
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient determination of non-steroidal anti-inflammatory drugs by micellar electrokinetic chromatography in wastewater†
Hanan
Alatawi
 a,
Anna
Hogan
a,
Ibtihaj
Albalawi
a,
Samia
Alsefri
a and
Eric
Moore
*ab
a,
Anna
Hogan
a,
Ibtihaj
Albalawi
a,
Samia
Alsefri
a and
Eric
Moore
*ab
aSchool of Chemistry, University College Cork, Cork, Ireland. E-mail: e.moore@ucc.ie
bTyndall National Institute, Cork, Ireland
First published on 11th January 2023
Abstract
Recently, non-steroidal anti-inflammatory drugs (NSAIDs) have been increasingly used in humans and animals. Despite being effective against a wide variety of diseases, they pose a threat to aquatic environments. In the current work, a highly efficient, selective, and sensitive micellar electrokinetic chromatography (MEKC) method was developed for the determination of five NSAIDs in environmental water samples. The optimal separation BGE was 15 mM borate buffer (pH 9), 90 mM SDS, and 10% methanol at a separation voltage of 15 kV and a hydrodynamic injection of 10 mbar for 5 s. The results presented in this study provide a higher number of theoretical plates N > 780![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 with excellent RSDs of 0.1–1.5% and great sensitivity (3–15 μg L−1) for NSAIDs. To validate this method, the solid phase extraction method was optimized using two different cartridges (C18 and Oasis HLB); the results showed excellent recoveries (73–111.6%) for all the analytes in wastewater samples.
000 with excellent RSDs of 0.1–1.5% and great sensitivity (3–15 μg L−1) for NSAIDs. To validate this method, the solid phase extraction method was optimized using two different cartridges (C18 and Oasis HLB); the results showed excellent recoveries (73–111.6%) for all the analytes in wastewater samples.
1 Introduction
Recently, NSAIDs have been increasingly used in humans and animals. Despite being effective against a wide variety of diseases, they pose a threat to aquatic environments.1,2 Studies conducted in various countries around the world have shown that NSAIDs are the most frequently found pharmaceuticals in wastewater at concentrations ranging from 22.7 to 2747.3 μg L−1,3,4 as many of them are available in some countries without the need for a prescription. Additionally, during the pandemic, NSAIDs were commonly employed to treat COVID-19-associated fever, pain, and inflammation symptoms. As a consequence of this, it is highly probable that the COVID-19 pandemic has resulted in an increased demand for pharmaceuticals, and it is expected that this trend will continue even as highly transmissible variants are developed.5NSAIDs have a high solubility and a low rate of degradation in water, which allow them to pass through all-natural filtration processes. It has been well known that conventional wastewater treatment plants (WWTPs) were not made to completely remove these compounds because they were not intended to do so.6,7 Consequently, numerous pharmaceuticals are returned to the environment. It is critical to develop analytical methods that are efficient, simple, and reliable for detecting NSAIDs in environmental water.
Many analytical methods have been proposed to detect NSAIDs in environmental water matrices, including high-performance liquid chromatography-mass spectroscopy (HPLC-MS) and gas chromatography-mass spectroscopy (GC-MS).8,9 These techniques, however, are costly, necessitate sample preparation, and produce significant volumes of organic waste. Capillary electrophoresis (CE) has advantages over other chromatography techniques including the short duration of method development, the simple instrumentation, the small volumes of sample and electrolyte, and high efficiency. The interest in CE has led to the development of different modes, e.g., capillary zone electrophoresis (CZE), micellar electrokinetic chromatography (MEKC), and capillary isoelectric focusing (CIEF), which have different mechanisms to separate a wide range of analytes. In addition, CE allows the use of various detectors besides the common ultraviolet-visible (UV) detector, such as in mass spectrometry,10 laser-induced fluorescence detection,11 and electrochemical detection,12,13 extending its applicability. CE has been shown to be a useful tool in the determination of NSAIDs in environmental water.14–21
In the current work, a highly efficient, selective, and sensitive MEKC method was developed for the determination of NSAIDs in environmental water samples. The separation parameters were optimized by variation of the composition of the BGE (BGE concentration, pH, SDS concentration, and methanol percentage). The results presented in this study provide a higher number of theoretical plates N (780000) with excellent RSDs 0.1–1.5% and great sensitivity (3–15 μg L−1) for NSAIDs. To validate this method, the solid phase extraction (SPE) method was optimized using two different cartridges (C18 and Oasis HLB) to obtain excellent recoveries for all the analytes in wastewater sample.
2 Experimental
2.1 Instrumentation
MEKC using an Agilent CE 7100 (Waldbronn, Germany) was used to separate five NSAIDS and detect them with a diode array UV/vis detector. Fused-silica capillaries with an id of 50 μm and od of 375 μm were used for the separations (Composite Metal Services Ltd, Shipley, BD17 7AD, UK). To allow the UV/vis light to pass through the capillary, a short length of 5 mm of the capillary was stripped of the polyimide layer by using an Innova TECH burner. The burned polyimide residues were eliminated by wiping the exposed section of the fused silica capillary with soft tissue wet with methanol. The new capillary was preconditioned with 1 M sodium hydroxide (NaOH) for 30 min, deionized (DI) water for 30 min, and running buffer for 30 min. The samples were injected at a pressure of 10 mbar for 5 s using hydrodynamic injection. All experiments were carried out at 15 kV. ChemStation CE software (Agilent Technologies) was used to control the CE instrument and to collect the analytical signals. A pH meter (Metrohm 654) with a microelectrode (Metrohm 6.0234.100) was used for all pH measurements.2.2 Chemicals
Analytical reagent grade chemicals ibuprofen (IB), ketoprofen (KET), diclofenac sodium (DIC), paracetamol (PAC), sodium hydroxide (NaOH), hydrochloric acid (HCl), sodium dodecyl sulfate (SDS), sodium tetraborate decahydrate, disodium hydrogen phosphate, acetate buffer, acetonitrile, ammonium hydroxide (NH4OH) solution 33%, acetone and methanol were purchased from Sigma-Aldrich (Dublin, Ireland). Ultrapure DI water with a resistivity of 18.2 MΩ cm was obtained from a Milli-Q (Millipore, Molsheim, France) water purification system.2.3 Sample preparation
Stock standard solutions of background electrolytes (BGEs) were prepared at concentrations of 100 mM in DI water for borate, acetate, and phosphate buffers. All buffer solutions were appropriately diluted to the required concentration. Different concentrations of SDS were weighed and added to the borate buffer. Individual stock standard solutions of PAC, IB, KET, ASA, and DIC were prepared in methanol at concentrations of 100 mg L−1 and stored in a refrigerator at 4 °C prior to their use. A working standard solution of 5 mg L−1 of each analyte was prepared in methanol. A mixed standard solution of the five NSAIDs was prepared at a concentration of 0.75 mg L−1 in water daily. Before the background electrolyte and analyte samples were transferred to sample vials, they were sonicated to remove air bubbles and filtered through 0.2 μm regenerated cellulose syringe filters.2.4 Wastewater samples
Effluent wastewater samples were collected from County Cork, Ireland. They were filtered using 0.45 μm filter paper (Millipore, Ireland) and stored in a refrigerator (4 °C) until analysis.Two different SPE cartridges, an Oasis HLB 6 cc (200 mg) column (Waters, Ireland) and a Sep-Pak C18 3 cc column (Waters, Ireland), were used to extract NSAID compounds from wastewater samples. Both cartridges were preconditioned with 3 mL methanol and 6 mL DI water before loading the samples (for the C18 cartridge, the pH of DI water was adjusted to 2.5 with HCl, and it was necessary to keep the four NSAID analytes in their protonated forms, whereas for the Oasis HLB cartridge the pH of DI water was not adjusted). It should be noted that 10 mL of wastewater (for the C18 cartridge, the pH of wastewater was adjusted to 2.5, and for the Oasis HLB cartridge, the pH of wastewater was not adjusted) was spiked with 100 μg L−1 of each NSAID (IB, DIC, KET, PAC and ASA). A constant loading rate of 1 drop/s was used to load the spiked wastewater sample for maximum retention. After passing the spiked sample through the cartridge, 1 mL of methanol was added to wash the cartridge. Subsequently, the cartridge was washed with 2 mL of methanol–water (10:90 v/v) and 1 mL of 2% acetic acid in 20% of methanol in water (Oasis HLB and C18). Washing with a 2% water solution of acetic acid will remove traces of water-soluble impurities that are potentially still at the sorbent.22 Finally, to elute the target analytes from the sorbents, the optimal solvent found suitable for both cartridges is 1 mL of methanol. The eluates were evaporated to dryness using a gentle stream of nitrogen and were then dissolved in 1 mL of methanol. To ensure consistency, the same method used for spiked wastewater sample preparation was used for the non-spiked wastewater sample.
3 Results and discussion
3.1 BGE pH optimization
It can be noticed that (Fig. 1) the migration time orders of the solutes obtained by MEKC were completely altered from those obtained by CZE. MEKC separation is based on the differential partition of analytes between micelles and aqueous buffer, whereas CZE separation is based on differences in the analytes' electrophoretic mobilities.24 In the presence of SDS in the BGE the analytes get separated due to the combination of electrophoretic mobility and differential interaction of the analyte with negatively charged SDS.27 Therefore, the migration behavior of the solute depends on several properties, including hydrophobicity and the degree of dissociation in the solution.24,27,28 The different ionization constants of the five NSAID analytes affect how well they separate. For example, PAC (pKa 9.5) is a zwitterion, which means that it has both acidic and basic groups. Its acidic protons are ionized in the alkaline medium of borate BGE (pH 9), which gives their molecules a strong negative charge that repels the negatively charged surface of SDS. PAC came first due to its lower molecular weight (151.71 g mol−1), followed by the four acidic NSAIDs that appeared in the following order: the most acidic ASA (pKa 3.15), KET (pKa 4), IB (pKa 4.6), and finally DIC (pKa 4.15) which has the highest molecular weight among the four (318.1 g mol−1) (see Table S1†).29 DIC gave a longer migration time, presumably owing to stronger interactions of the larger, more hydrophobic analyte with the pseudo-stationary micelles.28,30 While in CZE (Fig. S1†), ASA was the last eluted analyte due to its mass-to-charge ratio, the additional separation mechanism provided by micellar solubilization in MEKC allows for much more precise separations.
 | ||
| Fig. 1 MEKC electropherograms show the separation of NSAIDs at different pH (A) 8.5, (B) 9, and (C) 9.5; peaks: 1-PAC, 2-ASA, 3-KET, 4-IB, and 5-DIC at a concentration of 750 μg L−1 each. Capillary: 50 μm id, 31.5 cm effective length and 40 cm total length; BGE: borate buffer and 110 mM SDS; separation voltage: 15 kV. | ||
The pH of the BGE is a crucial parameter for optimizing the separation of ionizable analytes, as it affects the degree of dissociation of weak acids, which in turn affects their effective mobility.31 The effect of the BGE pH was examined over the 8.5–9.5 range using borate buffer with 110 mM SDS. In this study, increasing the pH had a moderate influence on analysis times. Fig. 1A and B show that at pH 8.5, the peaks of KET and IB started to resolve, and at pH 9, a better resolution was obtained. It was found that at pH greater than 9, the two peaks were co-eluted (Fig. 1C). Therefore, pH 9 was chosen as the optimal pH for this method.
3.2 BGE concentration
The concentration of BGE had a great impact on electrophoretic separation. Increasing the BGE concentration can cause a decrease in the electroosmotic flow (EOF) and electrophoretic mobility (μep), as well as an increase in the current generation and Joule heating, resulting in an increase or decrease in separation efficiency and resolution.32The effect of different concentrations of borate buffer (10, 15, 20, 25, and 30 mM) on the μep is illustrated in (Fig. 2). The μep was calculated according to the equations given below:
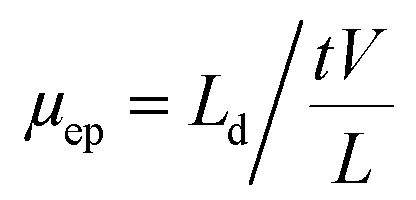
 | ||
| Fig. 2 Effect of BGE concentration on the electrophoretic mobility of the selected NSAIDs. | ||
3.3 SDS concentration
The effect of the addition of SDS with different concentrations (50, 70, 90, and 110 mM) to the BGE was studied. It was found that the addition of SDS to the BGE resulted in a significant increase in both the resolution and peak shapes of all analytes, as shown in (Fig. S2†). The migration time was also affected by the addition of SDS. The results demonstrated that with an increase in the SDS concentration, the resolution and migration time increased. Fig. S2C† shows that a concentration of 90 mM SDS yielded the best resolution. When the SDS concentration was increased to 110 mm, no further improvement was observed (Fig. S2D†). Therefore, the optimal concentration of SDS is 90 mm.3.4 Organic modifier
The addition of an organic modifier to the BGE has been reported as a crucial factor for enhancing separation selectivity, efficiency, and resolution.33,34 The presence of an organic modifier influences the distribution of analytes between the aqueous phase and micelles; additionally, it can affect the charge of ionizable groups.24,26,29 It is worth noting that the presence of methanol can change the order migration of KET and ASA (Fig. 3A and B), which could be explained by the fact that methanol reduced the partition coefficient between the micelle and the bulk solution.24 | ||
| Fig. 3 MEKC electropherograms show the effect of methanol percentages on the separation of NSAIDs (A) 0, (B) 5%, (C) 10%, and (D) 15% (v/v); peaks: 1-PAC, 2-ASA, 3-KET, 4-IB, and 5-DIC at a concentration of 750 μg L−1 each; capillary: 50 μm id, 31.5 cm effective length and 40 cm total length; BGE: 15 mM borate buffer, pH 9, 90 mM SDS; separation voltage: 15 kV. | ||
To study the effect of organic solvent on the separation of the selected analytes, different percentages of methanol (0, 5, 10, and 15% v/v) were tested. The total analysis times increased as the methanol percentage increased, as shown in Fig. 3. This behavior could be attributed to changing the zeta potential and decreasing the EOF; methanol could improve separation efficiency.35 The baseline resolution of the analytes was achieved with good resolution and high efficiency by using a BGE containing 10% (v/v) methanol (Fig. 3C). Although the addition of methanol to the BGE had a great effect on baseline resolution, increasing the percentage of methanol to 15% (v/v) resulted in co-eluted peaks. Therefore, 10% (v/v) was chosen as the optimal percentage of methanol.
3.5 Effect of the injection plug
The influence of the hydrodynamic injection plug (10, 20, 30, 40, and 50 mbar) and time (5, 8, and 10 s) on the efficiency was studied. The number of theoretical plates (N) and the resolution (R) were calculated according to the equations given below:| N = 16(t/W)2 |
| R = 2(t2 − t1)/(W1 + W2) |
As can be seen in Fig. S3†, when the injection plug increased, the number of theoretical plates decreased. Even though the larger injection plug and longer time provide high sensitivity, the resolution and efficiency decreased. For this reason, 10 mbar for 5 s was preferred for higher theoretical plates and obtained good sensitivity.
3.6 Effect of the voltage
The effect of applied voltage on the separation was investigated by increasing the separation voltage from 10–25 kV and calculating the number of theoretical plates and resolution of the studied compounds. The effect of the applied voltage on the migration time is illustrated in Fig. S4†. As expected, when the separation voltage increased, the analysis time decreased. However, the separation efficiency and resolution were reduced, and this behaviour is contrary to that expected in CE but could be attributable to an increase in the current. This is likely due to Joule heating problems at these high applied voltages, which can lead to temperature increases, temperature gradients, and peak broadening.36–38 In addition, it was found that increasing the separation voltage caused an increase in the noise of the baseline, which in turn resulted in a decrease in the signal-to-noise ratio (SNR) (Fig. S4C and D†). The net effect of these changes is a decrease in column efficiency32 (Fig. 4). Based on these experiments, 15 kV was chosen for better resolution (resolution > 2) without loss of separation efficiency due to Joule heating (number of theoretical plates is 192628.4, 544638.4, 591575, 677700.7, and 784655.6 for PAC, KET, ASA, IB, and DIC respectively at 15 kV).
 | ||
| Fig. 4 MEKC electropherograms show the effect of voltage on efficiency. Capillary: 50 μm id, 31.5 cm effective length and 40 cm total length; BGE: 15 mM borate buffer, pH 9, 90 mM SDS, 10% methanol; hydrodynamic injection: 10 mbar/5 s. | ||
| Analytes | Linearity range (μg L−1) | R 2 | LOD (μg L−1) | RSDn=6 (peak area) | RSDn=6 (migration time) |
|---|---|---|---|---|---|
| ASA | 25–400 | 0.9977 | 12.5 | 1.3 | 0.2 |
| DIC | 12.5–200 | 0.9999 | 3 | 0.5 | 0.3 |
| IB | 25–400 | 0.9993 | 12.5 | 1.3 | 0.23 |
| KET | 12.5–200 | 0.9981 | 6.2 | 0.8 | 0.2 |
| PAC | 31.5–500 | 0.997 | 15.2 | 1.5 | 0.5 |
To achieve a high extraction efficiency for the five NSAID compounds, an optimization of the SPE method was performed using a standard solution (DI water). The parameters optimized were sample pH and elution solvent.
The effect of sample pH was investigated on two different SPE sorbents C18 and Oasis HLB, by loading 10 mL of spiked DI water into the cartridges. The pH of the spiked DI water sample was tested at pH 2.5 and pH 7. Due to the acidic nature of NSAIDs, both ionic and non-ionic structures may be present in water samples of varying pH. So, it is important to evaluate the effect of pH (acidic pH and neutral pH of water) on extraction efficiency.
The results shown in Fig. 5A indicated that acidic pH obtained higher recoveries for the target compounds. This was an expected phenomenon for the reversed-phase mode. When a water sample is acidified to a pH lower than the pKa value of the target compounds, the acids become non-ionized, allowing them to adsorb via hydrophobic interactions.44,45 Further increases in the sample pH to 7 resulted in decreased peak areas as the targeted analytes started to be deprotonated into their ionic forms at higher pH values.
 | ||
| Fig. 5 Effect of sample pH on extraction of NSAIDs; (A) C18 and (B) Oasis HLB. Capillary: 50 μm id, 31.5 cm effective length and 40 cm total length; BGE: 15 mM borate buffer, pH 9, 90 mM SDS, 10% methanol; separation voltage: 15 kV; injection: 10 mbar/5 s. | ||
On the basis of the physicochemical properties of sorbents, as well as the polarity and solubility of ASA and PAC, it was assumed that ASA would be retained on the C18 cartridges if the solution had a pH of 2.5, while PAC was not eluted at both pHs; this can be explained by the fact that PAC is a very polar small molecule that interacts weakly with the C18 sorbent, leading to poor retention.
The combination of hydrophilic-lipophilic polymers in the Oasis HLB column allows for the extraction of acidic, neutral, and basic analytes over a broad pH range, including neutral pH. The results have shown that (Fig. 5B) all selected drugs were eluted with Oasis HLB cartridges with different recovery values. As can be seen in Fig. 5B, PAC obtained excellent recovery at pH 7, whereas the rest of the compounds’ acidic pH was preferable. Under both pH conditions evaluated, Oasis HLB provided good results for the majority of the compounds. Still, in comparison to C18, the latter was more efficient for KET, IB, and DIC, yielding higher recoveries.
Further experiments were conducted to enhance the recovery values of the rest of the compounds. To ensure complete elution of the compounds adsorbed on the SPE cartridge, a variety of solvent conditions were investigated. The most used elution solvents are methanol and acetonitrile.39 Thus, the elution solvents tested were methanol, acetonitrile, NH4OH, and acetone. As illustrated in Fig. 6A and B each elution solvent studied was able to elute a specific amount of a particular compound. However, elution of both cartridges C18 and Oasis HLB with methanol resulted in higher recoveries for all analytes. Based on these results, methanol was selected as the optimal elution solvent for both columns.
 | ||
| Fig. 6 Effect of elution solvent on extraction of NSAIDs; (A) C18 and (B) Oasis HLB. Capillary: 50 μm id, 31.5 cm effective length and 40 cm total length; BGE: 15 mM borate buffer, pH 9, 90 mM SDS, 10% methanol; separation voltage: 15 kV; injection: 10 mbar/5 s. | ||
The calibration curves for spiked wastewater samples were plotted using the peak area of the analytes versus the concentration, and each concentration was taken in triplicate. The calibration plots of each analyte prepared at 31–500 μg L−1 for PAC and ASA, 6–100 μg L−1 for DIC and KET, and 15–240 μg L−1 for IB were observed to be linear for spiked wastewater samples, with correlation coefficients R2 > 0.99. The LODs were 3, 3, 7.5, 15, and 15 μg L−1 for DIC, KET, IB, ASA, and PAC, respectively. The repeatability of SPE-MEKC was good, with (n = 6) RSDs of 0.5–8.4%.
To validate the above-described method, wastewater was spiked with the target analytes at different concentrations using two columns, C18 and Oasis HLB (Fig. 7A and B). As can be seen from Fig. 7B the five NSAIDs were extracted using Oasis HLB cartridges, and excellent recoveries were obtained for PAC and ASA (122.4 and 65.2%), respectively. However, the recoveries for IB, KET, and DIC were less than 41%. In comparison to the Oasis HLB cartridge, C18 obtained excellent extraction recoveries of KET, IB, and DIC (91, 83.8, and 118%), respectively. The Oasis HLB sorbent has an advantage over the C18 sorbent for extracting all the analytes; however, with poor recovery for KET, IB, and DIC (Table S2).† Therefore, C18 was selected for the three analytes to obtain excellent recoveries.
 | ||
| Fig. 7 Electropherograms of SPE for wastewater (A) C18 and (B) Oasis HLB; peaks: 1-PAC, 2-KET, 3-ASA, 4-IB, and 5-DIC. Capillary: 50 μm id, 31.5 cm effective length and 40 cm total length; BGE: 15 mM borate buffer, pH 9, 90 mM SDS, 10% methanol; separation voltage: 15 kV; injection: 10 mbar/5 s. | ||
As illustrated in Table 2, the recoveries obtained with the C18 cartridge for KET, IB, and DIC in two spiked concentrations (25 and 50 μg L−1) of wastewater were in the range of 72.6–111.6%, with RSD < 12% (n = 3), indicating excellent recoveries of these compounds. For PAC and ASA, the Oasis HLB cartridge was employed to determine their recoveries in different concentrations of wastewater samples, with good recoveries greater than 70% with RSD < 13.5% (n = 3).
| Recovery (RSD%) | |||
|---|---|---|---|
| Spiked concentration | Analyte | Wastewater sample | |
| C18 | Oasis HLB | ||
| 50 μg L−1 | IB | 111.6 (9.4%) | — |
| KET | 73.1 (6.7%) | — | |
| DIC | 93.4 (9.8%) | — | |
| 25 μg L−1 | IB | 81.1 (12%) | — |
| KET | 76.5 (9%) | — | |
| DIC | 72.6 (8.4%) | — | |
| 50 μg L−1 | ASA | — | 83.6 (10%) |
| PAC | — | 113.7 (7.4%) | |
| 25 μg L−1 | ASA | — | 73 (13.1%) |
| PAC | — | 80.8 (11.4%) | |
The SPE-MEKC method has been reported by Macià. A et al.18 They developed MEKC-stacking online pre-concentration in combination with offline SPE for the determination of NSAIDs (IB, fenoprofen, naproxen, and KET) in mineral water. They obtained better LODs (0.1–1.2 μg L−1), which should be attributed to the combination of online (stacking) and offline (SPE) pre-concentration. Even though low LODs were obtained, low precision also occurred owing to the electrokinetic sample injection. The results presented in this study provide excellent RSDs of 0.5–8.4% and good LODs (3–15 μg L−1), which are comparable with their concentrations in wastewater.3,4 The proposed method achieved excellent recoveries and can be used to determine the studied analytes in environmental applications.
4 Conclusion
The present work describes a highly efficient, selective, and sensitive method of using MEKC-UV for the determination of NSAIDs in wastewater. The effect of each parameter on the separation and efficiency was examined. Under optimal conditions, this study provides a higher number of theoretical plates of N > 780000 with excellent RSDs of 0.1–1.5% and great sensitivity (3–15 μg L−1) for NSAIDs. The method was validated using two different SPE cartridges (C18 and Oasis HLB); the results showed excellent recoveries (73–111.6%) for all the analytes in wastewater samples. The developed method based on MEKC-UV can be used to determine the studied analytes in environmental applications.
Author contributions
Hanan Alatawi: data curation, formal analysis, investigation, methodology, validation, visualization, conceptualization, project administration, resources writing – original draft, writing – review & editing. Ibtihaj Albalawi: methodology, validation, visualization, investigation, review & editing. Samia Alsefri: investigation, review & editing. Anna Hogan: validation, investigation, methodology, supervision, visualization, review & editing. Eric Moore: supervision, project administration, visualization, conceptualization, resources, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Ministry of Higher Education of Saudi Arabia for the financial support. The authors would also like to thank the School of Chemistry and University College Cork for providing the laboratory facilities.References
- T. Brodin, S. Piovano, J. Fick, J. Klaminder, M. Heynen and M. Jonsson, Philos. Trans. R. Soc., B, 2014, 369(1656), 20130580 CrossRef PubMed.
- A. Pereira, L. Silva, C. Laranjeiro, C. Lino and A. Pena, Molecules, 2020, 25, 1796 CrossRef CAS PubMed.
- A. K. Thalla and A. S. Vannarath, Environ. Monit. Assess., 2020, 192, 1–13, DOI:10.1007/s10661-020-08576-9.
- U. G. J. Zur, A. Pinski, A. Marchlewicz, K. Hupert-Kocurek and D. Wojcieszynska, Environ. Sci. Pollut. Res., 2018, 25, 21498–21524 CrossRef PubMed.
- W. Gwenzi, R. Selvasembian, N. A. O. Offiong, A. E. D. Mahmoud, E. Sanganyado and J. Mal, Environ. Chem. Lett., 2022, 20, 1275–1294 CrossRef CAS PubMed.
- R. Kumar, A. K. Sarmah and L. P. Padhye, J. Environ. Manage., 2019, 233, 649–659 CrossRef CAS PubMed.
- Y. Luo, W. Guo, H. H. Ngo, L. D. Nghiem, F. I. Hai, J. Zhang, S. Liang and X. C. Wang, Sci. Total Environ., 2014, 473–474, 619–641 CrossRef CAS PubMed.
- I. Reinholds, I. Pugajeva, D. Zacs, E. Lundanes, J. Rusko, I. Perkons and V. Bartkevics, Environ. Monit. Assess., 2017, 189, 568 CrossRef CAS PubMed.
- C. Hao, X. Zhao and P. Yang, TrAC, Trends Anal. Chem., 2007, 26, 569–580 CrossRef CAS.
- H. Helena, V. Ivona, Ř. Roman and F. František, J. Sep. Sci., 2022, 45, 305–324 CrossRef CAS PubMed.
- Y. Huang, X. Han, X. Yu, S. Wang and H. Zhai, Chromatographia, 2021, 84, 861–868 CrossRef CAS.
- H. Alatawi, A. Hogan, I. Albalawi, S. Alsefri, E. O. Sullivan-carroll, Y. Wang and E. Moore, Electrophoresis, 2022, 1–9 Search PubMed.
- H. Alatawi, A. Hogan, I. Albalawi, E. O'Sullivan-Carroll, Y. Wang and E. Moore, Electrophoresis, 2021, 1–8 Search PubMed.
- I. Maijó, F. Borrull, C. Aguilar and M. Calull, J. Liq. Chromatogr. Relat. Technol., 2012, 35, 2134–2147 CrossRef.
- M. Dawod, M. C. Breadmore, R. M. Guijt and P. R. Haddad, J. Chromatogr. A, 2008, 1189, 278–284 CrossRef CAS PubMed.
- M. Villar Navarro, M. Ramos Payán, R. Fernández-Torres, M. A. Bello-López, M. Callejón Mochón and A. Guiráum Pérez, Electrophoresis, 2011, 32, 2107–2113 CrossRef CAS PubMed.
- P. Lacina, Fresenius Environ. Bull., 2012, 21, 3312–3317 Search PubMed.
- A. Macià, F. Borrull, M. Calull and C. Aguilar, J. Chromatogr. A, 2006, 1117, 234–245 CrossRef PubMed.
- S. M. Ahmad, C. Almeida, N. R. Neng and J. M. F. Nogueira, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1008, 115–124 CrossRef CAS PubMed.
- M. Dawod, M. C. Breadmore, R. M. Guijt and P. R. Haddad, J. Chromatogr. A, 2009, 1216, 3380–3386 CrossRef CAS PubMed.
- A. Macià, F. Borrull, M. Calull and C. Aguilar, Chromatographia, 2006, 63, 149–154 CrossRef.
- S. Sunaric, M. Petkovic, M. Denic, S. Mitic and A. Pavlovic, Acta Pol. Pharm., 2013, 70, 403–411 CAS.
- A. Cifuentes and H. Poppe, Electrophoresis, 1995, 16, 516–524 CrossRef CAS PubMed.
- G. Hancu, B. Simon, A. Rusu, E. Mircia and Á. Gyéresi, Adv. Pharm. Bull., 2013, 3, 1–8 Search PubMed.
- F. Welder and C. L. Colyer, J. Chem. Educ., 2001, 78, 1525–1527 CrossRef CAS.
- M. G. Donato, E. van den Eeckhout, W. van den Bossche and P. Sandra, J. Pharm. Biomed. Anal., 1993, 11, 197–201 CrossRef CAS PubMed.
- R. Sekar, P. Ravi Prasad and M. Vairamani, Indian J. Chem. Technol., 2003, 10, 603–606 CAS.
- W. Muijselaar, Micellar Electrokinetic Chromatography: Fundamentals and Applications, 2022, vol. 1 Search PubMed.
- M. E. El-Kommos, N. A. Mohamed and A. F. Abdel Hakiem, J. Pharm. Anal., 2013, 3, 53–60 CrossRef PubMed.
- M. M. Bushey and J. W. Jorgenson, J. Microcolumn Sep., 1989, 1, 125–130 CrossRef CAS.
- S. Furlanetto, S. Lanteri, S. Orlandini, R. Gotti, I. Giannini and S. Pinzauti, J. Pharm. Biomed. Anal., 2007, 43, 1388–1401 CrossRef CAS PubMed.
- H. J. Issaq, I. Z. Atamna, G. M. Muschik and G. M. Janini, Chromatographia, 1991, 32, 155–161 CrossRef CAS.
- S. A. C. Wren and R. C. Rowe, J. Chromatogr. A, 1992, 609, 363–367 CrossRef CAS.
- Y. J. Lee, W. E. Price and M. M. Sheil, Analyst, 1995, 120, 2689–2694 RSC.
- Y. L. Chen and S. M. Wu, Anal. Bioanal. Chem., 2005, 381, 907–912 CrossRef CAS PubMed.
- X. Xuan and D. Li, J. Micromech. Microeng., 2004, 14, 1171–1180 CrossRef CAS.
- X. Cao, A. Hogan and E. Moore, J. Forensic Sci., 2019, 64, 1213–1220 CrossRef CAS PubMed.
- J. W. Jorgenson and K. DeArman Lukacs, J. High Resolut. Chromatogr., 1981, 4, 230–231 CrossRef CAS.
- D. Sadutto and Y. Picó, Molecules, 2020, 25, 5204 CrossRef CAS PubMed.
- A. Macià, F. Borrull, M. Calull, F. Benavente, E. Hernández, V. Sanz-Nebot, J. Barbosa and C. Aguilar, J. Sep. Sci., 2008, 31, 872–880 CrossRef PubMed.
- Y. P. Huang, R. C. Robinson, F. F. G. Dias, J. M. L. N. de Moura Bell and D. Barile, Foods, 2022, 11, 340 CrossRef CAS PubMed.
- N. Gilart, R. M. Marcé, F. Borrull and N. Fontanals, J. Sep. Sci., 2012, 35, 875–882 CrossRef CAS PubMed.
- H. Khazri, S. Ben Hassine, I. Ghorbel-Abid, R. Kalfat and M. Trabelsi-Ayadi, Environ. Forensics, 2019, 20, 121–128 CrossRef CAS.
- L. M. Madikizela and L. Chimuka, Water SA, 2017, 43, 264–274 CrossRef CAS.
- L. Maldaner and I. C. S. F. Jardim, Talanta, 2012, 100, 38–44 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ay01807a |
| This journal is © The Royal Society of Chemistry 2023 |