
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn environmentally friendly approach for the characterization of construction materials: determination of trace, minor, and major elements by slurry sampling high-resolution continuum source graphite furnace atomic absorption spectrometry†
Beatriz
Gómez-Nieto
 *a,
Carmen
Isabel-Cabrera
a,
María Jesús
Gismera
a,
María Teresa
Sevilla
a,
Jesús R.
Procopio
a and
María Isabel
Sánchez de Rojas
b
*a,
Carmen
Isabel-Cabrera
a,
María Jesús
Gismera
a,
María Teresa
Sevilla
a,
Jesús R.
Procopio
a and
María Isabel
Sánchez de Rojas
b
aDepartamento de Química Analítica y Análisis Instrumental, Facultad de Ciencias Universidad Autónoma de Madrid, Avda. Francisco Tomás y Valiente 7, Madrid 28049, Spain. E-mail: beatriz.gomez@uam.es
bDepartamento de Cementos y Reciclado de Materiales, Instituto de Ciencias de la Construcción Eduardo Torroja (IETcc-CSIC), C/Serrano Galvache 4, Madrid 28033, Spain
First published on 2nd February 2023
Abstract
A slurry sampling method was developed for the fast determination of Pb, Ni, Fe, and Mn in construction materials by high-resolution continuum source graphite furnace atomic absorption spectrometry (HR-CS GFAAS). For sample introduction into the GF, stable slurries were prepared by sonicating 10 mg of ground solid sample in 10.0 mL of 1% (v/v) Triton X-100 and 1% (v/v) HNO3 solution for 1.0 min. The determination of the four elements was carried out in three measurement runs, with Ni and Fe being determined simultaneously. The HR-CS GFAAS measurements were performed using analytical lines with adequate sensitivity, considering the content of each element in the material: Pb at 283.306 nm (42%), Mn at 403.080 nm (6.7%), Ni at 232.003 nm (100%) and Fe at 232.036 nm (1.4%). The pyrolysis and atomization temperatures and the use of chemical modifiers were optimized using both aqueous standards and slurry samples. At optimal conditions, samples with concentrations of Pb from 1.5 to 80 μg g−1, Ni from 4.0 to 75 μg g−1, Mn from 2.0 to 600 μg g−1, and Fe from 0.15 to 60 mg g−1 could be determined using a unique sample suspension. To assess the validity of the method, a fly ash certified reference material (CRM) was analysed using the slurry sampling HR-CS GFAAS method; this CRM and the construction material samples were also analysed by HR-CS GFAAS after the digestion of the samples. The obtained results using both methods were statistically comparable (Student's paired t-test for two independent methods at a 95% confidence level) demonstrating the suitability of the proposed method.
1. Introduction
The determination of metal and non-metal elements in construction materials, such as cement, ceramic bricks, mortars, or concrete, is of great importance for monitoring the quality of the products. Over the last few decades, research on the incorporation of different wastes (e.g., biomass ashes, construction, and demolition wastes) as supplementary materials for preparing more environmentally friendly concretes and mortars has increased. This strategy for waste reuse appears to be a suitable alternative to limit the use of natural resources and reduce the environmental problems associated with landfill disposal. The waste materials should be characterized before their use because some elements can influence the properties of the construction materials and/or have a negative environmental effect.1–4 In this context, the development of fast, accurate, and more environmentally friendly methods to determine different elements in construction materials is of great interest to simplify the analytical procedures to characterize these materials.Inductively coupled plasma optical emission or mass spectrometry (ICP OES or ICP MS), and atomic absorption spectroscopy (AAS) with Flame or graphite furnace atomizers (FAAS or GFAAS) are among the most widely used techniques for the determination of elements in construction materials after the adequate pre-treatment of the sample and release of the target analytes into solution.2–5 The digestion of construction materials is not an easy task and requires the use of toxic and corrosive chemicals such as HNO3, HCl and/or HF, which is not in accordance with the principles of green analytical chemistry.6–9 In addition, this step is usually time-consuming and increases the risk of sample contamination and/or analyte loss. In this context, analytical methods based on the direct analysis of solid samples are considered excellent alternatives. Among them, methods based on X-ray Fluorescence (XRF) are commonly used for the direct determination of minor and major elements in construction materials but they present limitations to determining elements at trace levels.4,10,11
GFAAS is a commonly used technique for determining minor and trace elements in liquid samples but it can also be used to carry out direct measurements on solid or slurry samples.12–16 GFAAS methods for the direct analysis of solid samples have undergone significant development since the arrival of high-resolution continuum source AA spectrometers (HR-CS AAS) due to the higher background correction capability of these instruments.17–19 Using these spectrometers, the main or secondary absorption lines and the wings of the absorption lines can be selected for the analytical measurements, and therefore the sensitivity for each element can be adjusted to the expected concentration level in the sample.20–22 The main drawback of direct solid sampling GFAAS methods is the greater imprecision compared to GFAAS methods with liquid sample introduction due to the intrinsic inhomogeneity of solid samples and the small amount of sample introduced into the graphite tube for the analysis. This limitation can be reduced by introducing the solid sample as a slurry since larger amounts of the sample can be used to prepare the suspension. This sampling approach combines the advantages of solid and liquid sampling, reducing the analysis time, minimising the inherent risk of contamination, and facilitating the sample introduction into the GF atomizer.12 To ensure representative results in slurry sampling GFAAS methods, homogeneous and stable suspensions (at least for the time taken to carry out the GFAAS measurements) must be prepared. Thus, factors that can influence the suspension stability, such as particle size, type of dispersing media, sample mass to dispersant volume ratio, and the mixing time and instrument used for their preparation, must be optimized.12,13,16
This work aims to develop a new and more environmentally friendly method for the determination of Ni, Pb, Fe, and Mn in cement-based and ceramic-based construction materials by slurry sampling HR-CS GFAAS. For this purpose, the slurry preparation conditions were optimized, evaluating the parameters that can affect their stability, and the GFAAS measurement conditions were established for the determination of the four elements using a unique sample suspension. The method was successfully applied to analyse different construction materials. In our opinion, this new method based on HR-CS GFAAS could be considered an interesting, reliable, and green analytical approach for the direct and rapid determination of Pb, Ni, Mn, and Fe elements present in construction materials at very different concentration levels.
2. Experimental
2.1. Reagents and solutions
Ultrapure water (UPW) with a resistivity of 18.2 MΩ cm obtained from a Milli-Q Direct water purification system (Merck Millipore, Germany) was used throughout. Sodium dodecyl sulphate (SDS), Triton X-100, and tetramethylammonium hydroxide (TMAH) (Fluka, Switzerland) were evaluated as dispersants for the preparation of the slurries. Trace analysis grade hydrofluoric acid of 47–51% (HF) (Fluka, Switzerland) and nitric acid of 65% (HNO3) (Scharlau, Spain) were used to carry out the microwave digestion of solid samples. HNO3 was also used to prepare aqueous standard solutions and/or slurries. All reagents were used as received without further purification.Multi-element standard solutions of Pb, Ni, Fe, and Mn were prepared from individual commercial 1.000 g L−1 standard solutions for AAS (Scharlau, Spain). The standard solutions were prepared daily by the adequate dilution of the stocks with 1% (v/v) HNO3 solution. A 1% (v/v) palladium (Pd) solution was prepared by dilution with ultrapure water from a commercially available Pd matrix modifier solution of palladium nitrate (Pd(NO3)2) of 10 g L−1 of Pd (Fluka, Switzerland). A commercially available TraceCert® 1000 mg L−1 silicon standard solution for AAS (Sigma-Aldrich, Germany) was used to create the SiO reference spectrum.
2.2. Samples and certified reference material
The 1633b certified reference material (CRM), trace elements in coal fly ash (National Institute of Standards and Technology, NIST, USA), was used to assess the validity of the method. This material was available as a fine powder with a particle size ≤90 μm.The cement sample was a commercially available Ordinary Portland Cement (OPC), (European Standard 197-1 CEM I/42.5R cement) supplied by Lafarge (Spain). The ceramic material was 100% ceramic waste (CW) (Portland-Valderrivas, Spain) from crushed and ground bricks and tiles. Different cement–mortar samples were provided by the Recycling Materials Group of the Department of Cement and Recycling Material (Eduardo Torroja Institute for the Construction Sciences of CSIC), (Madrid, Spain). Details on mortar preparation are included in the ESI.† All samples were ground and sieved through a nylon sieve of 32 μm mesh size before slurry preparation.
2.3. Instrumentation
An analytical microbalance model AX205DR (Mettler Toledo, Switzerland) with a sensitivity of ±0.01 mg was used for weighing the solid samples or CRM. The slurries were prepared using three different instruments: a Wizard IR Vortex mixer (Velp Scientifica, Italy), an Elma Sonic P30HS model ultrasonic bath (ELMA, Germany) operated at 100% power and 80 kHz, and a Sonopuls HD 2070 model ultrasonic homogenizer, 70 W, 20 kHz (Bandelin, Germany), equipped with a titanium microtip of 2 mm in diameter, 195 mm in length and 253 mm in maximum width. A Specord205 UV spectrophotometer (Analytik Jena, Germany) and 10 mm PMMA UV grade cuvettes were used for the transmittance measurements in the slurry stability studies. Wet digestion of the solid samples was performed with a microwave sample preparation system (Perkin Elmer-Anton PAAR Multiwave, Austria) equipped with perfluoroalkoxy (PFA) MF 100 model digestion vessels.The determination of metals was performed with a ContrAA 700 HR-CS AA spectrometer (Analytik Jena, Germany). This spectrometer is equipped with a transversely heated graphite furnace atomization unit and an MPE 60 liquid furnace autosampler (Analytik Jena, Germany). Pyrolytically coated pin-platform graphite tubes (Part No. 407-A81.025, Analytik Jena, Germany) were used in all measurements. High-purity argon (Ar) (99.9992%) (Nippon Gases, Madrid, Spain) was used as the purge and protective gas in all GFAAS measurements.
2.4. Procedures
All plastic and glassware used to prepare or store solutions and slurry samples were cleaned by soaking in a 10% (v/v) HNO3 solution for at least 24 hours, and then they were thoroughly rinsed with ultrapure water.| Stage | Temperature (°C) | Ramp (°C s−1) | Hold time (s) |
|---|---|---|---|
| Drying 1 | 80 | 6 | 20 |
| Drying 2 | 90 | 3 | 20 |
| Drying 3 | 110 | 5 | 10 |
| Pyrolysis 1 | 350 | 50 | 20 |
| Pyrolysis 2 | 900 (Pb) | 300 | 10 |
| 1200 (Ni and Fe) | |||
| 1400 (Mn) | |||
| Atomization | 1900 (Pb) | 1500 | 10 |
| 2500 (Ni and Fe) | |||
| 2200 (Mn) | |||
| Cleaning | 2450 (Pb) | 500 | 5 |
| 2650(Ni and Fe) | |||
| 2450 (Mn) |
The MPE 60 liquid furnace autosampler was used to inject slurries of solid samples and standards onto the pin-platform graphite tube and prepare the diluted standard solutions used to obtain the calibration curves. To carry out the determination of Ni, Fe, Mn, and Pb in the samples, approximately 1 mL of the slurry was transferred into a clean autosampler cup immediately after its preparation and automatically injected into the graphite tube platform. An injection volume of 20 μL was selected for the measurements of standards and samples. All determinations were performed in triplicate.
3. Results and discussion
3.1. Optimization of slurry preparation conditions
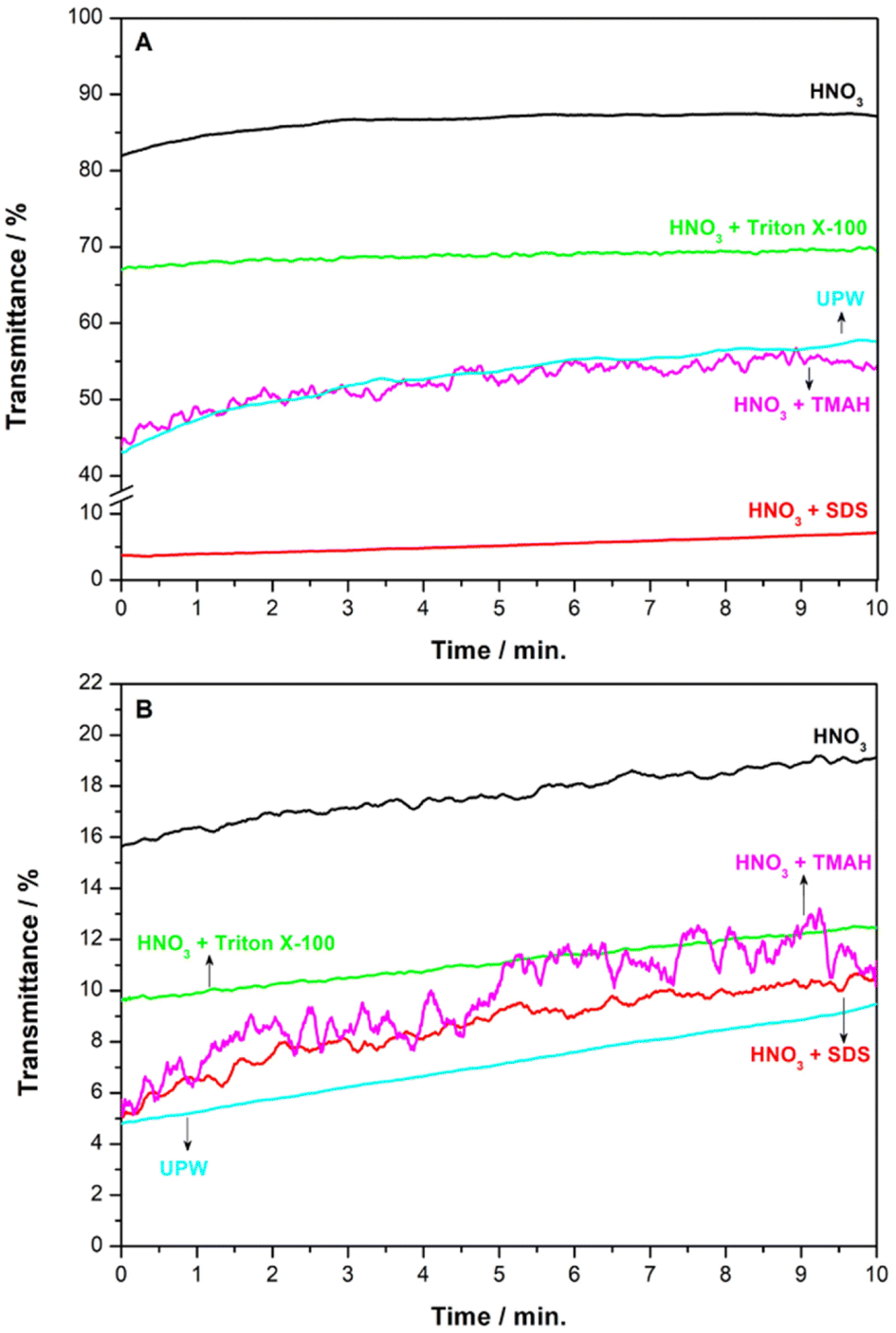 | ||
| Fig. 1 Mean values (n = 3) of transmittance percentage over time for OPC (A) and CW (B) slurries with 0.10% of solid sample prepared in 1% HNO3 (black), UPW (blue), 1% HNO3 and 1% Triton X-100 (green), 1% HNO3 and 1% TMAH (pink), and 1% HNO3 and 1%SDS (red). Mixing conditions: 1 minute in an ultrasonic bath (100% power; 80 kHz). | ||
As seen in Fig. 1A and B, the initial % T value in UPW for OPC (between 40 and 45%) was higher than that observed for the CW suspension (about 5%), which could indicate a better dispersion of a higher amount of the solid for CW as compared to OPC. For both materials, the initial % T values in 1% HNO3 are higher than in UPW, and the variability in the initial % T values (expressed as the relative standard deviation (RSD, %)) among three replicates of suspensions in 1% HNO3 (between 5–10%) was lower than that for replicates of suspensions in UPW (around 15%). This increase in the initial % T is because HNO3 produces a partial solubilization of the material with a reduction of the particle size and, therefore, the characteristics of the suspensions change. This effect was more pronounced in OPC, a relatively soluble material in acid media, with an initial % T greater than 80% for the suspension in 1% HNO3. Using the mixed medium of 1% HNO3 + 1% TMAH, the characteristics of the OPC and CW suspensions did not change significantly as compared to the suspensions prepared using UPW. However, less reproducible suspensions were prepared using this combination of reagents (RSD% > 20%). In 1% HNO3 + 1% SDS, the initial % T value for the OPC suspension decreased to about 5%, whereas no significant changes were observed for CW suspensions. This decrease in the initial % T values of OPC slurries indicates that the surfactant may prevent the solubilization of the material by HNO3. However, inhomogeneous suspensions with agglomerates of particles were observed with the naked eye using this mixed medium and RSD% values between 10–15% of the initial % T values were found, indicating that unstable and irreproducible suspensions were prepared using this combination of reagents. For OPC and CW suspensions in the mixed medium 1% HNO3 + 1% Triton X-100, stable and reproducible suspensions without significant variation in the % T values over the 10 minutes of study and with RSD% values < 2.0% were obtained. Considering these results, this medium was selected as optimal for preparing the OPC and CW slurries for GFAAS measurements.
3.2. Optimization of HR-CS GFAAS measurement conditions
 | ||
| Fig. 2 Time–wavelength resolved absorbance spectra obtained in the vicinity of the Ni main line (232.003 nm) and Fe secondary line (232.036 nm) for the (A) OPC slurry and (B) CW slurry before LSBC correction; (C) the OPC slurry and (D) CW slurry after subtraction of the SiO reference spectrum with LSBC; (E) SiO reference spectrum. Conditions: pyrolysis temperature of 1200 °C; atomization temperature of 2500 °C. | ||
 | ||
| Fig. 3 (Left) Pyrolysis (circles) and atomization (squares) curves for standard solutions of 10 μg L−1 Ni (A), 0.70 mg L−1 Fe (C), 200 μg L−1 Mn (E) and 10 μg L−1 of Pb (G) without modifier (close symbols) and with 5 μL of 1% (v/v) Pd modifier solution (open symbols). (Right) Pyrolysis (circles) and atomization (squares) curves for 0.10% (w/v) slurries of OPC (black) and CW (red) for Ni (B), Fe (D), Mn (F) and Pb (H) without modifier (close symbols) and with 5 μL of 1% (v/v) Pd modifier solution (open symbols). Error bars correspond to the standard deviation of n = 3 measurements. | ||
The atomization curves for Ni, Fe, Mn, and Pb in aqueous standards, and OPC and CW slurries were established by setting the selected pyrolysis temperatures of each analyte and varying the atomization temperature from 1800 °C, 2000 °C, or 1300 °C up to 2600 °C for Mn, Ni and Fe, and Pb, respectively. According to the results, (see Fig. 3E and F), a temperature of 2200 °C was selected for the atomization of Mn. For Ni (see data in Fig. 3A and B) and Fe (see Fig. 3C and D) the highest integrated absorbance values were obtained for temperatures of 2500 °C and 2400 °C, respectively. Considering that the Ni amount in OPC and CW is lower than the Fe content and these elements are determined simultaneously in the slurries, a common atomization temperature of 2500 °C was chosen to carry out the simultaneous determination of both elements. Given the results obtained for Pb, using the Pd matrix modifier (see Fig. 3G and H, open symbols), a temperature of 1900 °C was selected for Pb atomization. The final temperature programs for the determination of Ni, Fe, Mn, and Pb are shown in Table 1.
3.3. Calibration and figures of merit
Calibration curves for Pb, Ni, Fe, and Mn were established using multielement standard solutions prepared in 1% Triton X-100 containing 1% HNO3. The analytical signal used for quantification purposes was the sum of the integrated absorbance value of one or more pixels of the absorption line of each element. The number of pixels to quantify each metal was selected considering the amount of the element expected in the samples. In this way, for Pb and Ni, the sum of the integrated absorbance value of three pixels (the central pixel plus the adjacent ones, CP ± 1) of the absorption line was chosen as the analytical signal, while for Fe and Mn, just one pixel (the central pixel, CP) of the absorption line profile was selected as the analytical signal for quantification purposes. The use of the absorbance values at the absorption line wings, the so-called side pixel registration approach, was also evaluated to further extend the linear working range of Fe and Mn. Fig. 4 shows the calibration curves obtained for Pb, Ni, Fe, and Mn (see the figure legend for the pixels selected to establish the different calibration curves). | ||
| Fig. 4 Calibration curves obtained for aqueous standards of (A) Pb, (B) Ni, (C) Fe, and (D) Mn. Pixels for quantification: CP (only the central pixel); CP ± 1 (central pixel plus two adjacent ones; ∑(±3)) (sum of only the pixels in the lateral positions plus three); ∑(±4) (sum of only the pixels in the lateral positions plus four). Error bars correspond to the standard deviation of n = 3 measurements. | ||
Table 2 summarizes the figures of merit of the method for the four studied elements. The limits of detection (LODs) and quantification (LOQs) were respectively calculated as three times and ten times the standard deviation obtained for ten repetitive measurements of the blank solution (1% Triton X-100 containing 1% HNO3) divided by the slope of the calibration curve. The characteristic concentration (C0), defined as the content of analyte that produces an analytical signal of 0.0044 s, was also calculated. The upper limit of the linear range (ULLR) was graphically calculated as the analyte concentration at which the deviation of the slope was less than ±5% of that of the ideal linearity plots.
| ELEMENT | Pixel | Slope | r | C 0 | LOD | LOQ | ULLR |
|---|---|---|---|---|---|---|---|
| a Maximum concentration assayed. | |||||||
| Pb | CP ± 1 | 0.0097 ± 0.0002 s L μg−1 | 0.9991 | 0.45 μg L−1 | 0.50 μg L−1 | 1.5 μg L−1 | 80 μg L−1a |
| Ni | CP ± 1 | 0.0114 ± 0.0003 s L μg−1 | 0.9986 | 0.40 μg L−1 | 1.1 μg L−1 | 3.8 μg L−1 | 75 μg L−1a |
| Fe | CP | 0.0851 ± 0.0003 s L mg−1 | 0.9972 | 0.050 mg L−1 | 0.030 mg L−1 | 0.11 mg L−1 | 5.0 mg L−1 |
| ±3 | 0.0052 ± 0.0002 s L mg−1 | 0.9965 | 0.84 mg L−1 | 1.2 mg L−1 | 3.9 mg L−1 | 60 mg L−1 | |
| Mn | CP | 0.00208 ± 0.0004 s L μg−1 | 0.9990 | 2.12 μg L−1 | 0.50 μg L−1 | 1.7 μg L−1 | 300 μg L−1 |
| ±3 | 0.00114 ± 0.00003 s L μg−1 | 0.9971 | 3.86 μg L−1 | 1.2 μg L−1 | 4.0 μg L−1 | 400 μg L−1 | |
| ±4 | 0.00028 ± 0.00001 s L μg−1 | 0.9989 | 15.9 μg L−1 | 5.5 μg L−1 | 18 μg L−1 | 600 μg L−1 | |
The calibration curves of Pb and Ni were linear over the entire concentration range assayed (see Fig. 4A and B). For Mn and Fe, ULLRs of 300 μg L−1 and 5.0 mg L−1, respectively, were obtained using the central pixel (CP) for signal quantification. The ULLR of Mn can be increased up to 400 μg L−1 or 600 μg L−1 using the sum of the integrated absorbance values of the side pixels ∑(±3) or ∑(±4), respectively, as the analytical signal (see Fig. 4D). For Fe, the ULLR can be increased up to 60.0 mg L−1 using ∑(±3) (see Fig. 4C). The instrumental LODs and LOQs values for Ni and Pb were similar to those previously reported for these elements using HR-CS GFAAS.23,25–27 For the secondary lines used for Fe and Mn determination, for comparison, we did not find literature data using only the CP or the side pixel registration approach as the analytical signal. The sensitivity, LODs and LOQs of these elements could be improved, if necessary, to analyse samples with a lower content of Fe or Mn using CP ± 1 and/or a more sensitive analytical line for the determination. The relative LODs and LOQs in the materials were calculated, considering the percentage of solid sample in the slurry. The relative LOD values for Pb, Ni, Mn, and Fe were 0.50 μg g−1, 1.1 μg g−1, 0.50 μg g−1, and 0.030 mg g−1, respectively. The relative LODs for Pb and Ni were about 10 times lower than those reported for XRF methods.10,11 For Mn, the relative LOD value was better or similar to those reported for XRF methods, while for Fe, a higher value was obtained.10,11 These results for Mn and Fe were expected since secondary lines with relatively low sensitivity (6.7% for Mn and 1.4% for Fe) were selected for the determination of these elements, which are usually found in construction materials in higher amounts, and we wanted to determine the four elements in the same slurry of the solid sample.
The trace element coal fly ash 1633b CRM was analysed using the proposed method. For this purpose, slurries of this CRM with 0.1% of the solid sample were prepared in triplicate. Due to the high content of Ni and Fe in this CRM, the determination of these elements was carried out using suspensions with 0.04% of the solid sample. The determination of Fe content in the CRM slurry sample was performed using the side pixel registration approach (∑(±3)). No significant differences were observed between the concentrations of Pb, Ni, Fe, and Mn obtained by the proposed direct slurry sampling method and the certified values according to the Student's t-test at a 95% confidence level (see Table 3). The relative standard deviation (RSD, %) values were 8.5% for Pb, 9.0% for Ni, 6.8% for Fe and 5.0% for Mn.
| Sample | Pb (μg g−1) | Ni (μg g−1) | Fe (mg g−1) | Mn (μg g−1) | ||||
|---|---|---|---|---|---|---|---|---|
| Slurry | MW digestion | Slurry | MW digestion | Slurry | MW digestion | Slurry | MW digestion | |
| a Certified value: 68 ± 1 μg g−1 Pb; 120 ± 2 μg g−1 Ni; 78 ± 2 mg g−1 Fe; 132 ± 2 μg g−1 Mn. b Determination using 0.04% of solid sample in the slurry. | ||||||||
| CRMa | 59 ± 5 | 64 ± 4 | 110 ± 10b | 114 ± 9 | 74 ± 5b | 80 ± 6 | 121 ± 6 | 120 ± 8 |
| OPC | 14.5 ± 0.6 | 14 ± 1 | 11.7 ± 0.5 | 12.3 ± 0.8 | 21 ± 2 | 19 ± 2 | 314 ± 6 | 310 ± 13 |
| CW | 26 ± 1 | 25.8 ± 0.8 | 17.2 ± 0.6 | 16.7 ± 0.6 | 53 ± 5 | 51 ± 3 | 490 ± 16 | 494 ± 14 |
| Mortar 1 | 18 ± 1 | 18.8 ± 0.9 | 13.1 ± 0.4 | 13.6 ± 0.3 | 3.8 ± 0.2 | 4.0 ± 0.2 | 203 ± 9 | 195 ± 5 |
| Mortar 2 | 27 ± 2 | 25 ± 1 | 21 ± 1 | 19.3 ± 0.6 | 6.0 ± 0.3 | 6.1 ± 0.4 | 224 ± 18 | 206 ± 2 |
| Mortar 3 | 21 ± 1 | 21.8 ± 0.5 | 12.7 ± 0.9 | 12.5 ± 0.7 | 3.9 ± 0.2 | 3.5 ± 0.2 | 226 ± 10 | 222 ± 3 |
3.4. Analysis of real samples
The developed method was applied to determine Pb, Ni, Fe, and Mn in OPC, CW, and cement–mortar samples. For this purpose, slurries of the samples were prepared in triplicate. The results are shown in Table 3. The determination of Mn in OPC and CW samples was performed using the side pixel registration approach (∑(±3) and ∑(±4), respectively). For Fe determination in OPC and CW samples, ∑(±3) was used as the analytical signal. The concentrations of Pb in these samples were lower than the maximum level established by the Finland Government Decree for the use of certain wastes in earth construction.28To probe the validity of the method, all the samples and the CRM were digested (see Section 2.2.4.) and the Pb, Ni, Fe, and Mn contents were determined in the obtained solutions by HR-CS GFAAS. As can be seen in Table 3, the concentrations found using the proposed slurry sampling method were in good agreement (Student's t-test for two independent samples at a 95% confidence level) with those found in the samples after digestion and HR-CS GFAAS measurement in the solutions. These results demonstrate that the proposed method can be successfully applied in the determination of Pb, Mn, Ni, and Fe in cement, cement-additives as CW, and cement–mortar samples without the need to dissolve the samples.
3.5. Comparison with other sample preparation procedures
Table 4 compares some characteristics of the proposed slurry preparation procedure with those reported in other works devoted to the analysis of solid samples with similar inorganic matrixes using digestion or slurry sampling methods. Concentrated HNO3 alone, or combined with other corrosive reagents such as HF and HCl, is usually used for the treatment of inorganic matrix samples even in some slurry preparation procedures. Most of those procedures need temperatures higher than room temperature and about 20–60 minutes to digest the sample or to prepare the slurries. The sample treatment proposed in this work can be considered more environmentally friendly and in accordance with the principles of Green Analytical Chemistry as it avoids the use of corrosive and toxic chemicals such as concentrated mineral acids (HF, HCl or HNO3) and significantly reduces the time required for sample preparation and thereby, the energy consumption. Moreover, the conditions used to prepare the slurries in this work lead to the preparation of slurries that are stable for at least 10 minutes, avoiding the need to resuspend the sample between each replicate measurement; in most of the reported slurry sampling methods, the sample suspension must be shaken just before the measurement of each replicate.| Solid sample | Sample preparation procedure | Reference | |||
|---|---|---|---|---|---|
| Method | Reagents | Temperature | Time a | ||
| a Extraction time and/or mixing time for slurry preparation. b MW digestion program: 2 min at 250 W + 2 min at 400 W + 30 min at 650 W (Ta: 230 °C; P: 20 bars). 18 min at 0 W for cooling. | |||||
| Cement and clinker | Digestion | 65% HNO3 | 80 °C | 24 h | 5 |
| Geological materials and urban particulate matter | MW digestion | 65% HNO3 + 50% HF | 230 °C b | 51 min | 6 |
| Cement | Slurry sampling | 1% HNO3 + 1–5% HF + 10% EtOH | 25 °C | 25 min | 13 |
| Ceramic tableware | Slurry sampling | 65% HNO3 | 40 °C | 20 min | 16 |
| Bones | Slurry sampling | 5% HNO3 + 10% glycerol | 25 °C | 2 min | 17 |
| Geological materials | Slurry sampling | 65% HNO3 + 37% HCl +50% HF | 60 °C | 60 min | 18 |
| Road dust | Slurry sampling | 1% HNO3 | 25 °C | 34 min | 19 |
| Cement, mortar, and ceramic residues | Slurry sampling | 1% HNO3 + 1% Triton X-100 | 25 °C | 1 min | This work |
4. Conclusions
A new slurry sampling HR-CS GFAAS method was developed to determine Pb, Ni, Fe, and Mn in ceramic waste (CW), cement (OPC), and cement–mortar samples. Stable suspensions for carrying out the GFAAS measurements were prepared by mixing 10.0 mg of the solid powder sample with 10.0 mL of a 1% (v/v) Triton X-100 in 1% (v/v) HNO3 solution in an ultrasonic bath for 1.0 minutes. The analysis was performed using a unique suspension of the construction material and the metals were determined at concentrations between 1.5 and 80 μg g−1 for Pb, 4.0 and 75 μg g−1 for Ni, 2.0 and 600 μg g−1 for Mn and 0.15 and 60 mg g−1 for Fe. In comparison with methods based on XRF, 10 times better LODs and LOQs for Pb and Ni were achieved with the developed slurry sampling HR-CS GFAAS method. The developed method does not need the use of HF and the sample preparation requires less than 5 minutes. In our opinion, the proposed method could be a useful, simple, and environmentally friendly alternative tool for the routine analyses of construction materials since the use of corrosive reagents and the generation of toxic and hazardous wastes is minimized without deterioration of the analytical performance. This slurry sampling procedure could be used to develop new analytical methods for the determination of other trace elements, and minor and/or major elements in construction material samples after the proper optimization of the GFAAS temperature program and evaluation of spectral interferences.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript: B. Gómez-Nieto: investigation, conceptualization, writing-original draft, and editing. C. Isabel-Cabrera: investigation. M. J. Gismera: conceptualization, supervision, writing-original draft, and editing. M. T. Sevilla: conceptualization, supervision, writing-original draft, and editing. J. R. Procopio: reviewing, editing, and funding acquisition. M. I. Sanchez de Rojas: reviewing, editing, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research received funding from Spanish Ministry of Science and Innovation (project BIA2016-76643-C3-1-R). Carmen Isabel thanks to Community of Madrid and European Social Fund for the contract PEJ-2019-TL/IND-14286 through the Youth Employment Initiative (YEI).References
- Z. Huang, K. Liu, J. Duan and Q. Wang, A review of waste-containing building materials: Characterization of the heavy metal, Constr. Build. Mater., 2021, 309, 125107 CrossRef CAS.
- E. Asensio, C. Medina, M. Frías and M. I. S. de Rojas, Characterization of Ceramic-Based Construction and Demolition Waste: Use as Pozzolan in Cements, J. Am. Ceram. Soc., 2016, 99, 4121–4127 CrossRef CAS.
- N. Mijatović, A. Terzić, L. Pezo, L. Miličić, A. Milosavljević and D. Živojinović, Novel Approach for Determination of Potentially Toxic Elements via ICP–OES in Aqueous Solutions of Building Materials with Industrial Byproduct Addition, Sci. Sintering, 2019, 51, 429–444 CrossRef.
- D. P. Zhang, D. S. Xue, Y. H. Liu, B. Wan, Q. Guo and J. J. Guo, Comparative Study of Three Mixing Methods in Fusion Technique for Determining Major and Minor Elements Using Wavelength Dispersive X-ray Fluorescence Spectroscopy, Sensors, 2020, 20, 5325 CrossRef CAS PubMed.
- J. O. Ogunbileje, V. M. Sadagoparamanujam, J. I. Anetor, E. O. Farombi, O. M. Akinosun and A. O. Okorodudu, Lead, mercury, cadmium, chromium, nickel, copper, zinc, calcium, iron, manganese and chromium (VI) levels in Nigeria and United States of America cement dust, Chemosphere, 2013, 90, 2743–2749 CrossRef CAS PubMed.
- V. Sandroni, C. M. M. Smith and A. Donovan, Microwave digestion of sediment, soils and urban particulate matter for trace metal analysis, Talanta, 2003, 60, 715–723 CrossRef CAS PubMed.
- W. Zhang and Z. Hu, Recent advances in sample preparation methods for elemental and isotopic analysis of geological samples, Spectrochim. Acta, Part B, 2019, 160, 105690 CrossRef CAS.
- S. Garrigues and M. de la Guardia, Non-invasive analysis of solid samples, TrAC, Trends Anal. Chem., 2013, 43, 161–173 CrossRef CAS.
- D. L. Rocha, A. D. Batista, F. R. P. Rocha, G. L. Donati and J. A. Nóbrega, Greening sample preparation in inorganic analysis, TrAC, Trends Anal. Chem., 2013, 45, 79–92 CrossRef CAS.
- L. K. Andersen, T. J. Morgan, A. K. Boulamanti, P. Álvarez, S. V. Vassilev and D. Baxter, Quantitative X-ray Fluorescence Analysis of Biomass: Objective Evaluation of a Typical Commercial Multi-Element Method on a WD-XRF Spectrometer, Energy Fuels, 2013, 27, 7439–7454 CrossRef CAS.
- M. J. Gutiérrez-Ginés, J. Pastor and A. J. Hernández, Assessment of field portable X-ray fluorescence spectrometry for the in situ determination of heavy metals in soils and plants, Environ. Sci.: Processes Impacts, 2013, 15, 1545–1552 RSC.
- S. L. C. Ferreira, M. Miró, E. G. P. da Silva, G. D. Matos, P. S. dos Reis, G. C. Brandao, W. N. L. dos Santos, A. T. Duarte, M. G. R. Vale and R. G. O. Araujo, Slurry Sampling—An Analytical Strategy for the Determination of Metals and Metalloids by Spectroanalytical Techniques, Appl. Spectrosc. Rev., 2010, 45, 44–62 CrossRef CAS.
- I. López-García, E. Navarro, P. Viñas and M. Hernández-Córdoba, Rapid determination of lead, cadmium and thallium in cements using electrothermal atomic absorption spectrometry with slurry sample introduction, Fresenius. J. Anal. Chem., 1997, 357, 642–646 CrossRef.
- D. J. Butcher, Innovations and developments in graphite furnace atomic absorption spectrometry (GFAAS), Appl. Spectrosc. Rev., 2021, 1–18 Search PubMed.
- R. C. Machado, D. F. Andrade, D. V. Babos, J. P. Castro, V. C. Costa, M. Aurelio Sperança, J. Augusto Garcia, R. R. Gamela and E. R. Pereira-Filho, Solid sampling: advantages and challenges for chemical element determination—a critical review, J. Anal. At. Spectrom., 2020, 35, 54–77 RSC.
- E. Q. Oreste, A. O de Souza, C. C. Pereira, D. H. Bonemann, M. A. Vieira and A. S. Ribeiro, Evaluation of sample preparation methods for the determination of Cd, Cr and Pb in ceramic tableware by graphite furnace atomic absorption spectrometry, Anal. Lett., 2020, 53, 436–458 CrossRef CAS.
- L. Husáková, T. Šídová, L. Ibrahimová, M. Svízelová and T. Mikysek, Direct determination of lead in bones using slurry sampling and high-resolution continuum source electrothermal atomic absorption spectrometry, Anal. Methods, 2019, 11, 1254–1263 RSC.
- P. Zhang, Z. Yang, T. Yu, Q. Hou, X. Xia, G. Gao and R. Yang, Determination of Pb in Geological Materials by Heat Extraction Slurry Sampling ET-AAS, At. Spectrosc., 2020, 41, 205–210 CAS.
- S. S. Oliveira, V. S. Ribeiro, T. S. Almeida and R. G. O. Araujo, Quantification of ytterbium in road dust applying slurry sampling and detection by high-resolution continuum source graphite furnace atomic absorption spectrometry, Spectrochim. Acta, Part B, 2020, 171, 105938 CrossRef CAS.
- J. Briceño, M. A. Belarra, K. A. C. D. Schamphelaere, S. Vanblaere, C. R. Janssen, F. Vanhaecke and M. Resano, Direct determination of Zn in individual Daphnia magna specimens by means of solid sampling high-resolution continuum source graphite furnace atomic absorption spectrometry, J. Anal. At. Spectrom., 2010, 25, 503–510 RSC.
- B. Gómez-Nieto, M. J. Gismera, M. T. Sevilla and J. R. Procopio, Fast sequential multi-element determination of major and minor elements in environmental samples and drinking waters by high-resolution continuum source flame atomic absorption spectrometry, Anal. Chim. Acta, 2015, 854, 13–19 CrossRef PubMed.
- B. Gómez-Nieto, V. Motyzhov, M. J. Gismera, J. R. Procopio and M. T. Sevilla, Fast-sequential determination of cadmium and copper in milk powder and infant formula by direct solid sampling high-resolution continuum source graphite furnace atomic absorption spectrometry, Microchem. J., 2020, 159, 105335 CrossRef.
- B. Gómez-Nieto, M. J. Gismera, M. T. Sevilla and J. R. Procopio, Simultaneous and direct determination of iron and nickel in biological solid samples by high-resolution continuum source graphite furnace atomic absorption spectrometry, Talanta, 2013, 116, 860–865 CrossRef PubMed.
- B. M. Soares, R. F. Santos, R. C. Bolzan, E. I. Muller, E. G. Primel and F. A. Duarte, Simultaneous determination of iron and nickel in fluoropolymers by solid sampling high-resolution continuum source graphite furnace atomic absorption spectrometry, Talanta, 2016, 160, 454–460 CrossRef CAS PubMed.
- F. R. Adolfo, P. C. do Nascimento, G. C. Leal, D. Bohrer, C. Viana and L. M. de Carvalho, Simultaneous determination of Fe and Ni in guarana (Paullinia cupana Kunth) by HR-CS GF AAS: Comparison of direct solid analysis and wet acid digestion procedures, J. Food Compos. Anal., 2020, 88, 103459 CrossRef CAS.
- H. Tinas, N. Ozbek and S. Akman, Direct solid sampling determination of lead in cheese varieties by high resolution continuum source graphite furnace atomic absorption spectrometry, Anal. Methods, 2017, 9, 6365–6370 RSC.
- H. Tinas, N. Ozbek and S. Akman, Determination of lead in flour samples directly by solid sampling high resolution continuum source graphite furnace atomic absorption spectrometry, Spectrochim. Acta, Part B, 2018, 140, 73–75 CrossRef CAS.
- Government Decree Concerning the Recovery of Certain Wastes in Earth Construction 591/2006, Finland, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ay02036j |
| This journal is © The Royal Society of Chemistry 2023 |