
A highly sensitive LC-MS/MS method for the determination and quantification of a recently identified N-nitrosamine impurity in the sitagliptin phosphate monohydrate active pharmaceutical ingredient†
Zhen
Wang
 *a,
Shujun
Hu
b,
Xiaoying
Wu
b,
Zuwei
He
b,
Chunlong
Ke
b and
Miaomiao
Hu
b
*a,
Shujun
Hu
b,
Xiaoying
Wu
b,
Zuwei
He
b,
Chunlong
Ke
b and
Miaomiao
Hu
b
aDepartment of Chemistry, Yuquan Campus, Zhejiang University, Hangzhou 310058, China. E-mail: 896698921@qq.com
bChemical Process Research and Development, Zhejiang Tianyu Pharmaceutical Co., Ltd, Taizhou, 318020, China
First published on 16th December 2022
Abstract
A liquid chromatography-tandem mass spectrometry (LC-MS/MS) method was used for the quantification of 7-nitroso-3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (NTTP) in the sitagliptin phosphate monohydrate active pharmaceutical ingredient. Chromatographic separation was achieved using an Agilent-ZORBAX SB-C18 column, with 0.01 mol L−1 ammonium formate in water as mobile phase A and acetonitrile as mobile phase B in gradient elution mode at a 0.4 mL min−1 flow rate. Quantification of impurities was carried out using triple quadrupole mass detection with electrospray ionization in the multiple reaction monitoring mode. The method was fully validated with good linearity over the concentration range of 0.098–0.925 ppm of the sitagliptin phosphate monohydrate test concentration for NTTP. The correlation coefficient obtained in each case was >0.998. The recoveries were found to be satisfactory over the range between 80.0 and 120.0% for NTTP. The developed method was able to quantitate NTTP at a concentration level of 0.98 ng mL−1 (0.098 ppm with respect to 10 mg mL−1 sitagliptin phosphate monohydrate).
Introduction
N-Nitrosamines (NAs) are internationally recognized as a class of strong carcinogens according to the International Agency for Research on Cancer (IARC). N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA) are classified as the most potent carcinogens of the N-nitrosamines, and N-nitrosodibutylamine (NDBA), N-nitrosopiperidine (NPIP) and N-nitrosopyrrolidine (NPYR) are listed as potential carcinogens.1,2 The Environmental Protection Agency (EPA) of the United States considers that NDMA at extremely low concentrations (0.7 ng L−1) can cause cancer, and the risk index of NAs for human health damage is grade B2.3Given the great harm to human health that can be caused by NAs, the risk of NAs in active pharmaceutical ingredients (API) has been strictly controlled by regulatory agencies and pharmaceutical industries. The ICH M7 (R1) guideline4 defines N-nitrosamines as substances of the “cohort of concern” for which limits in medicinal products refer to the so-called substance-specific acceptable intake (AI) (the Threshold of Toxicological Concern, TTC, of 1.5 μg per day cannot be applied), which is associated with a negligible risk (theoretical excess cancer risk of <1 in 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 over a lifetime of exposure). In addition to the conventional genotoxic impurities (GTIs) described in the ICH M7 guideline, the European Medicines Agency (EMA)5,6 and the United States Food and Drug Administration (FDA)7,8 have released specific documents regarding regulatory issues related to the risk assessment and control strategy of NAs in new drug applications as well as approved drugs. According to the latest updated documents, N-nitrosamine precursors of secondary or tertiary amines from starting materials, intermediates and API should also be considered comprehensively in the risk assessment.
000 over a lifetime of exposure). In addition to the conventional genotoxic impurities (GTIs) described in the ICH M7 guideline, the European Medicines Agency (EMA)5,6 and the United States Food and Drug Administration (FDA)7,8 have released specific documents regarding regulatory issues related to the risk assessment and control strategy of NAs in new drug applications as well as approved drugs. According to the latest updated documents, N-nitrosamine precursors of secondary or tertiary amines from starting materials, intermediates and API should also be considered comprehensively in the risk assessment.
Recently, an N-nitrosamine impurity (Fig. 1) named 7-nitroso-3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (NTTP) was identified in sitagliptin phosphate monohydrate, the active pharmaceutical ingredient of an approved drug by EMA and FDA for the treatment of patients with type 2 diabetes. In the latest nitrosamine guidelines9 of EMA, the specific acceptable intake of NTTP recommended in this official document was 37 ng per day, according to the structure-activity-relationship (SAR)/read-across approach using the TD50 of N-nitroso-1,2,3,6-tetrahydropyridine as the point of departure. Considering the maximum daily dose of sitagliptin phosphate tablets (100 mg per day),10 the NTTP concentration must be controlled in sitagliptin phosphate monohydrate at concentrations lower than 0.37 ppm.
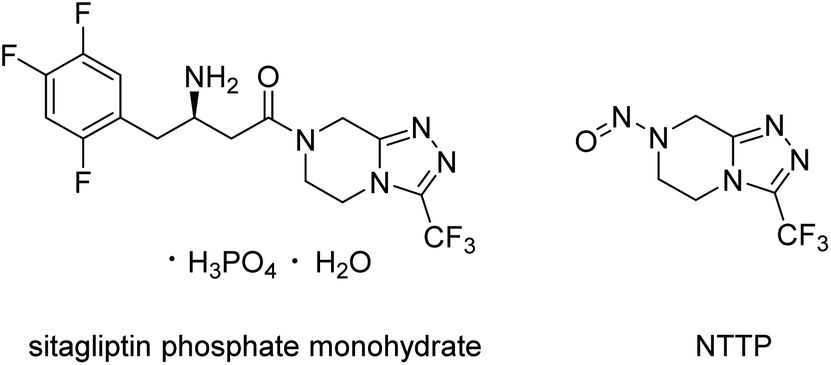 | ||
| Fig. 1 Structures of sitagliptin phosphate monohydrate and nitrosamine impurity (NTTP). | ||
Owing to the extremely limited time of the impurity identification, there was not a specific and sensitive method for the determination and quantification of NTTP in the sitagliptin phosphate monohydrate reported in the literature except for related substance methods.11–14 Herein, we would like to present the development of an LC-MS/MS method for the determination of NTTP in sitagliptin phosphate monohydrate, as well as validation of the method with respect to the specificity, limit of detection (LOD), the limit of quantification (LOQ), linearity, repeatability, accuracy and robustness, in accordance with ICH recommended conditions.
Experimental
Reagents and chemicals
All reagents used were of analytical grade with the highest purity of >99.8%. Methanol and acetonitrile were purchased from Merck (Darmstadt, Germany). The ammonium formate eluent additive for LC-MS was purchased from SIGMA-ALDRICH. Milli-Q water was used throughout the analysis and collected from a Milli-Q reverse osmosis system (Milli-Q®, HX 7040). Sitagliptin phosphate monohydrate and NTTP were synthesized and analysed at Tianyu Pharm., China (IR, TGA, NMR, MS and HPLC analyses).Mobile phase preparation
Mobile phase solution A was prepared by adding 0.01 mol of ammonium formate to 1000 mL of water. Acetonitrile was used as mobile phase solution B. Both mobile phase solutions were degassed and stored for further use at ambient temperature. Solvents were prepared before each series of analyses.Preparation of sample and standard solutions
A stock mixture of NTTP (0.05 mg mL−1) was prepared by dissolving suitable amounts of NTTP in 100% methanol as the diluent. From this solution, the diluted stock solution of 50 ng mL−1 was prepared by dilution with methanol. A series of calibration standards were prepared by diluting a 50 ng mL−1 solution to obtain the following final concentrations of 0.98, 1.85, 3.70, 5.55, and 9.25 ng mL−1.Solutions for recovery determination were prepared by accurately weighing 100 mg of sitagliptin phosphate monohydrate in 10 mL volumetric asks (in triplicate). By adding an appropriate amount of NTTP to the sitagliptin phosphate monohydrate solutions, 9.25 ng mL−1 (0.925 ppm), 3.7 ng mL−1 (0.37 ppm) and 0.98 ng mL−1 (0.098 ppm) solutions of NTTP with respect to 10 mg mL−1 sitagliptin phosphate monohydrate were prepared. All solutions prepared for analytical recovery were prepared in triplicate to ensure repeatability.
Operating conditions of LC-MS/MS
Chromatographic analysis was performed on an Agilent 1260 HPLC system equipped with a quad pump and an autosampler and an Agilent LC-MS/MS 6470 TripleQuad with an electrospray interface. The analytical column used in the LC-MS/MS study was ZORBAX SB-C18 (150 × 4.6 mm, 1.8 μm) (Agilent Co. Ltd, USA) employed in gradient mode using 0.01 mol L−1 of ammonium formate in water as mobile phase A and acetonitrile as mobile phase B at a flow rate of 0.4 mL min−1. The column oven temperature was maintained at 30 °C and the sample cooler temperature was fixed at 20 °C. The sample injection volume was 5.0 μL. The LC gradient program (time/% solution A) was set as follows: 0.00/60, 7.00/40, 9.00/10, 14.00/10, 14.01/60 and 20.00/60. The positive ESI probe was operated in MRM mode for the quantification of NTTP in the form of protonated ions (M + H)+ at m/z 222.0. Of the two different collision energy voltages used for development, the entrance potential (EP) was maintained at 5 V. The ion spray voltage (V) was maintained at 3500 V. Collision energy (120 V) was applied on NTTP. The curtain gas flow, ion source gas 1 (GS1) and ion source gas 2 (GS2), and nebulizer gas pressures were maintained at 45 psi. All parameters of LC and MS were controlled using Agilent MassHunter version 1.0.29.0.Method validation
The developed method was successfully validated in terms of specificity, repeatability, linearity, accuracy, LOD, LOQ, robustness and solution stability. Method validation for NTTP in sitagliptin phosphate monohydrate was conducted following ICH guidelines. Initially, individual solutions of NTTP were prepared (γ(impurity) = 0.98 ng mL; 0.098 ppm with respect to 10 mg mL−1 sitagliptin phosphate monohydrate) and their S/N ratios were calculated.The repeatability at the determined LOD and LOQ values was verified experimentally by injecting the same solutions three times. Next, the linearity of the method was evaluated from five concentration levels between the LOQ and a 9.25 ng mL−1 impurity concentration. Least squares linear regression analysis was employed to calculate the slope, intercept, and regression coefficient values. The specificity of the developed method was assessed with sitagliptin phosphate monohydrate tablets. Next, the accuracy of the method was calculated in triplicate at a 0.37 ppm concentration level by the standard addition method. The recoveries and RSD values were calculated for the impurities in three batches of the pure API. The robustness of the method was tested by altering the mobile phase flow rate and column temperature. Further, the analysis of the sample solution at different intervals of time was compared against fresh samples to evaluate the stability of impurities in the sample solution.
Results and discussion
Method development
This study was conducted to develop a sensitive and selective LC-MS/MS method that can separate and quantify NTTP in the sitagliptin phosphate monohydrate active pharmaceutical ingredient.A few columns were tested to obtain the most appropriate peak shape and separation. There was a greater overlap between NTTP and API peaks when using the InfinityLab Poroshell 120 EC-C18 column, and poor peak shapes were observed when using an InfinityLab Poroshell 120 PFP column. A ZORBAX SB-C18 column (150 × 4.6 mm, 1.8 μm) was found to be the most suitable regarding both peak shape and separation, as well as the response of analytes. The mobile phase was operated in gradient mode using 0.01 mol L−1 ammonium formate in water and acetonitrile (see the subsection on operating conditions of LC-MS/MS). Acetonitrile was chosen for its good elution efficiency. The flow rate of the mobile phase was maintained at 0.4 mL min−1, with the column temperature set at 30 °C. The retention times of NTTP were observed to be 5.22 min and the peak corresponding to sitagliptin phosphate monohydrate was eluted at 9.45 min. The chromatogram is given in the ESI,† Fig. S1.
Operating conditions of LC-MS/MS
The optimization of mass spectrometric ionization was aimed at developing a simple, rapid, sensitive, and stable analytical method for the determination of NTTP in the sitagliptin phosphate monohydrate API. Method development was carried out using LC-MS/MS for the detection and quantification of NTTP at a concentration level of 1 μg mL−1. During the early stages of method development, the signal intensity in the positive mode was found to be much higher than that in the negative mode, limiting the method development to the positive ESI source. To optimize the ESI conditions for NTTP, fragmentation was carried out using four different collision energy voltages (0 V, 40 V, 80 V and 120 V). The ion source parameters were optimized to obtain a good response and are shown in Table 1. The MS/MS spectra of NTTP at different collision energies are given in the ESI† (Fig. S2).| NAs | Precursor ion (m/z) | Product ion (m/z) | Collision energy (V) | Voltage (V) |
|---|---|---|---|---|
| NTTP | 222.0 | 192.1 | 0 | 5 |
| 222.0 | 69.0 | 120 | 5 |
Method validation
The optimized LC-MS/MS method was successfully validated in accordance with the ICH guidelines. Method validation was carried out in terms of its adequate selectivity, linearity, LOD and LOQ, accuracy, repeatability, recovery, and robustness.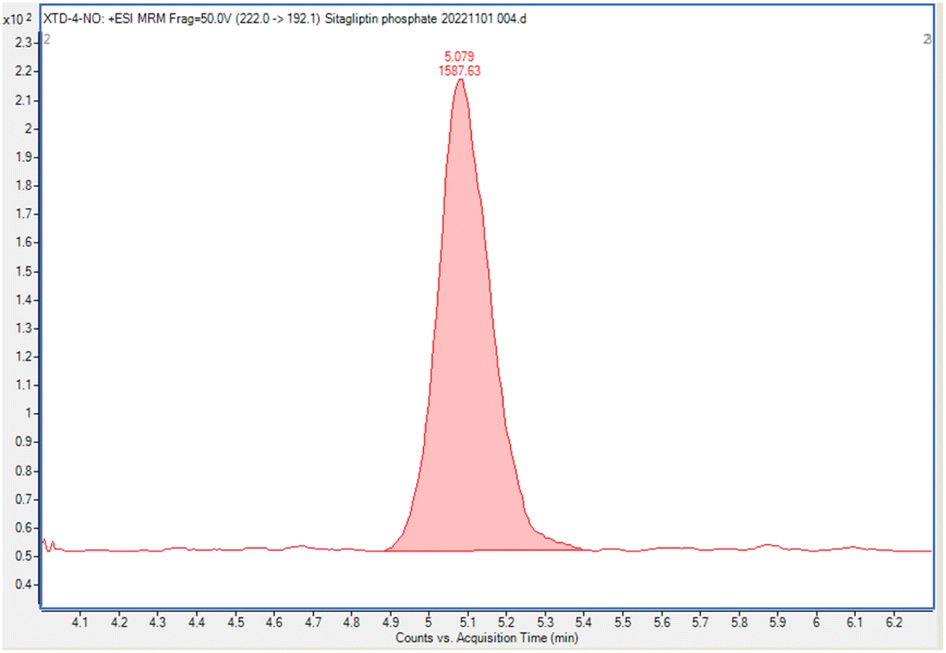 | ||
| Fig. 2 LC-MS/MS chromatogram of NTTP. | ||
| Parameter | NTTP |
|---|---|
| a Calculated from six determinations of the LOD and LOQ. b Calculated from three determinations of the LOQ and 0.37 ppm solutions. | |
| Calibration curve | y = 419.58x + 52.03 |
| Linearity range (ppm) | 0.098–0.925 |
| Correlation coefficient, γ2 | 0.9999 |
| S/Na (LOD, 0.037 ppm) | 18 |
| S/Na (LOQ, 0.098 ppm) | 47 |
| Accuracy (%) at LOQb | 104 |
| Accuracy (%) at specification levelb (0.37 ppm) | 103 |
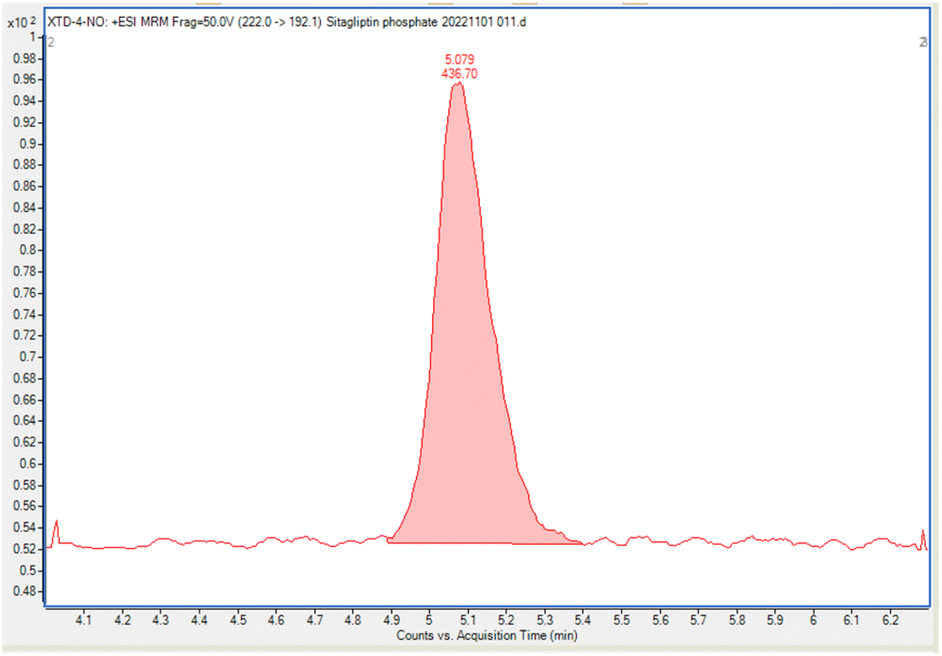 | ||
| Fig. 3 The chromatogram of solution of NTTP at 0.98 ng mL−1. | ||
| Impurity | Concentrationa (ppm) | Recovery in pure sampleb (%) | Recovery in formulated sampleb (%) |
|---|---|---|---|
| a The amount of NTTP spiked with respect to 10 mg mL−1sitagliptin phosphate monohydrate. b Mean ± RSD% for three determinations. | |||
| NTTP | 0.098 | 104.17 ± 2.96 | 80.83 ± 1.56 |
| 0.37 | 103.46 ± 2.40 | 107.81 ± 1.99 | |
| 0.925 | 97.73 ± 1.13 | 104.54 ± 1.28 | |
| Parameter | NTTP |
|---|---|
| Repeatability at 0.098 ppm (RSD%) | 3.89 |
| Repeatability at 0.370 ppm (RSD%) | 0.28 |
| Repeatability at 0.925 ppm (RSD%) | 0.31 |
Conclusions
In this study, we have developed a highly sensitive LC-MS/MS approach that is capable of quantifying NTTP in sitagliptin phosphate monohydrate using the positive ionization mode with multiple reaction monitoring (MRM). The method was validated as per ICH recommendations and it was found to be specific and linear over the specified concentration range. The determined LOD and LOQ values for NTTP were set very low and lie within the range of the 30% acceptable limit. The sample prepared in the analytical solution was found to be stable for at least 48 h. The method was fully validated and presents good linearity, accuracy, repeatability, and robustness. This method could be very useful for the determination of NTTP in sitagliptin phosphate monohydrate during its manufacture and product release.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Zhejiang Tianyu Pharmaceutical Co., Ltd. for financial support and quality control center for method development.References
- International Agency for Research on Cancer, IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Lyon, 1978, p. 17 Search PubMed.
- F. Liu, Y. Xian, J. Chen, H. Dong, H. Liu, X. Guo, M. Chen and H. Li, Anal. Methods, 2016, 8, 4245–4253 RSC.
- US Environmental Protection Agency, N-Nitrosodimethylamine (CASRN62-75-9), 2012, https://www.epa.gov/iris/subst/0045.htm Search PubMed.
- M7(R1) assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/m7r1-assessment-and-control-dna-reactive-mutagenic-impurities-pharmaceuticals-limit-potential Search PubMed.
- Questions and Answers for Marketing Authorisation Holders/applicants on the CHMP Opinion for the Article5(3) of Regulation (EC) No 726/2004 Referral on Nitrosamine Impurities in Human Medicinal Products, EMA/409815/2020 Rev.7.
- Angiotensin-II-receptor antagonists (sartans) Article 31 referral-CHMP assessment report, https://www.ema.europa.eu/en/documents/variation-report/angiotensin-ii-receptor-antagonists-sartans-article-31-referral-chmp-assessment-report_en.pdf Search PubMed.
- Control of nitrosamine impurities in human drugs, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/control-nitrosamine-impurities-human-drugs Search PubMed.
- Pharmaceutical analysis and characterization of nitrosamine impurities within angiotensin ii receptor blocker drug products, https://www.fda.gov/media/148748/download Search PubMed.
- Questions and answers for marketing authorisation holders/applicants on the CHMP Opinion for the Article 5(3) of Regulation (EC) No 726/2004 referral on nitrosamine impurities in human medicinal products, https://www.ema.europa.eu/en/documents/referral/nitrosamines-emea-h-a53-1490-questions-answers-marketing-authorisation-holders/applicants-chmp-opinion-article-53-regulation-ec-no-726/2004-referral-nitrosamine-impurities-human-medicinal-products_en.pdf Search PubMed.
- See the drug instruction manual of Januvia®.
- B. Pasquini, R. Gotti, M. Villar-Navarroc, M. Dousa, L. Renai, M. D. Bubbaa, S. Orlandini and S. Furlanetto, J. Pharm. Biomed. Anal., 2021, 202, 114163 CrossRef CAS PubMed.
- T. Ramesh, P. N. Rao and K. Suresh, Anal. Methods, 2014, 6, 223–228 RSC.
- D. P. Sonune and M. K. Mone, Int. J. Pharm. Sci. Res., 2013, 4, 3494–3503 Search PubMed.
- M. V. Dalawai, P. D. Sanasi and H. K. Sharma, Int. J. Pharm. Sci. Res., 2016, 7, 1493–1502 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ay01821g |
| This journal is © The Royal Society of Chemistry 2023 |