when available, as in the case of ERM-BC211 Rice, while for the other CRMs presented in Table 4, we used the values reported in collaborative trials organized by the EC or published data in review articles. These reference results were obtained using different extraction procedures and reagents (water, water–methanol mixture, NaOH in ethanol, HCl, HNO3, and HCl with separation in chloroform), and detection by various hyphenated techniques and non-chromatographic methods based on derivatization.4,13,25,53 The comparison of the measurement results with the certified values by Dunnett's method and the recoveries are presented in the ESI (Section 7, Tables S7 and S8†). Tukey's method indicated that there were no statistically significant differences between the results obtained for the employed preparation and analysis methods in the case of ERM-BC211 Rice, ERM-278k Mussel Tissue, BCR-627 Tuna Fish Tissue, and SRM 2976 Mussel Tissue (p < 0.05).| CRM | Certified or reference value ± Ua (mg kg−1) | Calib. method | Found value ± Ulabb (mg kg−1) | |||||
|---|---|---|---|---|---|---|---|---|
| Extraction in 10 mol L−1 HCl | Extraction in 0.28 mol L−1 HNO3 | Extraction in 0.01 mol L−1 HCl | Pooled results | |||||
| IMEP-41 procedurec | Separation by extraction in toluened | Without separationd | Without separationd | Without separationd | ||||
| a U is the expanded uncertainty (k = 2, 95% confidence level). b U lab – is the expanded uncertainty in laboratory (k = 2, n = 5 parallel measurements and 95% confidence level). c IMEP-41 procedure, prereduction with HBr and hydrazine sulfate, separation of iAs in toluene–1 mol L−1 HCl system, derivatization to arsine with NaBH4 in 1 mol L−1 HCl.25,53 d Prereduction with L-cysteine and derivatization to arsine with NaBH4 in 0.01 mol L−1 HCl and 0.2% L-cysteine. e Reference values from ref. 4, 25 and 53. f Reference values from ref. 59. g Reference values calculated as mean according to the results centralized by Petursdottir et al.3,13,33 h Reference values from ref. 52. i Significant differences (p < 0.05) using Tukey's method. | ||||||||
| ERM-BC211 Rice | 0.124 ± 0.011 | Ext. | 0.122 ± 0.025 | 0.117 ± 0.023 | 0.120 ± 0.016 | 0.117 ± 0.022 | 0.125 ± 0.037 | 0.120 ± 0.027 |
| Std. Ad. | 0.131 ± 0.019 | 0.114 ± 0.025 | 0.125 ± 0.026 | 0.114 ± 0.025 | 0.117 ± 0.044 | |||
| ERM-CE278k Mussel Tissue | 0.086 ± 0.008e | Ext. | 0.086 ± 0.009 | 0.081 ± 0.025 | 0.096 ± 0.016 | 0.105 ± 0.026 | 0.095 ± 0.026 | 0.094 ± 0.024 |
| 0.133 ± 0.048e | Std. Ad. | 0.085 ± 0.013 | 0.080 ± 0.026 | 0.105 ± 0.025 | 0.101 ± 0.033 | 0.101 ± 0.028 | ||
| BCR-627 Tuna Fish Tissue | 0.063 ± 0.027e | Ext. | 0.054 ± 0.014 | 0.054 ± 0.014 | 0.065 ± 0.014 | 0.055 ± 0.010 | 0.065 ± 0.008 | 0.060 ± 0.013 |
| Std. Ad. | 0.057 ± 0.011 | 0.054 ± 0.011 | 0.068 ± 0.014 | 0.065 ± 0.019 | 0.062 ± 0.015 | |||
| SRM 2976 Mussel Tissue | 0.110 ± 0.013f | Ext. | 0.101 ± 0.036 | 0.096 ± 0.034 | 0.110 ± 0.030 | 0.106 ± 0.030 | 0.109 ± 0.030 | 0.104 ± 0.027 |
| Std. Ad. | 0.098 ± 0.023 | 0.112 ± 0.031 | 0.105 ± 0.019 | 0.102 ± 0.014 | 0.103 ± 0.012 | |||
| IAEA-359 Cabbage | 0.091 ± 0.016e | Ext. | 0.092 ± 0.020 | 0.087 ± 0.018 | 0.081 ± 0.028 | 0.088 ± 0.017 | 0.108 ± 0.028 | 0.092 ± 0.024 |
| 0.074 ± 0.033e | Std. Ad. | 0.085 ± 0.026 | 0.102 ± 0.028 | 0.085 ± 0.026 | 0.092 ± 0.025 | 0.106 ± 0.028 | ||
| Tort-2 Lobster Hepatopancreas | 0.71 ± 0.04f | Ext. | 0.504 ± 0.090 | 0.500 ± 0.115 | 0.655 ± 0.135 | 0.615 ± 0.125 | 0.620 ± 0.135 | 0.555 ± 0.123 |
| 0.582 ± 0.081g | ||||||||
| 0.615 ± 0.086g | Std. Ad. | 0.493 ± 0.120 i | 0.510 ± 0.090 | 0.615 ± 0.115 | 0.525 ± 0.165 | 0.610 ± 0.128 | ||
| 0.544 ± 0.162h | ||||||||
| CS-M-3 Mushroom | Ext. | 0.378 ± 0.072 | 0.385 ± 0.073 | 0.375 ± 0.068 | 0.354 ± 0.081 | 0.364 ± 0.077 | 0.368 ± 0.130 | |
| Std. Ad. | 0.354 ± 0.204 | 0.352 ± 0.212 | 0.374 ± 0.164 | 0.360 ± 0.108 | 0.380 ± 0.129 | |||
| Pooled recovery (%) | Ext. | 92 ± 22 | 90 ± 26 | 107 ± 19 | 102 ± 21 | 107 ± 24 | 100 ± 23 | |
| Std. Ad. | 92 ± 22 | 94 ± 24 | 105 ± 22 | 97 ± 28 | 104 ± 25 | 98 ± 24 | ||
| 92 ± 22 | 92 ± 25 | 106 ± 21 | 100 ± 25 | 106 ± 25 | 99 ± 24 | |||
The analysis of the ERM-BC211 Rice sample provided a recovery in the range of 92–106% with trueness of 13–38% (k = 2) and no significant differences were observed between the results obtained by the external calibration and standard addition. Dunnett's method indicated there was no statistically significant difference, irrespective of the sample preparation and analysis against the certified value with 97% recovery and 21–25% trueness (p > 0.05).
In the case of the CRMs with no certified iAs content as shown above, we used the reference values calculated by us based on the centralized results reported by Llorente-Mirandes et al.,4 namely, ERM-CE278K Mussel Tissue (0.086 ± 0.008 mg kg−1),4,25,53 BCR-627 Tuna Fish (0.063 ± 0.027 mg kg−1)4,25,53 and SRM 2976 Mussel Tissue (0.110 ± 0.013 mg kg−1).59 Tukey's and Dunnett's methods did not show significant differences between the results of our methods and reference results, respectively. Compared to these values, our analyses gave pooled recovery in the range of 93–110% and trueness of 20–27%. In a collaborative trial to assess the accuracy of HG-AAS for iAs determination, the reported result for the analysis of ERM-CE278K Mussel Tissue by alternative hyphenated techniques was 0.133 ± 0.048 mg kg−1 iAs with recovery in the range of 153.7% ± 57.6%.4,53 Compared to this reference value, our results were significantly different (p < 0.05) and were not included in the calculation of the pooled recovery.
In the case of IAEA-359 Cabbage, a significant difference between the results obtained using the extraction of iAs in 10 mol L−1 HCl and in 0.01 mol L−1 HCl, without separation, external calibration, and prereduction with 0.2% L-cysteine followed by derivatization to arsine in 0.01 mol L−1 HCl and 0.2% L-cysteine was observed (marked in bold in Table 4). In the case of IAEA-359 Cabbage, Dunnett's method did not reveal any significant difference with the result reported in the IMEP-41 collaborative trial based on HG-AAS, extraction in concentrated HCl, selective separation of iAs in a chloroform–1 mol L−1 HCl system, prereduction with HBr and hydrazine sulfate and derivatization with NaBH4 in 1 mol L−1 HCl.25,53 Consequently, the pooled recovery and trueness were in the range of 103% ± 22% for external calibration and 103% ± 28% for standard addition, respectively. The comparison of the amount of 0.091 ± 0.016 mg kg−1 iAs reported in the IMEP-41 by HG-AAS with that found by hyphenated techniques of 0.074 ± 0.033 mg kg−1 gave the recovery of 81.6% ± 38.7%.24,25,51,53 In this case, Dunnett's method revealed significant differences for extraction in 10 mol L−1 HCl and separation in toluene (standard addition) and extraction in 0.01 mol L−1 HCl without separation (external and standard addition method). The significant difference for this reference value and wide confidence interval for accuracy were attributed to the polyatomic interference of 40Ar35Cl+ on the 75As+ isotope in the HG-ICP-MS method, foaming of sample during HG, or the fact that the chloroform was not purified by filtration before the back-extraction in HCl solution.
In the case of Tort-2 Lobster Hepatopancreas, one of the most frequently analyzed CRMs for the iAs fraction, a significant difference was also observed between the results obtained for extraction in 10 mol L−1 HCl, as previously described (external calibration), and other methods and highlighted in bold in Table 4. Therefore, the results obtained by extraction in 10 mol L−1 HCl without separation in toluene, prereduction and derivatization with L-cysteine and external calibration were not included in the mean calculation. Our method gave recoveries in the range of 85–113% and 80–107%, and trueness of 18–31% against the average concentrations of 0.582 ± 0.081/0.615 ± 0.086 mg kg−1 reported by Petursdottir et al. by hyphenated techniques and HG-AAS with/without separation in chloroform.13 A similar recovery in the range of 91–120% and trueness of 18–31% were obtained versus 0.544 ± 0.162 mg kg−1 iAs reported in the IMEP-32 collaborative trial organized by the EC, aiming to validate the HG-AAS method for the determination of iAs in seafood.52 In all cases, Tukey's and Dunnett's methods revealed no significant differences related to the results previously mentioned. Instead, significant differences appeared between our obtained by applying the IMEP-41 procedure and that based on extraction in 10 mol L−1 HCl, separation in toluene and derivatization in the presence of L-cysteine for both calibration approaches and the method using extraction in 0.28 mol L−1 HNO3 (without separation and standard addition) versus the reference value of 0.71 ± 0.04 mg kg−1 obtained by LC-ICP-MS.59
The concentrations of iAs in the range 0.352–0.385 mg kg−1 found by us in CRM CS-M-3 Mushroom Powder were similar to that reported in the IMEP-116 collaborative trial organized by the EC and were not significantly different among methods, according to Tukey's test.55
It should be noted that both sample preparation procedures using extraction in 0.28 mol L−1 (2% v/v) HNO3 or 0.01 mol L−1 HCl, especially the latter using 0.01 mol L−1 HCl for derivatization to arsine are more economical than the procedures proposed in the collaborative trials based on extraction in 10 mol L−1 HCl and derivatization in 1 mol L−1 HCl.25,53 In addition, the two preparation procedures did not lead to non-spectral interferences from the mineral matrix for the reasons shown in the case of tAs determination. The recoveries of iAs in the food samples were between 93% ± 26% and 111% ± 25% by external calibration, similar to that in the standard addition approach (94% ± 24% and 109% ± 25%) and that obtained in the extraction in concentrated HCl with or without separation in toluene (93% ± 26%, 99% ± 24%, 108% ± 18% and 106% ± 23%), (k = 2). The only problem in the extraction procedures in concentrated or diluted HCl was sample foaming during HG as a result of the reaction between proteins and NaBH4, when the separation in toluene was not applied. As shown above, foaming was eliminated by the addition of 0.05% (v/v) antifoaming agent in NaBH4 solution. The composition of the mineral matrix in the CRMs and food test samples is presented in the ESI (Section 6, Tables S4–S6†).
Analysis of real samples using HG-HR-CS-QTAAS
Once validated, the HG-HR-CS-QTAAS method was applied for the analysis of common foods of animal and vegetable origin, which can be sources of iAs in the human body. Sample preparation and derivatization to arsine were carried out similar to CRMs. The results for tAs and iAs are presented in Table 5. No significant differences were found between the results obtained by the procedures used for sample preparation, prereduction and derivatization according to Tukey's method (p < 0.05). For iAs determination in foods, the extraction in 0.28 mol L−1 HNO3 or 0.01 mol L−1 HCl, without separation in toluene, and then derivatization in 0.01 mol L−1 HCl medium in the presence of 0.2% (m/v) L-cysteine was the best choice. The precision of the tAs and iAs measurements by HG-HR-CS-QTAAS based on combined standard uncertainties (uc = Ulab/2) was 8.7–32.0%. The HG-HR-CS-QTAAS method fulfils the criteria in the AOAC guide for the quantification of As, given that the precision was generally better than 30%.67 The weight of iAs from tAs in food was 19% ± 15% in the rice and rice-based preparations for children, 14% ± 11% in foods of terrestrial animal origin and 5% ± 4% in fish muscle (ESI, Section 8, Fig. S9†). It has been previously reported that rice mostly contains dimethylarsinic acid (DMA), which is weakly reactive in the derivatization stage, enabling the selective determination of iAs without mutual interference from DMA in the derivatization step.23,31,33,63,64 It has been also shown that rice does not contain monomethylarsonic acid (MMA), which is more reactive to derivatization, or its fraction is insignificant, causing no interference in the determination of iAs in this type of food in HG-based procedures. Thus, good agreement was found between the results for iAs by HG-AAS and hyphenated HPLC-ICP-MS techniques.22,31,33,63,64 Arsenic is contained in fish mainly as non-toxic organic species (arsenobetaine), which is not reactive to derivatization, provided that the As–carbon bond is not broken.13| Sample | Calib. method | tAs | iAs | |||||
|---|---|---|---|---|---|---|---|---|
| Extraction in 10 mol L−1 HCl | Extraction in 0.28 mol L−1 HNO3 | Extraction in 0.01 mol L−1 HCl | Pooled results | |||||
| IMEP-41 procedureb | Separation by extraction in toluenec | Without separationc | Without separationc | Without separationc | ||||
| a U lab – is the expanded uncertainty in laboratory (k = 2, n = 5 parallel measurements and 95% confidence level). b IMEP-41 procedure, prereduction with HBr and hydrazine sulfate, separation of iAs in toluene–1 mol L−1 HCl system, derivatization to arsine with NaBH4 in 1 mol L−1 HCl.25,53 c Prereduction with L-cysteine and derivatization to arsine with NaBH4 in 0.01 mol L−1 HCl and 0.2% L-cysteine. d PP1 – rice with carrot and vitamins. e PP2 – organic preparation with rice and corn flour, tapioca and vitamins. f PP3 – rice with vegetables, chicken, and rapeseed. g RSD – is the relative standard deviation for combined uncertainty and n = 5 parallel measurements. | ||||||||
| Fish muscle | ||||||||
| Tilapia | Ext. | 0.18 ± 0.05 | 0.029 ± 0.012 | 0.023 ± 0.006 | 0.022 ± 0.011 | 0.027 ± 0.006 | 0.026 ± 0.006 | 0.025 ± 0.009 |
| Std. Ad. | 0.027 ± 0.013 | 0.027 ± 0.014 | 0.027 ± 0.013 | 0.025 ± 0.016 | 0.028 ± 0.013 | 0.027 ± 0.014 | ||
| Hake | Ext. | 5.29 ± 1.36 | 0.044 ± 0.018 | 0.041 ± 0.018 | 0.041 ± 0.016 | 0.042 ± 0.017 | 0.045 ± 0.018 | 0.043 ± 0.017 |
| Std. Ad. | 0.042 ± 0.022 | 0.049 ± 0.023 | 0.042 ± 0.022 | 0.048 ± 0.025 | 0.052 ± 0.020 | 0.047 ± 0.022 | ||
| Carp | Ext. | 0.37 ± 0.10 | 0.014 ± 0.005 | 0.012 ± 0.006 | 0.013 ± 0.005 | 0.018 ± 0.007 | 0.015 ± 0.006 | 0.014 ± 0.006 |
| Std. Ad. | 0.017 ± 0.009 | 0.015 ± 0.008 | 0.016 ± 0.008 | 0.016 ± 0.006 | 0.019 ± 0.010 | 0.017 ± 0.008 | ||
| Trout | Ext. | 0.53 ± 0.10 | 0.008 ± 0.004 | 0.010 ± 0.004 | 0.009 ± 0.004 | 0.010 ± 0.004 | 0.009 ± 0.004 | 0.009 ± 0.004 |
| Std. Ad. | 0.007 ± 0.003 | 0.007 ± 0.003 | 0.010 ± 0.005 | 0.008 ± 0.003 | 0.008 ± 0.003 | 0.008 ± 0.003 | ||
|
||||||||
| Meat | ||||||||
| Chicken 1 | Ext. | 0.095 ± 0.034 | 0.025 ± 0.008 | 0.024 ± 0.012 | 0.026 ± 0.008 | 0.018 ± 0.005 | 0.023 ± 0.007 | 0.023 ± 0.008 |
| Std. Ad. | 0.020 ± 0.011 | 0.021 ± 0.010 | 0.024 ± 0.007 | 0.019 ± 0.007 | 0.021 ± 0.008 | 0.021 ± 0.009 | ||
| Chicken 2 | Ext. | 0.052 ± 0.017 | 0.007 ± 0.004 | 0.007 ± 0.002 | 0.007 ± 0.002 | 0.005 ± 0.002 | 0.005 ± 0.002 | 0.006 ± 0.003 |
| Std. Ad. | 0.007 ± 0.003 | 0.008 ± 0.005 | 0.006 ± 0.003 | 0.006 ± 0.003 | 0.006 ± 0.003 | 0.007 ± 0.003 | ||
| Pork | Ext. | 0.131 ± 0.049 | 0.009 ± 0.003 | 0.008 ± 0.003 | 0.007 ± 0.002 | 0.010 ± 0.004 | 0.010 ± 0.003 | 0.009 ± 0.003 |
| Std. Ad. | 0.009 ± 0.004 | 0.010 ± 0.006 | 0.008 ± 0.003 | 0.009 ± 0.003 | 0.009 ± 0.003 | 0.009 ± 0.004 | ||
| Pork liver | Ext. | 0.141 ± 0.047 | 0.020 ± 0.006 | 0.022 ± 0.006 | 0.023 ± 0.005 | 0.020 ± 0.009 | 0.019 ± 0.008 | 0.021 ± 0.007 |
| Std. Ad. | 0.018 ± 0.007 | 0.024 ± 0.008 | 0.018 ± 0.008 | 0.018 ± 0.006 | 0.017 ± 0.006 | 0.019 ± 0.007 | ||
|
||||||||
| Rice and baby food | ||||||||
| Brown rice | Ext. | 0.30 ± 0.06 | 0.051 ± 0.013 | 0.054 ± 0.011 | 0.052 ± 0.009 | 0.056 ± 0.015 | 0.052 ± 0.017 | 0.053 ± 0.013 |
| Std. Ad. | 0.052 ± 0.022 | 0.053 ± 0.020 | 0.054 ± 0.014 | 0.050 ± 0.018 | 0.053 ± 0.029 | 0.052 ± 0.021 | ||
| White rice | Ext. | 0.17 ± 0.05 | 0.047 ± 0.014 | 0.062 ± 0.017 | 0.046 ± 0.011 | 0.041 ± 0.014 | 0.063 ± 0.031 | 0.052 ± 0.019 |
| Std. Ad. | 0.044 ± 0.020 | 0.057 ± 0.024 | 0.054 ± 0.026 | 0.046 ± 0.015 | 0.058 ± 0.018 | 0.052 ± 0.021 | ||
| White rice | Ext. | 0.22 ± 0.07 | 0.030 ± 0.013 | 0.026 ± 0.010 | 0.026 ± 0.016 | 0.022 ± 0.007 | 0.024 ± 0.009 | 0.026 ± 0.011 |
| Std. Ad. | 0.028 ± 0.011 | 0.027 ± 0.013 | 0.024 ± 0.006 | 0.023 ± 0.006 | 0.028 ± 0.013 | 0.026 ± 0.010 | ||
| PP1d | Ext. | 0.39 ± 0.09 | 0.048 ± 0.016 | 0.054 ± 0.015 | 0.048 ± 0.013 | 0.058 ± 0.016 | 0.046 ± 0.011 | 0.051 ± 0.014 |
| Std. Ad. | 0.041 ± 0.016 | 0.053 ± 0.022 | 0.043 ± 0.017 | 0.058 ± 0.028 | 0.044 ± 0.013 | 0.048 ± 0.020 | ||
| PP2e | Ext. | 0.12 ± 0.03 | 0.047 ± 0.014 | 0.048 ± 0.017 | 0.047 ± 0.011 | 0.045 ± 0.014 | 0.047 ± 0.019 | 0.047 ± 0.015 |
| Std. Ad. | 0.049 ± 0.016 | 0.053 ± 0.019 | 0.045 ± 0.014 | 0.051 ± 0.017 | 0.055 ± 0.024 | 0.051 ± 0.018 | ||
| PP3f | Ext. | 2.77 ± 0.79 | 0.019 ± 0.007 | 0.024 ± 0.008 | 0.020 ± 0.005 | 0.024 ± 0.007 | 0.024 ± 0.011 | 0.022 ± 0.008 |
| Std. Ad. | 0.020 ± 0.008 | 0.022 ± 0.010 | 0.022 ± 0.012 | 0.028 ± 0.013 | 0.026 ± 0.011 | 0.024 ± 0.011 | ||
| RSDg (%) | 9.8–18.8 | 12.7–28.6 | 10.2–31.3 | 8.7–30.8 | 11.1–32.0 | 11.5–27.4 | 12.6–26.5 | |
Conclusions
The results of this study demonstrated that the HG-HR-CS-QTAAS method is versatile and reliable for tAs and iAs quantification in common foodstuffs without limitations in terms of nature and matrix composition. Besides the very good figures of merit, this method has wide applicability for diverse matrices and provides As quantification by external calibration under identical derivatization conditions to arsine, irrespective of the sample matrix. However, in the case of some CRMs, Tukey's and Dunnett's methods indicated the existence of significant differences in terms of iAs found using extraction in 10 mol L−1 HCl without separation and that applying separation in toluene. No significant differences were observed when performing the extraction in 0.28 mol L−1 HNO3 or 0.01 mol L−1 HCl without separation, respectively. Thus, it was demonstrated that under the optimized working conditions for HG (0.01 mol L−1 HCl in the presence of 0.2% (m/v) L-cysteine), the separation of the iAs fraction by extraction in toluene before quantification is not necessary. The extraction in 0.28 mol L−1 nitric acid or 0.01 mol L−1 HCl used also as the medium for derivatization in the presence of L-cysteine in both cases was found to be suitable for iAs determination without the inconvenience of non-spectral interference coming from the mineral matrix. This method consumes low amounts of HCl, but needs rigorous control of both the NaOH concentration in the NaBH4 solution as a stabilizer and pH in the sample before derivatization. Purging the reaction cell with an Ar stream before the addition of the NaBH4 solution removed oxygen from the gas–liquid separator, while drying of the Ar–arsine stream limited water vapor entering QTA, resulting in a substantial improvement in the repeatability and sensitivity. However, the literature data indicated significant differences between the iAs reference values (not certified) in the CRMs. The validation of the HG-HR-CS-QTAAS method for the determination of the iAs fraction in foodstuffs also considered Tukey's and Dunnett's statistical multiple comparison tests. The validation results together with the attractive LODs for tAs and iAs in food represent a promising starting point for the further development of methods applicable in official control laboratories, and can also increase the interest in HG-HR-CS-QTAAS instrumentation for tAs quantification and iAs speciation.Author contributions
Lucia Chirita: investigation, methodology, resources, funding acquisition, data curation, formal analysis, writing – original draft. Eniko Covaci: formal analysis, visualization, data curation, software, writing – original draft. Michaela Ponta: validation, writing – original draft. Tiberiu Frentiu: conceptualization, supervision, funding acquisition, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant of Ministry of Research and Innovation, Romania, project CNFIS-FDI-2022-0179, within PNCDI III and project financed by the European Commission, number POCU/380/6/13/123886.References
- IARC Working Group on the Evaluation of Carcinogenic Risk to Humans, Arsenic, Metal, Fibers and Dusts, International Agency for Research on Cancer (IARC Monographs on the Evaluation of Carcinogenic Risk to Humans), Lyon, France, vol. 100C, 2012, https://monographs.iarc.who.int/wp-content/uploads/2018/06/mono100C.pdf, accessed 1 September 2022 Search PubMed.
- S. McSheehy, P. Pohl, D. Velez and J. Szpunar, Anal. Bioanal. Chem., 2002, 372, 457–466 CrossRef CAS PubMed.
- A. H. Petursdottir, J. J. Sloth and J. Feldmann, Anal. Bioanal. Chem., 2015, 407, 8385–8396 CrossRef CAS PubMed.
- T. Llorente-Mirandes, R. Rubio and J. F. Lopez-Sanchez, Appl. Spectrosc., 2017, 71, 25–69 CrossRef CAS PubMed.
- European Commission (EC), Commission Regulation (EU) No 2015/1006 amending Regulation (EC) No 1881/2006 as regards maximum levels of inorganic arsenic in foodstuffs, Off. J. Eur. Union, 2015, L161, 14–16 Search PubMed.
- European Food Safety Authority (EFSA), Dietary exposure to inorganic arsenic in the European population, EFSA J., 2014, 12, 3597 Search PubMed , 1–68..
- European Commission (EU), Commission Recommendation (EU) 2015/1381 on the monitoring of arsenic in food, Off. J. Eur. Union, 2015, L213, 9–10 Search PubMed.
- D. Arcella, C. Cascio and J. A. G. Ruiz, EFSA J., 2021, 19, e6380 Search PubMed.
- Commission Regulation (EC), 1881/2006 setting maximum levels for certain contaminants in foodstuffs, issued by the European Commission, Off. J. Eur. Communities, 2006, L364, 5–24 Search PubMed.
- C. D. B. Amaral, J. A. Nobrega and A. R. A. Nogueira, Talanta, 2013, 115, 291–299 CrossRef CAS PubMed.
- A. Dhillon, M. Nair and D. Kumar, Anal. Methods, 2015, 7, 10088–10108 RSC.
- F. Ardini, G. Dan and M. Grotti, J. Anal. At. Spectrom., 2020, 35, 215–237 RSC.
- A. H. Petursdottir, H. Gunnlaugsdottir, E. M. Krupp and J. Feldmann, Food Chem., 2014, 150, 353–359 CrossRef CAS PubMed.
- N. Zhang, K. Shen, X. M. Yang, Z. X. Li, T. K. Zhou, Y. Zhang, Q. L. Sheng and J. B. Zheng, Food Chem., 2018, 264, 462–470 CrossRef CAS PubMed.
- X. Hu, Z. Cao, W. Sun, H. Yang, P. Xu and Z. Zhu, Anal. Methods, 2016, 8, 6150–6157 RSC.
- P. M. Leal, E. V. Alonso, M. M. L. Guerrero, M. T. S. Cordero, J. M. C. Pavon and A. G. de Torres, Talanta, 2018, 184, 251–259 CrossRef PubMed.
- R. Kashanaki, H. Ebrahimzadeh and M. Moradi, Anal. Methods, 2017, 9, 3121–3127 RSC.
- R. A. Zounr, M. Tuzen and M. Y. Khuhawar, J. AOAC Int., 2018, 101, 593–600 CrossRef CAS PubMed.
- X. Wang, G. Xu, P. Chen, Y. Sun, X. Yao, Y. Lv, W. Guo and G. Wang, RSC Adv., 2018, 8, 16858–16865 RSC.
- I. Lopez-Garcia, J. J. Marin-Hernandez and M. Hernandez-Cordoba, Talanta, 2018, 181, 6–12 CrossRef CAS PubMed.
- A. V. Zmozinski, T. Llorente-Mirandes, I. C. F. Damin, J. F. Lopez-Sanchez, M. G. R. Vale, B. Welz and M. M. Silva, Talanta, 2015, 134, 224–231 CrossRef CAS PubMed.
- J. Werner, T. Grzeskowiak, A. Zgola- Grzeskowiak and E. Stanisz, TrAC, Trends Anal. Chem., 2018, 105, 121–136 CrossRef CAS.
- Y. Huang, J. H. Shan, B. Fan, Y. He, S. M. Xia, Y. F. Sun, J. Lu, M. Wang and F. Z. Wang, Anal. Methods, 2015, 7, 8896–8900 RSC.
- A. D'Ulivo and R. Sturgeon, Vapor generation techniques for trace element analysis – Fundamental aspects, Elsevier, Amsterdam, 1st edn, 2022 Search PubMed.
- I. Fiamegkos, F. Cordeiro, P. Robouch, D. Velez, V. Devesa, G. Raber, J. J. Sloth, R. R. Rasmussen, T. Llorente-Mirandes, J. F. Lopez-Sanchez, R. Rubio, F. Cubadda, M. D'Amato, J. Feldmann, A. Raab, H. Emteborg and M. B. de la Calle, Food Chem., 2016, 213, 169–179 CrossRef CAS PubMed.
- C. S. Huber, M. G. R. Vale, M. B. Dessuy, M. Svoboda, S. Musil and J. Dedina, Talanta, 2017, 175, 406–412 CrossRef CAS PubMed.
- A. Khaligh, H. Z. Mousavi, H. Shirkhanloo and A. Rashidi, RSC Adv., 2015, 5, 93347–93359 RSC.
- A. Elik, A. Demirbas and N. Altunay, Anal. Methods, 2019, 11, 3429–3438 RSC.
- G. Ma, S. Liu, J. Sun and X. Duan, Spectrochim. Acta, Part B, 2018, 150, 38–42 CrossRef CAS.
- B. Chen, W. T. Corns, P. B. Stockwell and J.-H. Huang, Anal. Methods, 2014, 6, 7554–7558 RSC.
- M. Welna, P. Pohl and A. Szymczycha-Madeja, Food Anal. Methods, 2019, 12, 581–594 CrossRef.
- P. M. Leal, E. V. Alonso, M. M. L. Guerrero, M. T. L. Cordero, J. M. C. Pavon and A. G. de Torres, Talanta, 2018, 184, 251–259 CrossRef PubMed.
- A. H. Petursdottir, N. Friedrich, S. Musil, A. Raab, H. Gunnlaugsdottir, E. M. Krupp and J. Feldmann, Anal. Methods, 2014, 6, 5392–5396 RSC.
- M. M. L. Guerrero, E. V. Alonso, J. M. C. Pavon, M. T. S. Cordero and A. G. de Torres, J. Anal. At. Spectrom., 2016, 31, 975–984 RSC.
- A. I. G. de las Torres, M. S. Moats, G. Rios, A. R. Almansa and D. Sanchez-Rodas, Anal. Methods, 2020, 12, 1943–1948 RSC.
- J. Proch and P. Niedzielski, Talanta, 2020, 208, 120395 CrossRef CAS PubMed.
- P. Montoro-Leal, J. C. Garcia-Mesa, I. Morales-Benitez, A. Garcia de Torres and E. Vereda Alonso, Talanta, 2021, 235, 122769 CrossRef CAS PubMed.
- J. Sun, Z. G. Yang, H. W. Lee and L. Wang, Anal. Methods, 2015, 7, 2653–2658 RSC.
- P. Alava, T. Van de Wiele, F. Tack and G. Du Laing, Anal. Methods, 2012, 4, 1237–1243 RSC.
- A. Pell, A. Marquez, R. Rubio and J. F. Lopez-Sanchez, Anal. Methods, 2013, 5, 2543–2550 RSC.
- B. M. Freire, V. da Silva Santos, P. de Carvalho Ferreira Neves, J. M. Oliveira Souza Reis, S. S. de Souza, F. Barbosa Jr and B. L. Batista, Anal. Methods, 2020, 12, 2102–2113 RSC.
- P. Petrov, S. Cowen and H. Goenaga-Infante, Anal. Methods, 2021, 13, 3641–3648 RSC.
- B. Welz, Anal. Bioanal. Chem., 2005, 381, 69–71 CrossRef CAS PubMed.
- M. Resano and E. Garcia-Ruiz, Anal. Bioanal. Chem., 2011, 399, 323–330 CrossRef CAS PubMed.
- B. Welz, S. Mores, E. Carasek, M. G. R. Vale, M. Okruss and H. Becker-Ross, Appl. Spectrosc. Rev., 2010, 45, 327–354 CrossRef CAS.
- L. Chirita, E. Covaci, A. Mot, M. Ponta, A. Gandea and T. Frentiu, J. Anal. At. Spectrom., 2021, 36, 267–272 RSC.
- M. Schneider, H. R. Cadorim, B. Welz, E. Carasek and J. Feldmann, Talanta, 2018, 188, 722–728 CrossRef CAS PubMed.
- E. R. Pereira, T. S. de Almeida, D. L. G. Borges, E. Carasek, B. Welz, J. Feldmann and J. D. C. Menoyo, Talanta, 2016, 150, 142–147 CrossRef CAS PubMed.
- S. Hesse, T. Ristau and J. W. Einax, Microchem. J., 2015, 123, 42–50 CrossRef CAS.
- A. C. Valdivia, M. M. L. Guerrero, E. I. V. Alonso, J. M. C. Pavon and A. G. de Torres, Microchem. J., 2018, 138, 109–115 CrossRef.
- J. Kratzer, B. Docekal, U. Heitmann and J. Dedina, J. Anal. At. Spectrom., 2011, 26, 2230–2237 RSC.
- J. Sloth, F. Cordeiro Raposo, R. Rasmussen, R. Hedegaard, P. Emteborg, I. Verbist, J. Danier and M. De La Calle Guntinas, IMEP-32: Determination of Inorganic Arsenic in Animal Feed of Marine Origin: A Collaborative Trial Report, EUR 24938 EN, Publications Office of the European Union, Luxembourg, 2011. p. JRC66416 Search PubMed.
- I. Fiamegkos, F. Cordeiro, V. Devesa, D. Velez, P. Robouch, H. Emteborg, H. Leys, A. Cizek-Stroh and M. B. de la Calle, IMEP-41: Determination of inorganic As in food Collaborative Trial Report, JRC Technical Reports, Joint Research Center, Institute for Reference Materials and Measurements, Geel, Belgium, January, 2015, p. JRC94325 Search PubMed.
- M. B. de la Calle, T. Linsinger, H. Emteborg, J. Charoud-Got and I. Verbist, Report of the seventh interlaboratory comparison organized by the European Union – Reference laboratory for heavy metals in feed and food. IMEP-107: Total and inorganic As in rice, EUR 24314 EN, Publication Office of the European Union, Luxembourg, 2010, p. JRC57768 Search PubMed.
- F. R. Cordeiro, P. Robouch, H. Emteborg, J. Seghers, I. Fiamegkos, A. Cizek-Stroh and M. B. de la Calle, IMEP-116: Determination of total cadmium, lead, arsenic, mercury and inorganic arsenic in mushrooms – Interlaboratory Comparison Report, EUR 26214, Publication Office of the European Union, Luxembourg, 2013, p. JRC85158 Search PubMed.
- P. Dvorak, M. Talaba, J. Kratzer and J. Dedina, Chem. Sci., 2019, 10, 3643–3648 RSC.
- M. Lacko, K. Dryahina, P. Spanel, J. Kratzer, T. Matousek and J. Dedina, Anal. Chem., 2022, 94, 13163–13170 CrossRef CAS PubMed.
- J. Kratzer, M. Lacko, K. Dryahina, T. Matousek, P. Spanel and J. Dedina, Anal. Chim. Acta, 2022, 1190, 339256 CrossRef CAS PubMed.
- A. V. Zmozinski, T. Llorente-Mirandes, J. F. Lopez-Sanchez and M. M. da Silva, Food Chem., 2015, 173, 1073–1082 CrossRef CAS PubMed.
- T. Frentiu, S. Butaciu, E. Darvasi, M. Ponta, M. Senila, D. Petreus and M. Frentiu, Anal. Methods, 2015, 7, 747–752 RSC.
- L. Haghnazari, N. Mirzaei, H. Arfaeinia, K. Karimyan, H. Sharafi and N. Fattahi, Biol. Trace Elem. Res., 2018, 183, 173–181 CrossRef CAS PubMed.
- J.-H. Huang, G. Ilgen and P. Fecher, J. Anal. At. Spectrom., 2010, 25, 800–802 RSC.
- J.-H. Huang, P. Fecher, G. Ilgen, K.-N. Hu and J. Yang, Food Chem., 2012, 130, 453–459 CrossRef CAS.
- C. Cerveira, D. Pozebon, D. P. de Moraes and J. C. Silva de Fraga, Anal. Methods, 2015, 7, 4528–4534 RSC.
- J. W. Tukey, Biometrics, 1949, 5, 99–114 CrossRef CAS PubMed.
- C. W. Dunnett, J. Am. Stat. Assoc., 1955, 50, 1096–1121 CrossRef.
- C. Smith, S. Bhandari, C. Blake, D. Boaz, M. Briscoe, F. Cho, S. Christiansen, M. Clabaugh, M. Clarke, R. Clifford, M. Collison, X. J. Deng, B. Duruttya, D. Ellingson, P. Faison, J. Farrow, N. Gras, I. P. Ho, G. Hostetler, M. Huang, E. Konings, K. Laurvick, J. Lupean, F. Maniei, C. Mejia, J. Messerly, W. Mindak, C. Murphy, B. Nelson, J. Nelson, L. Pacquette, E. Phifer, E. Porter, R. Reba, J. Roberts, S. Royce, L. Santiago-Connolly, B. Schaneberg, L. Sheng, R. C. Shih, K. Stanley, T. Stilwater, J. Szpylka, S. Wall, L. Wood, J. Wubben, Y. Zhou, J. Zhu, B. Zombro, R. Zywicki and S. Coates, J. AOAC Int., 2019, 98, 1102–1103 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ay00142c |
| This journal is © The Royal Society of Chemistry 2023 |