
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiply charged ion profiles in the UHPLC-HRMS analysis of palytoxin analogues from Ostreopsis cf. ovata blooms†
Noemí Inmaculada
Medina-Pérez
 ab,
Francisco Javier
Santos
ac,
Elisa
Berdalet
b and
Encarnación
Moyano
*ac
ab,
Francisco Javier
Santos
ac,
Elisa
Berdalet
b and
Encarnación
Moyano
*ac
aDepartment of Chemical Engineering and Analytical Chemistry, University of Barcelona, Barcelona, Spain. E-mail: encarna.moyano@ub.edu
bDepartment of Marine Biology and Oceanography, Institute of Marine Sciences (ICM-CSIC), Barcelona, Spain
cWater Research Institute (IdRA), University of Barcelona, Barcelona, Spain
First published on 8th February 2023
Abstract
Analogues of palytoxin (PLTX), one of the most potent marine biotoxins, are produced by some species of the marine dinoflagellates of the genus Ostreopsis. The proliferation of these species in different coastal zones represents a potential threat of seafood poisoning in humans because the produced toxins can be transferred through marine food webs. Thus, the determination of the concentration of PLTX analogues (ovatoxins—OVTXs, ostreocins—OSTs and isobaric PLTX) in different matrices (seawater, marine fauna, etc.) is necessary to protect human health. This study is addressed to overcome some of the challenges that the chemical complexity of these molecules poses to their quantification by ultra-high-performance liquid chromatography high-resolution mass spectrometry-based techniques (UHPLC-HRMS). In particular, the mass spectra of the palytoxin analogues show the presence of a large number of ions (including mono- and multiply charged ions) whose nature, relative abundances and behavior can lead to quantitation errors if the correct ions are not selected. In this work, the variability of the PLTX and OVTX profiles under different instrument conditions, including the use of diverse electrospray generation sources and different quantitation methods, is studied. Moreover, the extraction protocol in seawater containing Ostreopsis sp. ovata cells is also evaluated. The use of a heated electrospray operating at 350 °C and a quantitative method including ions from different multiply charged species provides a more robust and reliable method for overcoming the problems due to the variability in the toxin's mass spectrum profile. A single MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (80:20, v/v) extraction is proposed as the best and reliable procedure. The overall method proposed was applied to quantify OVTXs (-a to -g) and iso-PLTX along the 2019 Ostreopsis cf. ovata bloom. The cells contained a total toxin concentration of up to 20.39 pg per cell.
:H2O (80:20, v/v) extraction is proposed as the best and reliable procedure. The overall method proposed was applied to quantify OVTXs (-a to -g) and iso-PLTX along the 2019 Ostreopsis cf. ovata bloom. The cells contained a total toxin concentration of up to 20.39 pg per cell.
1. Introduction
Certain benthic dinoflagellates, which grow preferentially attached to biotic and abiotic substrates in the seabed, are of special interest because some of them can produce toxic compounds that affect marine fauna and humans. In particular, Ostreopsis, a genus of tropical origin, has been increasing its biogeographic distribution to temperate waters since the beginning of the 21st century and producing recurrent blooms on certain Mediterranean beaches.1 Some Ostreopsis species can produce palytoxin analogues (ovatoxins—OVTXs, ostreocins—OSTs and isobaric palytoxin—iso-PLTX)2–7 that have been related, although not completely proven yet, to dramatic seafood poisonings in tropical areas.8 In the Mediterranean, iso-PLTX, OVTX-a and OVTX-b were found in the tissues of some bivalve (mussels, clams, and hairy mussels) and other marine fauna collected from Greek, French and Italian coasts, but seafood-borne poisonings have not been (luckily) detected in humans after their consumption.9–14 However, respiratory and cutaneous irritations, general malaise and other relatively mild symptoms have been documented in beach users of certain areas (in Algeria, Croatia, France, Italy and Spain) where Ostreopsis spp. proliferations occur.15–20 Massive mass mortalities of benthic marine fauna have also been found concurrently with high concentrations of Ostreopsis species. For this reason, the recurrence of Ostreopsis proliferation in touristic areas in summer and the potential risk of food poisoning have raised concern and incentivized monitoring to prevent impacts on human health.17,21 The biogeographic expansion of Ostreopsis spp., likely linked to climate change and anthropic pressures on the coasts, is also a matter of study,22 especially after the important bloom episodes in the Bay of Biscay (France) affecting between 700 and 3000 people in 2021.23Methodological advances in the last few decades have allowed the efficient and reliable development of extraction and analytical determination methods (chromatography and mass spectrometry) for the most common phycotoxins (saxitoxin, domoic acid, lipophilic toxins, and microcystins) whose presence in seafood and drinking water is strictly controlled by European regulations.24,25 However, the analogues produced by Ostreopsis have not yet been included in routine monitoring as blooms of these microalgae are still an emerging and poorly characterized issue that requires fundamental research of their dynamics and impacts on human health and the environment. Furthermore, it is also necessary to overcome some analytical methodology limitations related to the quantitative analysis of toxins produced by Ostreopsis in different matrices.
The extraction of PLTX analogues from samples or cultures of Ostreopsis spp. is conducted on cell pellets obtained by centrifugation or filtration (nylon filters). Toxins are extracted using mixtures of MeOH:H2O (50:50 or 80:20)2,7,12,26,27 with, sometimes, the addition of small percentages of acetic or formic acid to improve recovery.28–30 Despite some differences in the extraction used in the published studies, the obtained recoveries are similar to those reported by Ciminiello et al., i.e. 91–98%.7
Nowadays, PLTX analogues are mainly determined by mass spectrometry (MS)-based techniques, since they offer high detection capabilities and provide exhaustive structural information and accurate quantitative analysis with a relevant degree of selectivity. Different OVTX analogues (OVTX-a to -i) produced by Ostreopsis spp. have been identified by multiple-stage mass spectrometry (MSn) and high-resolution mass spectrometry (HRMS),3,5 and the isobaric palytoxin molecule produced by the microalga has also been identified as a structural isomer of the palytoxin produced by the soft coral Palythoa tuberculosa.2
PLTX analogues are chromatographically separated by UHPLC using reversed-phase C18 columns7,30 and detected by mass spectrometry using electrospray ionization (ESI) as an ionization technique and a hybrid mass spectrometer such as a linear ion trap-Orbitrap (LIT-Orbitrap).3,29,31 By electrospray, a large number of mono- and multiply charged ions are mainly generated by proton transfer in addition to adducts formed with monovalent and divalent cations (Na+, K+, Ca2+, Mg2+, and Fe2+). Additionally, some fragment ions because of in-source collision-induced dissociation (in-source CID) processes can also be generated. The ESI-HRMS spectrum profile is used as a fingerprint for the identification of both iso-PLTX and OVTXs produced by Ostreopsis cf. ovata.
So far, the quantitative estimation of iso-PLTX and OVTX analogues has been based on the selection of only two ions: [M + 2H+–H2O]2+ as representative of doubly charged ions and [M + H + Ca]3+ or [M + 2H + K]3+ for triply charged ions depending on the ion abundance observed.2,29,32,33 However, the large number of ions that can be generated by ESI from each molecule and the subsequent variation of the mass spectral profile28 (depending on the working conditions) can have relevant implications for the correct quantification of the concentration of each analogue.
In this work, two key aspects in the quantitative determination of the PLTX analogues produced by O. cf. ovata are studied: the variability of the PLTX analogue mass spectral profiles under different instrument conditions, including the use of different electrospray sources and different extraction solvents. The proposed method was then applied to determine the concentrations of PLTX analogues along the 2019 natural O. cf. ovata bloom.
2. Materials and methods
2.1. Chemicals and materials
The PLTX standard (from Palythoa tuberculosa) was purchased from Wako Chemicals GmbH (Germany) (lot number: SAH7036), and it was used to prepare standard calibration solutions. Water, acetonitrile (ACN), methanol, formic acid and acetic acid (LC-MS grade >98%) used to prepare mobile phases and standard solutions and as extraction solvents were purchased from Sigma-Aldrich (Steinheim, Germany).PLTX stock standard solution (100 μg mL−1) was prepared by weight in MeOH:H2O (50:50, v/v). PLTX calibration working solutions were prepared at concentration levels ranging from 0.005 to 3 μg mL−1 by dilution of the stock standard solution in MeOH:H2O (80:20, v/v). All standard working solutions were stored at −20 °C until their use.
Solvents used for the mobile phase were filtered through 0.22 μm nylon membrane filters (Whatman, Clifton, NJ, USA) before use. Nitrogen (99.95%) supplied by Linde (Barcelona, Spain) was used as the sheath gas and auxiliary gas in a heated electrospray ionization (HESI) source. GF/F glass fiber filters (25 mm diameter) were provided by Whatman (Clifton, NJ, USA). Neutral Lugol solution was prepared with I2 and IK, from Merck, Darmstadt, Germany.34
2.2. Instrumentation and analytical procedure
System I was a hybrid mass spectrometer equipped with a Q-Orbitrap (Q-Exactive Orbitrap, Thermo Fisher Scientific) mass analyzer and a heated electrospray of second-generation (HESI-II) as an ionization source, which operated in positive ion mode. The HESI-II source used nitrogen as sheath gas, sweep gas and auxiliary gas at flow rates of 60, 0 and 10 au (arbitrary units), respectively. The temperatures of the vaporizer and ion transfer tube were set at 350 °C (Method 1) or 25 °C (Method 2) and 275 °C, respectively. The spray voltage was +3.5 kV and the S-lens RF level was 70 au. The Q-Orbitrap mass spectrometer operated in positive ion mode and full scan MS mode (m/z 800–1500) at a mass resolution of 70000 at full width at half maximum (FWHM) (m/z 200). The Xcalibur software v2.1 (Thermo Fisher Scientific) was used to control the UHPLC-HRMS system and to acquire and process the mass spectrometry data.
System II was a hybrid mass spectrometer equipped with a LIT-Orbitrap (LTQ-Orbitrap Velos, Thermo Fisher Scientific) and a classical electrospray of a second-generation (ESI) source without thermal assistance (Method 3), which operated in positive ion mode. The ESI source used nitrogen as sheath gas, sweep gas and auxiliary gas at flow rates of 60, 0 and 10 au (arbitrary units), respectively. The temperature of the ion transfer tube was set at 275 °C. The spray voltage and the S-lens RF level were +3.5 kV and 70 au, respectively. The LIT-Orbitrap mass spectrometer operated in full scan MS mode (m/z 800–1500) at a mass resolution of 70000 at full width at half maximum (FWHM) (m/z 200). The Xcalibur software v2.1 (Thermo Fisher Scientific) was used to control the UHPLC-HRMS system and to acquire and process the MS data.
Analytical standards for iso-PLTX and OVTXs are not available; thus, their quantitative determination was based on PLTX equivalents. The calculation of the elemental formulae was based on the monoisotopic mass with a mass tolerance of <5 ppm and an isotope fit score of >80%. Extracted ion chromatograms for iso-PLTX and OVTXs were reconstructed using the integrated signal of the ions selected (Table S2†) from the different cluster ions observed for each toxin ([M + 3H–4H2O]3+, [M + 3H–3H2O]3+, [M + 3H–2H2O]3+, [M + 3H–H2O]3+, [M + H + Mg]3+, [M + H + Ca]3+, [M + H + Fe]3+, [M + 2H–3H2O]2+, [M + 2H–2H2O]2+, [M + 2H–H2O]2+, [M + 2H]2+, [M + H + Na]2+, and [M + H + K]2+). The limit of quantitation of the PLTX standard was estimated by injecting a blank sample filter extract spiked with the PLTX standard at low concentration levels and considering a signal-to-noise ratio (S/N) of 10.
:H2O (80:20, v/v) and an ice-cooled ultrasonic bath (5 min). The supernatant was centrifuged (3893 g, 4 °C, 5 min), filtered through a 0.22 μm nylon membrane and stored in amber glass vials at – 80 °C until their analysis by UHPLC-HRMS.
This extraction protocol was applied to evaluate extraction efficiency (EE%) and the matrix effect (ME%) using filters containing O. cf. ovata cells obtained during the bloom and proved to be free of any PLTX analog by previous analysis. These filters will be referred to as “blank sample filters”.
For the EE%, three blank sample filters were spiked with a 2 μg PLTX standard and submitted to the indicated extraction procedure, in order to obtain 1 μg mL−1 of the PLTX standard in the final extract. In parallel, three non-spiked blank sample filters were also extracted and the PLTX standard was added at a final concentration of 1 μg mL−1 in the obtained extracts and before the injection into the UHPLC-HRMS system. To evaluate the EE%, the signal of the PLTX standard in the two obtained extracts was compared.
The ME% has been evaluated by analyzing four different extracts of the non-spiked blank sample filters (post-extraction). The PLTX standard was added to four aliquots of each extract to obtain four spiked concentration levels (0.035, 0.200, 0.500 and 1 μg mL−1) that were injected into the UHPLC-HRMS system in triplicate. The obtained areas were interpolated into an external calibration PLTX curve to obtain the recovered concentration. The ME% was estimated by correlating the recovered vs. the spiked concentration of each extract [ME% = (1 − slope) × 100].
For clarity, Table S1† shows a summary of the analytical methods tested with an indication of the instrumentation and electrospray ionization systems used and the type of quantification approach applied.
2.3. Study site and Ostreopsis cf. ovata bloom sampling in 2019
The natural samples for this study were obtained from the benthic Ostreopsis cf. ovata bloom that occurred in the 2019 summer at the rocky beach of Sant Andreu de Llavaneres (Catalan coast of Spain, 41°33.13′N; 2°29.54′E). At this sampling site, the sedimentary substrate extends from the shoreline to at least 5–7 m depth and is densely colonized by macroalgae, mainly Ellisolandia elongata Ellis & Solander and Jania rubens (Linnaeus) J. V. Lamouroux. To these macroalgae, Ostreopsis adheres and proliferates during the summer-fall period.17,35 From July 19th to November 7th, samplings were performed at a very shallow depth (20–50 cm) at three points (A, B and C) separated by approximately 10 m among them, to monitor the O. cf. ovata bloom and characterize the temporal toxin variation.On every sampling day, ca. 10–20 g of fresh macroalgae were collected and transferred into a 250 mL plastic bottle and filled with a known volume (200–300 mL) of the surrounding seawater. Each collected sample was then vigorously shaken for 1 minute, and subsequently sieved through a 200 μm mesh. A subsample of the percolated seawater (125 mL), which contained O. cf. ovata and the accompanying microbiota, was preserved with 0.5 mL of Lugol solution to estimate the concentration of O. cf. ovata cells. Ostreopsis cf. ovata cell concentrations were estimated by using a 1 mL Sedgwick–Rafter counting cell chamber under a Leica-inverted light microscope. This is a suitable counting method to quantify the number of cells in the range of 105–106 cells per Lwater, with a 5% confidence limit. The sampling and counting method in this study had an estimated global variation coefficient (CV%) of 20%. The macroalgae were slightly wiped and weighed. Cell abundances were expressed as cells per g of fresh weight (FW) of macroalga. For toxin characterization, 5 to 30 mL subsamples of the percolated water were filtered through 25 mm GF/F filters and frozen at −80 °C until extraction. Water temperature and salinity were measured using a microprocessor conductimeter WTW (Model LF197).
Some samples collected during the bloom were used to conduct several methodological tests and are explained in the different sections of this paper.
3. Results and discussion
3.1. UHPLC-HRMS of PLTX analogues
Traditionally, the methods published describe the use of HPLC separations of PLTX analogues based on reversed-phase chromatography using C18 columns (Gemini C18) packed with 3 μm fully porous particles and ACN:H2O (with acetic acid) mobile phases.29,36–38 However, in the last decade, most methods proposed the use of UHPLC columns packed with superficially porous particles (Poroshell 120 EC-C18 and Kinetex C18 columns)5,14,39 and Ternon et al.40 used 1.8 μm fully porous particles for the first time .41,42 In the present work, an Hypersil GOLD C18 column packed with 1.9 μm fully porous particles is used to take advantage of the higher chromatographic efficiency it provides. The chromatographic separation of PLTX analogues was optimized in the present study by using an O. cf. ovata extract that contains most of the target compounds (Fig. 1).
 | ||
| Fig. 1 (a) UHPLC-HRMS chromatogram of a 2019 sample extract that contains the PLTX analogues (m/z of the extracted ion chromatogram is shown) (the ions listed are the monoisotopic mass of the most intense isotopes); (b) HRMS spectra corresponding to the PLTX standard at 1 μg mL−1; (c) zoomed view of two m/z areas to visualize the isotope cluster of doubly and triply charged ions, as an example (mass error < 5 ppm). | ||
Different gradient elution programs and different mobile phase composition (ACN:H2O with acetic or formic acid) were tested. The two most intense ions observed were both the triply charged [M + 3H–3H2O]3+ and the doubly charged [M + 2H–H2O]2+, which showed the same relative abundance when using both mobile phases (adding acetic or formic acid). However, by using formic acid the chromatographic peak was slightly narrower indicating an improvement in the chromatographic efficiency (7199 theoretical plates when using formic acid vs. 6741 theoretical plates when using acetic acid). This fact was probably due to the effect of the lower pKa of formic acid compared to that of acetic acid; the higher acidity favored the protonation of the silanol groups on the surface of the silica particles preventing their collateral interactions with protonated basic nitrogen on the toxin molecule. Furthermore, the slow step gradient elution (from 30% to 35% in 12 min, Section 2.2.1) using ACN:H2O (0.1% formic acid) allowed the baseline chromatographic separation of all compounds in less than 6 minutes (Fig. 1a). As can be seen in Fig. 1a, iso-PLTX elutes ca. 1 min earlier than the PLTX standard.
The chromatographic system was coupled to two mass spectrometer systems (System I and II) equipped with different generations of electrospray sources (Method 1, 2 or 3, detailed below). The study of the HRMS spectra of iso-PLTX and OVTXs revealed the formation of a large number of ions for each target compound. As an example, Fig. 1b shows the HRMS spectrum obtained for the PLTX chromatographic peak in the analysis of a PLTX standard solution (1 μg mL−1) by UHPLC-HRMS (HESI-II, Method 1 at 350 °C), and Fig. 1c shows zoomed views of the m/z areas of doubly and triply charged ions. Furthermore, Table S2† summarizes the most abundant ions (exact mass and monoisotopic mass of the most abundant isotope) observed in the UHPLC-HRMS analysis of an O. cf. ovata 2019 sample extract that contained the target marine biotoxins (iso-PLTX and OVTXs). The complex HRMS spectra include doubly and triply charged species originated by proton transfer mechanisms (e.g. [M + 2H]2+), often with subsequent in-source CID fragmentation (e.g. [M + 2H–H2O]2+ or [M + 3H–H2O]3+) and adduct ion formation with monovalent and divalent cations (e.g. [M + 2H + Na]2+, [M + H + K]2+ or [M + H + Ca]3+). These complex mass spectra, while challenging the quantitative analysis, are useful for the identification of PLTX congeners. Furthermore, some of these ions can be confused with other possible nearby isobaric ions, with differences in their m/z values lower than 0.005 indicating that it is necessary for a mass resolution higher than 400000 FWHM to distinguish these ions. In fact, the maximum available mass resolution in the Orbitrap instrument used was 140000 FWHM. For instance, the ion at 1359.7267 (the most abundant isotope) could be assigned to both [M + H + K]2+ (3.7 ± 1.4 ppm) or [M + Ca]2+ (1.5 ± 1.0 ppm) and the ion at 901.4914 (the most abundant isotope) could also be assigned to [M + H + Mg]3+ (2.5 ± 2.3 ppm) or [M + 2H + Na]3+ (2.3 ± 2.2 ppm). For the final assignment (Table S2†), the information obtained from the experiments where alkali ions were added by post-column addition (discussed in Section 3.2) was useful, which was in agreement with the results reported by Ciminiello et al.31 In preliminary tests, it was observed that the nature of ions and their relative abundances changed with working conditions. Thus, a detailed study of these variations using different instrument conditions and different generations of electrospray ionization sources was conducted. The PLTX standard solution used was freshly prepared from the stock standard solution for each injection at an identical concentration (1 μg mL−1). Fig. 2 shows the HRMS spectra of the PLTX standard analyzed at different times: on the day, one week and one year later (stock standard maintained frozen at −80 °C) and using the same instrument and source parameters after instrument maintenance and tuning. As shown in Fig. 2, the relative ion ratio of the different ions changed over time resulting in significant modification of the mass spectrum profile. In particular, focusing on the most abundant ions, [M + H + Ca]3+ at m/z 906.4828, [M + 2H–H2O]2+ at m/z 1331.2418 and [M + 3H–nH2O]3+ at m/z 887.8303, when the abundance of one of them decreased, the relative contribution of the other increased. These differences might depend on the state of the instrument (cleanliness and small variations in the voltages applied to the lenses, temperatures and gas flow rates) and the salinity of samples injected before the analysis of the PLTX standard. This is an important fact to consider, especially for those instruments that process samples of different natures and concentrations.
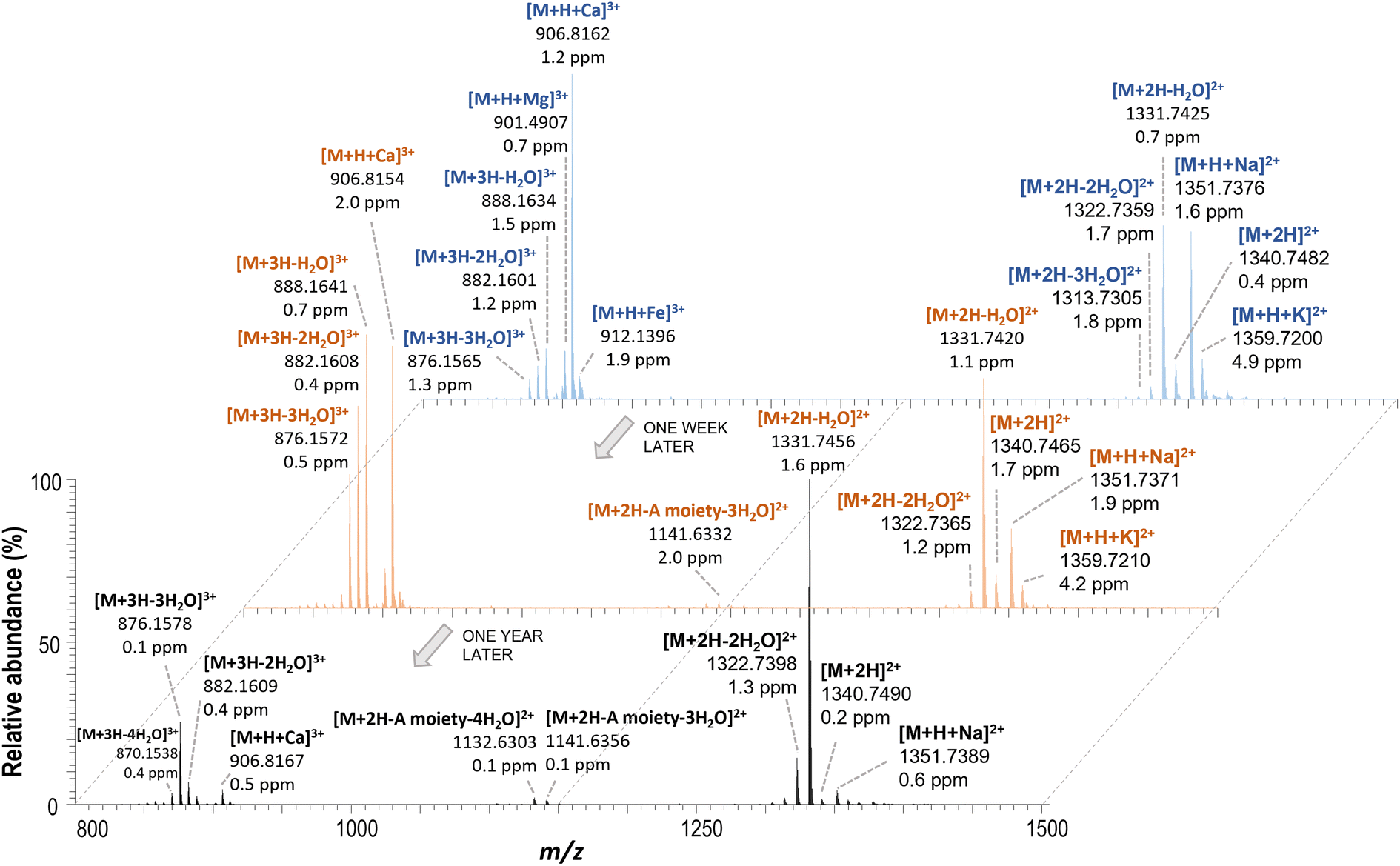 | ||
| Fig. 2 HRMS spectra of freshly prepared PLTX standard solutions (1 μg mL−1) from the stock solution made after purchasing the chemical and one week and one year later by using the same UHPLC-HRMS instrument conditions and electrospray source (ESI, Method 3). | ||
Moreover, the intensity of the different ions and their relative abundances also varied when the type of electrospray ionization source changed (Fig. 3). In the analysis of the PLTX standard, the intensity of all ions was higher when the HESI-II source was used with the vaporizer temperature set at 350 °C (Method 1, Table S1†). This may be due to the efficient desolvation/ionization of this electrospray source that produced a higher number of gas-phase ions. However, the abundance of fragment ions due to water loss increased, indicating that they could be related to possible thermal degradation. Nevertheless, HESI-II (Method 1, 350 °C) is preferred to the other electrospray sources that work at room temperature, because the ion responses are much higher. Therefore, these conditions were proposed for toxin analysis along the O. cf. ovata 2019 bloom.
 | ||
| Fig. 3 Intensity of ions detected in the UHPLC-HRMS analysis of the PLTX standard (1 μg mL−1) using HESI-II (Method 1 or 2) or ESI (Method 3). | ||
3.2. Effect of the presence of calcium/sodium on the toxin mass spectral profiles
To investigate the effect of salinity on the mass spectral profile of PLTX analogues, the post-column addition of Ca2+ or Na+ was performed and compared with that under control conditions. It was observed that the ionic species changed depending on the presence of alkali metal ions in the UHPLC-HRMS system (Fig. S1†). The base peak in the mass spectrum changed from the triply charged ion [M + H + Ca]3+ when Ca2+ was added (Fig. S1b†) to the doubly charged ion [M + H + Na]2+ when Na+ was added (Fig. S1c†), while ions such as [M + 2H+–H2O]2+ and [M + 3H–nH2O]3+ were still present but at different relative abundance. Furthermore, the intensity of the ions at m/z 1359.7267 assigned to [M + H + K]2+ and m/z 901.4914 assigned to [M + H + Mg]3+ remained unchanged or decreased with the addition of Ca2+ or Na+, respectively. These results compared to the control ones (Fig. S1a†) were in agreement with those of Ciminiello et al.31 that stated that the addition of divalent cations (Ca2+) favour the formation of doubly charged ions and the divalent cations (Na+) favour the formation of triply charged ions. These results evidence that small variations in salt composition and concentration, in both the sample and mobile phase or even in the mass spectrometer environment, could produce changes in the HRMS spectrum profile of these toxins, which is a relevant aspect for the determination of marine toxins.3.3. Toxin extraction
The literature shows that different protocols are being applied for the extraction of PLTX analogues from macroalgal samples. Essentially, the main differences between these protocols are in the methanol:water ratio and in the number of extraction steps used. In the present study, a double extraction vs. a single extraction using two extraction solvents, MeOH:H2O (50:50, v/v) and MeOH:H2O (80:20, v/v), were tested. For each condition and in triplicate, the same volume (20 mL) of the percolated seawater containing O. cf. ovata cells, obtained from the benthic samples (Section 2.3), was filtered through GF/F filters. The highest total toxin concentration in a sample (1037 ± 83 ng mL−1) was obtained with a double extraction in MeOH:H2O (80:20, v/v), while, the single extraction in MeOH:H2O (80:20, v/v) yielded only 98 ± 4%. In MeOH:H2O (50:50, v/v), the toxin extracted accounted for 53 ± 1% in one single step and 42 ± 9% in a double step. In consequence, a single extraction with MeOH:H2O (80:20, v/v) was proposed for further studies and EE% for these conditions (detailed in the Experimental section 2.2.3) was 95–116%. The ME% was evaluated as indicated in Section 2.2.3 and the correlation between the recovered and the spiked concentration is plotted in Fig. S2.† The slopes of the correlation curves for the four different extracts were between 0.86 and 1.02 indicating a ME% from −14% to 2%.
3.4. Effect of UHPLC-HRMS configuration systems on the quantitation of PLTX analogues
All the samples collected during the O. cf. ovata bloom in 2019 and extracted with the optimized extraction method (described in the Experimental section 2.2.3) were analyzed using System I (Methods 1 and 2) and System 2 (Method 3). Additionally, they were quantified with two ions (QM-A) or with the integrated signal of the most abundant isotope of all adduct ions (>5%) (QM-B), providing the results in picograms of each toxin per O. cf. ovata cell (pg per cell). A paired t-test at a 5% significance level was applied to evaluate the statistical differences between the three different methods used (Table S3†). The results showed that when using the quantitation method QM-A, there were significant differences (p-value < 0.05) between the three instrument methods (Methods 1–3). However, when using the acquisition method QM-B (the integrated signal of the most abundant isotopes of all adduct ions), there were no significant differences (p-value = 0.183) between instrumental Methods 1 and 2. This means that only the HESI-II source (Methods 1 and 2) provided comparable results. Furthermore, if the quantification methods are compared (QM-A vs. QM-B) using the three different methodologies, the p-values (using one or two tails) were always smaller for Method 1 (HESI-II at 350 °C), than those found when using Method 2 (HESI-II at room temperature) or Method 3 (ESI source), indicating that Method 1 was yielding more comparable results.As has been shown, the most abundant ions in the toxin profile can change in the ionization/mass spectrometry acquisition process, depending on the instrumental conditions used, but the ion profile can also change even between ovatoxins. Fig. 4 shows the abundance of OVTX-a (Fig. 4a) and OVTX-b (Fig. 4b) for two replicates (R1 and R2) of the same sample analyzed on different days. As can be observed, the abundance of the two most abundant ions ([M + 3H–3H2O]3+ and [M + 2H–2H2O]2+) for OVTX-a varies from one replicate (R1) to another (R2) despite continuing to be the most abundant in both replicates. Conversely, the nature of the two most abundant ions for OVTX-b changes from one replicate to another. In the first replicate (R1), the two most abundant ions are [M + 3H–3H2O]3+ and [M + 2H–2H2O]2+ but in the second replicate (R2) were [M + 2H–2H2O]2+ and [M + 2H]2+. The quantification procedure with a larger number of ions should be more robust since it considers these changes in the toxin mass spectral profile. When the intensity of the most abundant ion decreases, the intensity of another ion in the profile should increase. Using method QM-B, the maximum RSD% observed was 8% for the PLTX standard, while the RSD% was up to 28% when using method QM-A.
 | ||
| Fig. 4 Relative abundance of some ions of the (a) OVTX-a and (b) OVTX-b and between two different replicates (R1 and R2). | ||
3.5. UHPLC-HESI-HRMS quality parameters
Table 1 summarizes the quality parameters estimated for the analysis of PLTX in O. cf. ovata samples using the proposed UHPLC-HESI-HRMS (System I – Method 1) method. The limit of quantification (LOQ), based on a S/N of 10 and estimated by spiking a blank sample filter extract with the PLTX standard at low concentrations, was 6.5 ng mL−1. Precision (run-to-run and day-to-day) and trueness were calculated by analyzing 5 replicates of the blank sample filter extract spiked with the PLTX standard at two concentration levels (low level: 75 ng mg−1; medium level: 800 ng mL−1). As can be seen in Table 1, precision values were always lower than 3.4% (RSD, %) and trueness values (rel. error, %) lower than 9.7%. These quality parameters indicated the good performance of the method for the determination of the target marine toxins.| Concentration | % | |
|---|---|---|
| Run-to-run precision (RSD, %) | Low level (75 ng mL−1) | 1.3 |
| Medium level (800 ng mL−1) | 1.0 | |
| Day-to-day precision (RSD, %) | Low level (75 ng mL−1) | 1.6 |
| Medium level (800 ng mL−1) | 3.4 | |
| Trueness (rel. error, %) | Low level (75 ng mL−1) | 7.6 |
| Medium level (800 ng mL−1) | 9.7 | |
| LOQ | 6.5 ng mL−1 |
3.6. Study of the Ostreopsis cf. ovata bloom in 2019
Using the proposed method, the temporal evolution of the toxin concentration per cell along the 2019 O. cf. ovata proliferation was studied. Fig. 5a shows the temporary abundance of O. cf. ovata per gram of macroalga (an indicator of the intensity of the bloom) and the total toxin content per cell over time. It was observed that the toxin concentration was similar in the 3 collecting points during the proliferation (Fig. S3a†), so the average concentration (ABC) is used. During the sampling period, the water temperature and salinity were 15–27 °C and 37.6–38.2 g L−1, respectively (Fig. S3b†). | ||
| Fig. 5 (a) Temporal evolution of the O. cf. ovata bloom in 2019 (referred to cell concentration per gram of fresh weight of macroalgae), with the average toxin cell content variation along the bloom. (b) Cell toxin profile (%) of the O. cf. ovata bloom samples. | ||
The bloom developed exponentially (cells per gFW) from the beginning of the sampling period (early June) until July 23rd. At this moment, the stationary phase of the bloom begins with a subsequent decline phase. The maximum toxin cell concentration was found at the beginning of the stationary phase with values between 14.66 and 19.11 pg per cell and in the decline phase with values of 19.29 ± 1.10 pg per cell (Fig. 5a). Concentrations of the same order of magnitude have been reported for other Mediterranean O. cf. ovata strains (22–75 pg per cell).27,42
The OVTX analogues-a to -g were detected in most of the analyzed extracts (2019), while iso-PLTX was only detected on August 6th and September 3rd. In this period, the highest O. cf. ovata concentration (marked in Fig. 5a with a circle) was observed and the total toxin cell concentrations oscillated between 0.01 and 0.04 pg per cell within this period. In the rest of the samples, iso-PLTX was below the quantitation or detection limit. Regarding the toxin profile, the relative abundance of individual toxins was higher for OVTX-a and OVTX-b (abundance > 25%) followed by OVTX-c (3.06 ± 0.23%), OVTX-d (6.78 ± 0.67%), and OVTX-e (6.09 ± 1.18%), while the contribution of the other toxin analogues (OVTX-f, OVT-g and iso-PLTX) corresponds to the remaining 2.5% (Fig. 5b). In all samples, OVTX-a and OVTX-f were the major and minor components of the toxin profile, respectively.
The main challenge posed by the O. cf. ovata blooms is to establish the link between cell abundance and toxin risk. The data suggest that there is no direct link and the intracellular toxin concentration was not constant, suggesting that complex interacting metabolic, physiological and ecological factors may be at play. Understanding the factors that control toxins is key to estimating the health and environmental risks associated with O. cf. ovata proliferations. Such studies are underway by many research groups today.
4. Conclusions
In this work, the variability of the PLTX analogue mass spectral profiles under different instrumental conditions, including the use of different generations of electrospray sources, was studied. Our results show that the variability in the intensity of the different ions could change using different electrospray ionization sources, but the abundances of ions can also change when analyzing the same sample at different times. These differences might depend on the state of the instrument and the salinity of samples injected before the analysis of PLTX analogues. This fact should be especially considered for those instruments in which different types of samples are processed. Among the methods tested, the use of HESI-II (350 °C – Method 1) for the ionization process and the quantitation method that monitors multiple ions are recommended. This approach provided the best quantitative results since it allowed for the best ionization efficiency and considered the potential changes that could take place in toxin mass spectral profiles. Regarding the extraction method, the results indicated that a single extraction with MeOH:H2O (80:20, v/v) is enough for the efficient extraction of PLTX analogues from these samples. Finally, the method proposed in this study showed a good performance and it was successfully applied for the study and quantitation of PLTX and OVTXs from the O. cf. ovata proliferation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the different financing agencies that supported the study. N. I. M.-P. was granted the fellowship FI-SDUR2020-00254 (AGAUR, Government of Catalonia). Financial support: MICIU (Spain, project PGC2018-095013-B-I00), AGAUR (Generalitat de Catalunya, project 2017SGR-310) and the Water Research Institute (IdRA, University of Barcelona). Support was provided also by the CoCliME project, an ERA4CS network (ERA-NET) initiated by JPI Climate and co-financed by FORMAS (SE) and the European Union (Grant no. 690462), ERC-2019-CoG-863934-GlycoSpec (European Research Council Consolidator Grant). Furthermore, the authors want to thank Laia Viure (ICM-CSIC) for the field samplings and logistics. CoCliME is endorsed to the IOC/SCOR Program GlobalHAB and contributes to the implementation of its research plan, concerning in particular, toxin determination.Notes and references
- A. S. Pavaux, E. Berdalet and R. Lemée, Front. Mar. Sci., 2020, 7, 498 CrossRef.
- M. García-Altares, L. Tartaglione, C. Dell'Aversano, O. Carnicer, P. De La Iglesia, M. Forino, J. Diogène and P. Ciminiello, Anal. Bioanal. Chem., 2015, 407, 1191–1204 CrossRef PubMed.
- L. Tartaglione, E. Dello Iacovo, A. Mazzeo, S. Casabianca, P. Ciminiello, A. Penna and C. Dell'Aversano, Environ. Sci. Technol., 2017, 51, 13920–13928 CrossRef CAS PubMed.
- P. Ciminiello, C. Dell'Aversano, M. Forino and L. Tartaglione, Eur. J. Org. Chem., 2014, 7, 1357–1369 CrossRef.
- L. Tartaglione, A. Mazzeo, C. Dell'Aversano, M. Forino, V. Giussani, S. Capellacci, A. Penna, V. Asnaghi, M. Faimali, M. Chiantore, T. Yasumoto and P. Ciminiello, Anal. Bioanal. Chem., 2016, 408, 915–932 CrossRef CAS PubMed.
- P. Ciminiello, C. Dell'Aversano and M. Forino, in Phycotoxins: Chemistry and Biochemistry, ed. L. M. Botana and A. Alfonso, Wiley-Blackwell, 2nd edn, 2015, pp. 85–111 Search PubMed.
- P. Ciminiello, C. Dell'Aversano, E. Fattorusso, M. Forino, G. S. Magno, L. Tartaglione, C. Grillo and N. Melchiorre, Anal. Chem., 2006, 78, 6153–6159 CrossRef CAS PubMed.
- Y. Onuma, M. Satake, T. Ukena, J. Roux, S. Chanteau, N. Rasolofonirina, M. Ratsimaloto, H. Naoki and T. Yasumoto, Toxicon, 1999, 37, 55–65 CrossRef CAS PubMed.
- K. Aligizaki, P. Katikou, G. Nikolaidis and A. Panou, Toxicon, 2008, 51, 418–427 CrossRef CAS PubMed.
- R. Biré, S. Trotereau, R. Lemée, C. Delpont, B. Chabot, Y. Aumond and S. Krys, Harmful Algae, 2013, 28, 10–22 CrossRef.
- R. Biré, S. Trotereau, R. Lemée, D. Oregioni, C. Delpont, S. Krys and T. Guérin, Mar. Drugs, 2015, 13, 5425–5446 CrossRef PubMed.
- Z. Amzil, M. Sibat, N. Chomérat, H. Grossel, F. Marco-Miralles, R. Lemée, E. Nezan and V. Sechet, Mar. Drugs, 2012, 10, 477–496 CrossRef CAS PubMed.
- S. Accoroni, M. Ubaldi, S. Bacchiocchi, F. Neri, M. Siracusa, M. G. Buonomo, A. Campanelli and C. Totti, J. Mar. Sci. Eng., 2022, 10, 1402 CrossRef.
- P. Ciminiello, C. Dell'Aversano, E. Dello Iacovo, M. Forino and L. Tartaglione, Anal. Bioanal. Chem., 2015, 407, 1463–1473 CrossRef CAS PubMed.
- C. Brescianini, C. Grillo, N. Melchiorre, R. Bertolotto, A. Ferrari, B. Vivaldi, G. Icardi, L. Gramaccioni, E. Funari and S. Scardala, Eurosurveillance, 2006, 11, 03040 Search PubMed.
- P. Ciminiello, C. Dell'Aversano, E. Dello Iacovo, E. Fattorusso, M. Forino, L. Tartaglione, G. Benedettini, M. Onorari, F. Serena, C. Battocchi, S. Casabianca and A. Penna, Environ. Sci. Technol., 2014, 48, 3532–3540 CrossRef CAS PubMed.
- E. Berdalet, A. S. Pavaux, R. Abós-Herràndiz, M. Travers, G. Appéré, M. Vila, J. Thomas, L. de Haro, M. Estrada, N. I. Medina-Pérez, L. Viure, B. Karlson and R. Lemée, Harmful Algae, 2022, 119, 102320 CrossRef CAS PubMed.
- H. Illoul, F. Rodríguez, M. Vila, N. Adjas, A. A. Younes, M. Bournissa, A. Koroghli, N. Marouf, S. Rabia and F. L. K. Ameur, Cryptogam.: Algol., 2012, 33, 209–216 CrossRef.
- L. Tichadou, M. Glaizal, A. Armengaud, H. Grossel, R. Lemée, R. Kantin, J. L. Lasalle, G. Drouet, L. Rambaud, P. Malfait and L. De Haro, Clin. Toxicol., 2010, 48, 839–844 CrossRef PubMed.
- M. Pfannkuchen, J. Godrijan, D. Marić Pfannkuchen, L. Iveša, P. Kružić, P. Ciminiello, C. Dell'Aversano, E. Dello Iacovo, E. Fattorusso, M. Forino, L. Tartaglione and M. Godrijan, Environ. Sci. Technol., 2012, 46, 5574–5582 CrossRef CAS PubMed.
- A. S. Pavaux, E. Ternon, L. Dufour, S. Marro, M. P. Gémin, O. P. Thomas and R. Lemée, Aquat. Toxicol., 2020, 223, 105485 CrossRef CAS PubMed.
- P. A. Tester, R. W. Litaker and E. Berdalet, Harmful Algae, 2020, 91, 101655 CrossRef PubMed.
- N. Chomérat, E. Antajan, I. Auby, G. Bilien, L. Carpentier, M. N. de Casamajor, F. Ganthy, F. Hervé, M. Labadie, C. Méteigner, C. Paradis, M. Perrière-Rumèbe, F. Sanchez, V. Séchet and Z. Amzil, Mar. Drugs, 2022, 20, 461 CrossRef PubMed.
- EFSA, Scientific Opinion. Marine Biotoxins in Shellfish – Saxitoxin Group. Scientific Opinion of the Panel on Contaminants in the Food Chain (Question No EFSA-Q-2006-065E), EFSA J., 2009, 1019, 1–76 Search PubMed.
- EFSA, Scientific Opinion on Marine Biotoxins in Shellfish – Palytoxin group. EFSA Panel on Contaminants in the Food Chain (CONTAM), EFSA J., 2009, 7, 1393 CrossRef.
- F. Guerrini, L. Pezzolesi, A. Feller, M. Riccardi, P. Ciminiello, C. Dell'Aversano, L. Tartaglione, E. Dello Iacovo, E. Fattorusso, M. Forino and R. Pistocchi, Toxicon, 2010, 55, 211–220 CrossRef CAS PubMed.
- S. Accoroni, T. Romagnoli, F. Colombo, C. Pennesi, C. G. di Camillo, M. Marini, C. Battocchi, P. Ciminiello, C. Dell'Aversano, E. Dello Iacovo, E. Fattorusso, L. Tartaglione, A. Penna and C. Totti, Mar. Pollut. Bull., 2011, 62, 2512–2519 CrossRef CAS PubMed.
- P. Ciminiello, C. Dell'Aversano, E. Dello Iacovo, E. Fattorusso, M. Forino and L. Tartaglione, Toxicon, 2011, 57, 376–389 CrossRef CAS PubMed.
- P. Ciminiello, C. D. Aversano, E. Dello Iacovo, E. Fattorusso, M. Forino, L. Tartaglione, C. Battocchi, R. Crinelli, E. Carloni, M. Magnani and A. Penna, Chem. Res. Toxicol., 2012, 25, 1243–1252 Search PubMed.
- P. Ciminiello, C. Dell'Aversano, E. Dello Iacovo, E. Fattorusso, M. Forino, L. Tartaglione, T. Yasumoto, C. Battocchi, M. Giacobbe, A. Amorim and A. Penna, Harmful Algae, 2013, 23, 19–27 CrossRef CAS.
- P. Ciminiello, C. Dell'Aversano, E. Dello Iacovo, E. Fattorusso, M. Forino, L. Grauso and L. Tartaglione, J. Am. Soc. Mass Spectrom., 2012, 23, 952–963 CrossRef CAS PubMed.
- L. Pezzolesi, F. Guerrini, P. Ciminiello, C. Dell'Aversano, E. Dello Iacovo, E. Fattorusso, M. Forino, L. Tartaglione and R. Pistocchi, Water Res., 2012, 46, 82–92 CrossRef CAS PubMed.
- S. Vanucci, L. Pezzolesi, R. Pistocchi, P. Ciminiello, C. Dell'Aversano, E. Dello Iacovo, E. Fattorusso, L. Tartaglione and F. Guerrini, Harmful Algae, 2012, 15, 78–90 CrossRef CAS.
- P. Andersen and J. Throndsen, Estimating cell numbers, in Manual on Harmful Marine Microalgae, ed. G. M. Hallegraeff, D. M. Anderson and A. D. Cembella, UNESCO, Paris, 2004, pp. 99–129 Search PubMed.
- M. Vila, R. Abós-Herràndiz, J. Isern-Fontanet, J. Àlvarez and E. Berdalet, Sci. Mar., 2016, 80(S1), 107–115 CrossRef CAS.
- V. Giussani, F. Sbrana, V. Asnaghi, M. Vassalli, M. Faimali, S. Casabianca, A. Penna, P. Ciminiello, C. Dell'Aversano, L. Tartaglione, A. Mazzeo and M. Chiantore, Harmful Algae, 2015, 44, 46–53 CrossRef CAS.
- L. Pezzolesi, S. Vanucci, C. Dell'Aversano, E. Dello Iacovo, L. Tartaglione and R. Pistocchi, Harmful Algae, 2016, 55, 202–212 CrossRef CAS PubMed.
- O. Carnicer, M. García-Altares, K. B. Andree, L. Tartaglione, C. Dell'Aversano, P. Ciminiello, P. de la Iglesia, J. Diogène and M. Fernández-Tejedor, Harmful Algae, 2016, 57, 98–108 CrossRef CAS PubMed.
- S. Accoroni, T. Romagnoli, A. Penna, S. Capellacci, P. Ciminiello, C. Dell'Aversano, L. Tartaglione, M. Abboud-Abi Saab, V. Giussani, V. Asnaghi, M. Chiantore and C. Totti, J. Phycol., 2016, 52, 1064–1084 CrossRef CAS PubMed.
- E. Ternon, A. S. Pavaux, S. Marro, O. P. Thomas and R. Lemée, Harmful Algae, 2018, 75, 35–44 CrossRef CAS PubMed.
- E. Ternon, A. S. Pavaux, A. Peltekis, M. P. Gemin, C. Jauzein, B. Bailleul, R. Lemée and O. P. Thomas, Aquat. Ecol., 2022, 56, 475–491 CrossRef.
- C. Brissard, C. Herrenknecht, V. Séchet, F. Hervé, F. Pisapia, J. Harcouet, R. Lémée, N. Chomérat, P. Hess and Z. Amzil, Mar. Drugs, 2014, 12, 2851–2876 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ay02019j |
| This journal is © The Royal Society of Chemistry 2023 |