
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a designed comparison method based on isotope dilution liquid chromatography–tandem mass spectrometry for determining plasma renin activity and its clinical assessment of renin activity stability in plasma†
Zhenni
Liu
 ab,
Lizi
Jin
ab,
Jiangtao
Zhang
a,
Tianjiao
Zhang
ab,
Jie
Zeng
a,
Weiyan
Zhou
*a and
Chuanbao
Zhang
*ab
ab,
Lizi
Jin
ab,
Jiangtao
Zhang
a,
Tianjiao
Zhang
ab,
Jie
Zeng
a,
Weiyan
Zhou
*a and
Chuanbao
Zhang
*ab
aNational Center for Clinical Laboratories, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing Hospital/National Center of Gerontology, P. R. China. E-mail: wyzhou@nccl.org.cn; cbzhang@nccl.org.cn; Fax: +86 010-65132968; Tel: +86 010-58115059
bChinese Academy of Medical Sciences and Peking Union Medical College, No. 1 Dahua Road, Dongcheng District, Beijing 100730, P. R. China
First published on 6th January 2023
Abstract
Plasma renin activity (PRA) is recommended as the first screening indicator for primary aldosteronism. Immunoassays and liquid chromatography–tandem mass spectrometry (LC-MS/MS) methods have been developed for quantifying PRA, but the interchangeability across assays and laboratories was suboptimal, which predominantly related to the differences in the plasma incubation strategy. This study aims to establish and validate a designed comparison method based on LC-MS/MS. The sensitivity, matrix effect, precision, accuracy, and storage stability were validated according to the Clinical Laboratory Standard Institution (CLSI) C-62A guidelines. The plasma incubation procedure was optimized to achieve maximum PRA results. The short-term stability of PRA plasma was assessed at 4 °C and room temperature (RT) for specific time points. Differences from the baseline were calculated using a one-way analysis of variance. The designed comparison method for PRA measurement exhibits excellent performance characteristics. The results from the 2022 national external quality assessment scheme for PRA showed good consistency of the developed method with other LC-MS/MS methods (relative biases: −6.8% to 4.6%), which demonstrated the reliability of the established method. Two sets of generation buffers were optimized to maximize the renin activity. The acetate buffer was recommended to be used in laboratory practice due to better metrological sensitivity. PRA plasma is stable for one day at 4 °C and RT. In summary, a reliable, traceable, and reproducible LC-MS/MS method for determining PRA was well-established and validated. The recommended incubation protocol is hoped to reduce the discrepancy in Ang1 generation. The evaluated short-term stability for PRA plasma could provide flexibility in clinical practice.
Introduction
Primary aldosteronism (PA) is one of the most common causes of secondary hypertension, characterized by significantly elevated plasma aldosterone and decreased plasma renin activity (PRA) levels, hypokalemia, and hypertension.1–3 A timely, precise diagnosis and treatment could effectively improve the prognosis and life quality of PA patients. The Endocrine Society4 has recommended the aldosterone-to-renin ratio (ARR) as the first-line indicator for PA screening. Many studies suggested that ARR has a superior diagnosis efficacy to plasma potassium or aldosterone assays.3–5 PRA was measured as the denominator to calculate the ARR value, which plays an important role in the evaluation of disease conditions, especially in patients with slightly elevated plasma aldosterone levels.6,7Renin, an aspartate proteolytic enzyme released from the renal juxtaglomerular cells, substantially reflects PRA levels. Renin cleaves the endogenous substrate angiotensinogen (AGT) and produces angiotensin 1 (Ang1). PRA can be calculated as the amount of Ang1 produced per unit time during the incubation period, i.e., [(the concentration of Ang1 after incubation minus before incubation)/incubation time], with the unit of ng mL−1 h−1. This enzymatic assay can reflect the real condition and activity of individual renin–angiotensin systems since it takes into account the contributions of both enzyme (i.e., renin) and substrate (i.e., AGT) levels.8 The in vitro reaction of enzyme activity highly relied on an appropriate buffer system, since non-specific proteinase might hydrolyze Ang1 into other peptides (e.g., angiotensin 2 and angiotensin 1–7), and cause underestimation of PRA levels. Thus, proteinase inhibitors such as ethylenedinitrilotetraacetic acid (EDTA) and phenylmethylsulfonylfluoride (PMSF) were added to the incubated plasma to avoid the degradation or transformation of Ang1.9 Incubation strategies consist of pH buffers, proteinase inhibitors, incubation time, etc., which greatly influence the renin enzyme activity, and further produce variable results.
Accurate measurement of PRA was essential and troublesome. For advantages such as cost-effectiveness, high throughput, and practical simplicity, radioimmunoassay (RIA) and chemiluminescence immunoassay (CLIA) have been the mainstream assays for PRA determination for decades, but they also have several major drawbacks, e.g., cross-reactivity, antibody recognition sites, radioactive hazards, etc. Moreover, incubation procedures were extremely variable across different laboratories or manufacturers, since no (inter-) nationally recommended consensus is available. A comprehensive multicenter comparison was conducted among twelve European laboratories to determine the intra- and inter-laboratory reproducibility.8 A commercial diagnostic kit based on RIA for PRA assay was applied for the comparison. Citrate buffer (pH = 6) containing PMSF was added to each aliquot of plasma sample in duplicate, and then incubated at 37 °C and 4 °C for 90 min, respectively. Despite the use of uniform reagents and adherence to the instrument of the manufacturer, significant inter-laboratory coefficients of variation (CVs) were observed, particularly in low-concentration plasma (e.g., PRA: 0.14 ng mL−1 h−1; with a CV of 59.4%). The complex incubation details and inaccurate quantification may represent major obstacles to reproducibility and comparability among different assays and laboratories.
To improve the accuracy and reliability of PRA measurement, liquid chromatography–tandem mass spectrometry (LC-MS/MS) assays have been established.9–12 However, the lack of a harmonized incubation protocol hampered the interchangeability among different laboratories. According to the statistics from the 2021 National External Quality Assessment (EQA) Program organized by the National Center for Clinical Laboratories (NCCL, China, Available at: https://www.nccl.org.cn/mainCn), five EQA materials with a PRA concentration range of 1.88–13.63 ng mL−1 h−1 were provided for participating laboratories. Intra-assay CVs ranged from 23 to 66% (n = 12) for LC-MS/MS assays and 34 to 111% (n = 47) for CLIA assays. The mean biases between CLIA assays and LC-MS/MS methods ranged from −12.1% to 76.5%. Remarkable inconsistency existed among different assays and laboratories.
PRA assays are susceptible to environmental influences, such as temperature, cryoactivation of prorenin,13 plasma incubation conditions, non-specific proteinase degradation, etc. Therefore, it is necessary to achieve a consensus about plasma incubation for reducing the variations from the analytical process and further improving the harmonization status. In this study, we aim to establish a precise, reproducible, and reliable LC-MS/MS method for PRA measurement, and to evaluate the stability of PRA plasma.
Experimental
Chemicals and reagents
The Ang1 standard materials (AS-20627, 5 mg) with purity ≥95% and isotopic internal standard (IS) Ang1-[13C6, 15N] were purchased from AnaSpec Inc. (USA). The standard reference material of Ang1 (SRM 998, 0.5 mg per vial) was obtained from the National Institute of Standards and Technology (NIST, USA); the purity is 99.9% ± 0.1%. The certified Ang1 purity value was assessed by HPLC and confirmed indirectly by nuclear magnetic resonance (within the reported uncertainty). Phenylmethanesulfonyl fluoride (PMSF), soybean trypsin inhibitor (SBTI), zinc sulfate heptahydrate, bovine serum albumin (BSA), and ethylenedinitrilotetraacetic acid (EDTA) were purchased from Sigma-Aldrich (USA). Tris-base was obtained from Roche (Germany). Methanol, hexane, and formic acid were HPLC grade purchased from Fisher Scientific Co. (USA).Analytical equipment and supplies
Ultra-pure deionized water (≥18.2 MΩ cm) was prepared using a Millipore-Q water purifier (Billerica, MA, USA). A 6500 plus triple quadrupole mass spectrometer (AB Sciex, USA) coupled with a Waters ACQUITY UPLC Fl-I class system (Waters, USA) was used to perform the LC-MS/MS analysis. A Phenomenex Kinetex C18 column (2.6 μm, 100 mm × 2.1 mm) was purchased from Phenomenex (CA, USA). Oasis HLB cartridges used for solid-phase extraction (SPE) were obtained from Waters Corporation (USA).Preparation of stock and working solutions
All standard and IS solutions were gravimetrically prepared. For the preparation of the Ang1 standard stock solution, deionized water containing 10% formic acid was used to dissolve the Ang1 standard materials. The IS Ang1-[13C6, 15N] stock solution was prepared similarly. Ang1 working solution (17.6 ng g−1) was prepared in 1% BSA buffer solution from stock solutions; this buffer solution consists of 0.1 mol L−1 Tris in deionized water, adjusted to pH = 6 using glacial acetic acid. The IS working solution (10.7 ng g−1) was diluted with deionized water containing 10% formic acid. Aliquots (1 mL) of stock and working solutions were kept in polypropylene Protein LoBind® tubes (Eppendorf) and frozen at −70 °C.Plasma collection and sample preparation
Leftover EDTA plasma samples from outpatients or hospitalized patients who underwent PRA examination (unaffected or affected patients were both included) were collected from the endocrine laboratory of Beijing Hospital between September and December 2021 and were rapidly frozen at −70 °C. Two concentrations of quality controls (QCs) (i.e., 3.8 and 10.1 ng mL−1 h−1) for PRA were prepared using pooled residual plasma and analyzed in each run. QCs and plasma samples were prepared by the following procedure. The collection of residual plasma had been approved by the Ethics Committee of Beijing Hospital.Considering that the frozen plasma samples ought to be rapidly thawed at room temperature rather than in a refrigerator,13–15 plasma samples/calibrators/QCs were fully thawed at 37 °C for 3 min, to avoid cryoactivation and help to speed up the thawing process. The sample preparation consists of plasma incubation and extraction. Ang1 generation buffer consists of pH buffer and proteinase inhibitors. These proteinase inhibitors were prepared in 1 mol L−1 sodium acetate aqueous solution (adjusted to pH = 5.6–5.7 using glacial acetic acid), with the final concentrations of PMSF, SBTI, and EDTA of 5 mmol L−1, 200 mg L−1, and 50 mmol L−1, respectively. Particularly, it is better to add PMSF and SBTI to the inhibitor buffer before each use.
A bracketing calibration was used to quantify Ang1.16 Appropriate amounts of Ang1 standard and IS working solutions were exactly weighed using an electronic balance and fully mixed to produce the six-point bracketing calibrators with analyte-to-IS ratios ranging from 0.25 to 6. 60 μL of generation buffer was added to 200 μL of plasma, and the mixture was fully agitated and then incubated at 37 °C for 3 h. 5 μL of formic acid and 50 μL of IS solution were added to stop the incubation reaction. The mixture was equilibrated at 4 °C for 0.5 h.
Sample extraction comprised protein precipitation and solid-phase extraction (SPE). 800 μL of 0.1 mol L−1 zinc sulfate in methanol/water (v/v, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) was added to precipitate the protein. After vortex mixing and centrifugation at 4 °C (12000 rpm, 10 min), the supernatants were transferred to Oasis HLB cartridges which were preconditioned with methanol (1 mL) followed by water containing 5% formic acid (1 mL). The loaded cartridges were washed sequentially with 15% methanol/water (1 mL) and hexane (1 mL), then samples were eluted with methanol containing 1% formic acid (1.5 mL), and evaporated to dryness under nitrogen. The residuals were reconstituted with 60 μL of 20% methanol/water containing 2% formic acid. After centrifugation at 4 °C (12000 rpm, 10 min), 50 μL of residues was used for LC-MS/MS analysis. The injection volume was 15 μL.
:50) was added to precipitate the protein. After vortex mixing and centrifugation at 4 °C (12000 rpm, 10 min), the supernatants were transferred to Oasis HLB cartridges which were preconditioned with methanol (1 mL) followed by water containing 5% formic acid (1 mL). The loaded cartridges were washed sequentially with 15% methanol/water (1 mL) and hexane (1 mL), then samples were eluted with methanol containing 1% formic acid (1.5 mL), and evaporated to dryness under nitrogen. The residuals were reconstituted with 60 μL of 20% methanol/water containing 2% formic acid. After centrifugation at 4 °C (12000 rpm, 10 min), 50 μL of residues was used for LC-MS/MS analysis. The injection volume was 15 μL.
LC-MS/MS analysis
Chromatography was performed on a Kinetex C18 column which was maintained at 40 °C. The automatic sampler temperature is 10 °C. Mobile phase A contained 0.2% formic acid in deionized water and mobile phase B contained 0.2% formic acid in methanol. Initial conditions were 90:10 (v/v) mobile phase A: mobile phase B at a flow rate of 0.4 mL min−1 with the following linear gradient steps: 0.5 min, 10% B; 1.5 min, 95% B; 3.5 min, 95% B; 3.6 min, 10% B and 5.0 min, 10% B. The total run time was 5 min.
The column eluate was injected into a 6500 plus triple quadrupole mass spectrometer (AB Sciex, USA) maintained in electrospray positive ionization mode, with a source temperature of 450 °C and ionspray voltage of 5500 V. Multiple reaction monitoring (MRM) mode was used to analyze the mass transitions of Ang1. The most abundant transitions identified for Ang1 were m/z 433.2 → 647.4 as a quantifier and 433.2 → 619.4 as a qualifier. For Ang1-IS, m/z 437.6 → 660.5 was a quantifier, and 437.6 → 631.4 was a qualifier. Nitrogen was used as the curtain gas (CUR), nebulizer gas (GS1), auxiliary gas (GS2), and collision gas (CAD), and the pressures of the gases were set at 30, 40, 50, and 6 psi, respectively. Data are analyzed using Analyst 1.7 software (AB Sciex, USA). Typical chromatograms for Ang1 and Ang1-IS of an extracted plasma sample are shown in Fig. 1.
 | ||
| Fig. 1 A representative chromatogram of Ang1 and the IS extracted from a plasma sample. | ||
Quantitative method
Ang1 concentrations were calculated by comparing the analyte-to-IS ratios of the samples with those of the six-point calibrators, The mass fractions (ng g−1) were converted to mass concentrations (ng mL−1) by multiplying the density of the plasma measured with a density meter (DMA 4500 M, Anton Paar, Austria).PRA was calculated using the following equation:  (ng mL−1 h−1). Although it might be reasonable to subtract endogenous Ang1 (non-generated) from generated Ang1, many studies have proposed neglecting the blanks during measurements.12,13,15,17 Since the majority of endogenous Ang1 was usually below the limit of quantification, for the remaining measurable ones, endogenous Ang1 merely constitutes a minor proportion of generated Ang1 ranging from 5.6% to 8.5%, so they could exert little impact on the calculated results. Therefore, the calculation of PRA was simplified to the above equation.
(ng mL−1 h−1). Although it might be reasonable to subtract endogenous Ang1 (non-generated) from generated Ang1, many studies have proposed neglecting the blanks during measurements.12,13,15,17 Since the majority of endogenous Ang1 was usually below the limit of quantification, for the remaining measurable ones, endogenous Ang1 merely constitutes a minor proportion of generated Ang1 ranging from 5.6% to 8.5%, so they could exert little impact on the calculated results. Therefore, the calculation of PRA was simplified to the above equation.
Method validation
:1 and 3:1, respectively, and the CV ≤ 20% for 20 injections. The standard solutions were processed according to the sample preparation procedure and were analyzed to evaluate the LOQ and LOD of the LC-MS/MS method.
Secondly, we prepared four different solutions to conduct the matrix admixing experiment: (X) the pure analyte/IS mixed standard solution; (Y) adding the same amount of analyte/IS mixed standard solution to the extracted matrix; (Z) equal volume of the extracted matrix only; (W) adding the same amount of IS solution to the extracted matrix. Therefore, the original matrix effect can be calculated using the following eqn (1):
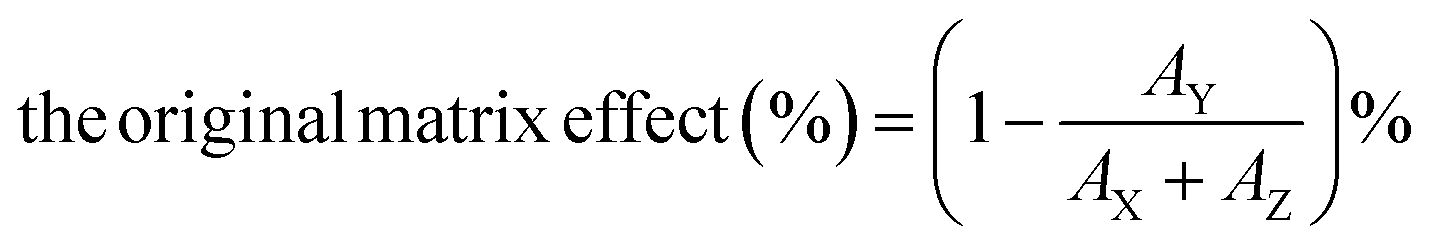 | (1) |
 | (2) |
The collected plasma samples were thawed and mixed to prepare a plasma pool, and then aliquoted into 0.6 mL per vial. Samples were kept at 4 °C and RT (20 °C) from 0.5 h to 7 days to evaluate the stability of the PRA plasma during short-term storage. Two vials were placed under each storage condition and at each time point, each vial was measured in two replicates and the averages of replicates (n = 4) were calculated as the final results. Acetate buffer was adopted to perform plasma incubation in the subsequent application.
The influence of multiple freeze-thaw stability was also examined. Samples were thawed at RT for 1 h and frozen again at −70 °C. This action was performed in respective tubes once (FT1), twice (FT2) or three times (FT3) on different days. After the multiple freeze–thaw, all samples were frozen at −70 °C until further analysis.
Plasma frozen at −70 °C was used as a baseline. Samples under all storage conditions and at all time points were in two replicates, and the averages of replicates were calculated as the final results. Percentage changes of more than 10% from baseline concentration were considered as the stability threshold. The percentage changes (%) were calculated as [(Tx − T0)/T0] × 100%, where Tx is the result measured at a specific time point at a given temperature, and T0 is the concentration of the baseline sample.
Livesey et al.21 suggested that they combined analytical imprecision and intra-individual biological variation as the total change limit (TCL), to evaluate the pre-analytical instability. To our knowledge, there is still no available meta-analysis or analytical performance specification updated for PRA. Hence, we chose 10% as the acceptable percentage change from the baseline concentration; this is generally used as a threshold for acceptable pre-analytical variability.22–24
Data were analyzed using SPSS Statistics 26.0 (IBM Corp, USA). One-way ANOVA was used to assess the significance of differences. p < 0.05 was regarded as statistical significance.
Results and discussion
Absolute recovery, limit of quantification (LOQ), limit of detection (LOD), and linearity
The absolute recovery of the LC-MS/MS method ranged from 82% to 90%. The LOQ and LOD values of the LC-MS/MS method were 0.05 ng g−1 and 0.02 ng g−1 with a CV of 8.7% (n = 20). The average slope, intercept, and correlation relationship (R) with their 95% confidence interval (CI) obtained from 11 inconsecutive calibration curves used for analysis during one month were 0.067 (0.064 to 0.070), 0.069 (0.048 to 0.087), and 0.999 (0.999 to 1.000), respectively.Matrix effect
The post-column infusion experiment verified that no apparent ion suppression or enhancement was observed near the retention time of the analyte or the IS peak. The matrix admixing experiment employed two plasma matrixes (low and high concentrations of PRA), and the results showed that the original matrix effect ranged from 10.3% to 41.0%, and the IS-calibrated matrix effect ranged from 2.6% to 5.2%, suggesting that the IS could efficiently compensate ion suppression for the LC-MS/MS method.Imprecision
The imprecision of the LC-MS/MS method for Ang1 and PRA measurement is shown in Table 1. The intra-run, inter-run, and total CVs for Ang1 quantification were 1.12–4.05%, 1.14–2.59%, and 1.05–4.81%, respectively. The intra-run, inter-run, and total CVs for PRA (acetate buffer) were 2.78–3.39%, 3.64–8.58%, and 4.58–9.22%, respectively. The intra-run, inter-run, and total CVs for PRA (Tris buffer) were 3.47–5.08%, 4.30–6.11%, and 5.53–7.94%, respectively.| Mean | Intra-run CV, % | Inter-run CV, % | Total CV, % | ||
|---|---|---|---|---|---|
| a The unit of Ang1 is ng mL−1. b The unit of PRA (both for acetate and Tris buffers) is ng mL−1 h−1; the medium and high plasma pools used in acetate and Tris buffers were the same, while the low plasma pools for the two buffers were different samples. | |||||
| Ang1a | Sample 1 | 0.81 | 4.05 | 2.59 | 4.81 |
| Sample 2 | 2.19 | 2.04 | 1.14 | 2.33 | |
| Sample 3 | 18.52 | 1.12 | — | 1.05 | |
| Sample 4 | 31.06 | 1.71 | 2.07 | 2.68 | |
| PRA (acetate)b | Low plasma pool-1 | 3.01 | 2.78 | 3.64 | 4.58 |
| Medium plasma pool | 5.43 | 3.39 | 8.58 | 9.22 | |
| High plasma pool | 31.29 | 2.99 | 7.87 | 8.42 | |
| PRA (Tris)b | Low plasma pool-2 | 2.44 | 4.08 | 5.90 | 7.18 |
| Medium plasma pool | 4.61 | 3.47 | 4.30 | 5.53 | |
| High plasma pool | 27.78 | 5.08 | 6.11 | 7.94 | |
Analytical recovery and accuracy
A standard spiking and recovery experiment showed that the analytical recoveries of the LC-MS/MS method ranged from 95.8% to 106.9% (Table 2). The biases between the designed comparison method and target values of 2022 national EQA materials (with the concentrations ranging from 1.51 to 10.06 ng mL−1 h−1) were −6.8% to 4.6%, which were within the bias criterion of 15%25 (1/2 total allowable error, i.e., 30%, which referred to the acceptable limit of the 2022 national EQA scheme).| Spiked samples | Added concentrations, ng mL−1 | Measured concentrations, ng mL−1 | Recoveries, % | Mean recovery, % | Coefficients of variation, % (n = 6) |
|---|---|---|---|---|---|
| Sample 1 | 0.40 | ||||
| Low | 3.83 | 3.88 | 96.3–105.0 | 101.4 | 3.0 |
| Medium | 6.94 | 7.07 | 99.9–104.9 | 101.8 | 1.6 |
| High | 23.31 | 23.70 | 98.4–106.9 | 101.7 | 2.8 |
| Sample 2 | 18.61 | ||||
| Low | 11.64 | 11.60 | 97.0–102.9 | 100.3 | 2.1 |
| Medium | 19.90 | 19.87 | 96.9–104.7 | 100.2 | 2.4 |
| High | 36.47 | 37.24 | 95.8–101.7 | 98.0 | 1.9 |
A reliable, robust, and traceable LC-MS/MS method was developed as the designed comparison method for Ang1 measurement. Ang1 is a basic polypeptide, and the addition of formic acid improves the solubility and stability of the standard material. 1% BSA solution and polypropylene Protein LoBind tubes help prevent peptide adsorption on the container surface. Ang1 was extracted and purified by protein precipitation and SPE, which produces a cleaner matrix and improves analytical reproducibility. We adopted the bracketing calibration with an isotopic ratio range of 0.25 to 6 and achieved satisfactory linearity for Ang1. All the standard calibrators and IS solutions were gravimetrically prepared. Differing from the common quantification method, which employed the volumetric method and linearity calibration with a wide concentration range, bracketing calibration allows laboratory technicians to adjust the amounts of samples and IS solutions weighed according to the actual concentration, especially for those beyond the isotopic ratio range; this could ensure that the area ratios fall within the measurable range to achieve superb linearity. No obvious matrix effect exists after the IS calibration. We adopted NIST SRM 998 as the available higher-order standard material to achieve the metrological traceability to the SI unit. The LC-MS/MS method exhibits excellent sensitivity, precision, and analytical recovery for Ang1 measurement, as well as good precision for the entire measuring procedure (i.e., plasma incubation plus Ang1 determination). The results showed that the imprecision was ≤5% for Ang1 quantification and ≤10% for the PRA assay. It is of note that the analytical recovery experiment was performed on Ang1 quantification but did not include the plasma incubation procedure. Actually, we tried to add Ang1 standard solution to the plasma and then performed plasma incubation; the ultimately generated Ang1 was measured by the developed LC-MS/MS method, but the results showed lower Ang1 levels in spiked plasma compared with the non-spiked plasma (ESI Table 1†). One possible explanation could be that increased production caused negative feedback to the activity of the renin enzyme and lowered the reaction rate. The reliability of the established LC-MS/MS method was demonstrated through the 2022 national EQA scheme for PRA, in which good consistency of the developed method with other LC-MS/MS methods (relative biases: −6.8% to 4.6%) was observed.
Optimization of plasma incubation buffers
The PRA levels elevated as the concentrations of Tris and acetate increased stepwise. When Tris and acetate concentrations reached 1 mol L−1, the PRA reached the maximum value (Fig. 2a); this is consistent in both low and high PRA plasma (Fig. 2b). Next, the optimal EDTA concentrations in two buffers were evaluated. 50 mM EDTA in 1 mol L−1 acetate buffer and 100 mM EDTA in 1 mol L−1 Tris buffer (Fig. 2c) supplemented with PMSF and SBTI generated higher PRA. A combination of 5 mM PMSF and 200 mg L−1 SBTI added to the optimized buffer would exert higher PRA results (Fig. 2d). | ||
| Fig. 2 The optimization of the incubation buffer. (a) PRA generated from different concentrations of acetate or Tris; (b) different concentrations of acetate or Tris buffers incubated with low and high levels of PRA plasma; (c) PRA generated from 1 mol L−1 acetate/Tris added with different concentrations of EDTA and/or PMSF + SBTI; (d) PRA generated from different concentrations of PMSF/SBTI. | ||
Little research was conducted on the influence of different buffers on Ang1 generation. We compared and optimized two mainstream buffer components9,12 to maximize the enzyme catalytic activity of renin and inhibit the non-specific degradation of Ang1. 1 mol L−1 Tris or acetate was chosen as the pH buffer to achieve optimal activity of the renin enzyme. There are two other reasons to support this result. First, the commercially available Tris buffer is generally set at 1 mol L−1, which is sufficient to meet the need to maintain pH constant. The higher concentration might cause incomplete dissolution and economic cost. Second, in previously published LC-MS/MS assays, 1 mol L−1 Tris/acetate buffer was the most commonly used incubation buffer.12,15,17,26 For achieving the harmonization of the incubation strategy, the widely employed buffer setting is easier to adopt, and fewer changes would be required for laboratory professionals, and thus might facilitate the harmonization process. pH 5.6–5.7 was the favorable range for proteinase inhibition.27 EDTA acts as a metalloprotease inhibitor to prevent the angiotensin-converting enzyme (ACE) from converting Ang1 to Ang2. In this study, 100 mmol L−1 EDTA in 1 mol L−1 Tris and 50 mmol L−1 EDTA in 1 mol L−1 acetate were adopted. But higher concentrations of EDTA (over 250 mmol L−1) were not evaluated, because we found that during the solvent preparation, when EDTA concentrations were higher than 250 mmol L−1, it is hard to achieve complete dissolution in Tris or acetate buffers despite an ultrasonic vibration over 30 min. In addition, the proteinase inhibitors (PMSF and SBTI) exert a positive influence on generated Ang1, with the appropriate concentrations of PMSF and SBTI being 5 mmol L−1 and 200 mg L−1, respectively (Fig. 2d); the add-on proteinase inhibitors might provide supplementary protection for proteinase degradation. The ultimate generation buffer settings are displayed in Fig. 2.
Two optimized generation buffers (i.e., acetate and Tris) for plasma incubation were developed, however, the generated Ang1 significantly differed between the two buffers. Ang1 level was approximately 1.1 to 1.3-fold higher in acetate than in Tris buffer. In the present study, considering the quantitative sensitivity, we prefer to recommend acetate as the inhibitor buffer and use it in the subsequent determination. Furthermore, it is suggested that the correlation and comparability between the generation buffers deserve further studies.
The different generation buffers adopted in CLIA and LC-MS/MS methods were observed. Commercial CLIA diagnostic kits usually adopted EDTA, dimercapto-propanol, and 8-hydroxyquinoline sulfate as metalloproteinase inhibitors, while LC-MS/MS methods9,12,28 utilized EDTA, PMSF, and SBTI. The difference in inhibitors, as well as the cross-reactivity and sensitivity of CLIA, may together cause poor consistency (−12.1% to 76.5%) between CLIA and LC-MS/MS assays. This issue may be addressed by harmonizing the inhibitors between different assays to minimize the bias source from plasma incubation.
This study proposed a reliable and robust incubation procedure for PRA assay. When the validated incubation strategy is introduced in LDT laboratories, the harmonization among LC-MS/MS assays would be improved greatly. After the application of the uniform protocol in LC-MS/MS laboratories, it is time to appeal for immunoassays to coordinate with this harmonized standard (e.g., modifying their kits), and eventually improve the comparability across various laboratories and assays.
Stability of Ang1 working solution and plasma samples
The short-term stability experiment of the Ang1 working solution (Table 3) showed that the concentrations of Ang1 working solution stored at 4 °C and RT at different time points showed little change, ranging from 19.5 ng mL−1 to 20.2 ng mL−1 compared with the baseline concentration being 19.9 ng mL−1 (percentage changes: −1.9% to 1.5%). Ang1 working solution is stable at 4 °C and RT for 3 days at least. The results of three freeze–thaw cycles for the Ang1 working solution were not significantly different from the baseline concentration (19.9 ng mL−1), with the change percentage ranging from −0.6% to −0.3% (p > 0.05).| Ang1 working solution, ng mL−1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| T 0 | TCL (10%) | Mean measured concentration (percentage difference, %), n = 4 | |||||||
| 2 h | 6 h | 12 h | 1 day | 36 h | 2 days | 3 days | |||
| a T 0, baseline concentration; TCL, total change limit. | |||||||||
| 4 °C | 19.9 | 17.9–21.9 | 19.9 | 20.0 | 19.9 | 19.8 | 19.7 | 19.9 | 19.7 |
| (+0.0%) | (+0.6%) | (+0.3%) | (−0.4%) | (−0.7%) | (+0.3%) | (−1.0%) | |||
| RT | 19.9 | 17.9–21.9 | 20.1 | 20.2 | 19.7 | 19.9 | 19.5 | 19.8 | 19.5 |
| (+1.0%) | (+1.5%) | (−0.7%) | (+0.2%) | (−1.9%) | (−0.4%) | (−1.6%) | |||
| Freeze–thaw | 1 cycle | 2 cycles | 3 cycles | ||||||
| 19.9 | 17.9–21.9 | 19.7 | 19.7 | 19.8 | |||||
| (−0.6%) | (−0.6%) | (−0.3%) | |||||||
| PRA plasma, ng mL−1 h | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T 0 | TCL (10%) | Mean measured concentration (percentage difference, %), n = 4 | ||||||||
| 0.5 h | 1 h | 2 h | 6 h | 12 h | 1 day | 3 days | 7 days | |||
| 4 °C | 8.3 | 7.5–9.1 | 8.8 | 8.5 | 8.7 | 8.0 | 8.6 | 8.5 | 10.4 | 10.8 |
| (+6.5%) | (+2.7%) | (+5.2%) | (-3.3%) | (+3.3%) | (+2.2%) | (+25.2%) | (+29.6%) | |||
| RT | 9.6 | 8.6–10.5 | 9.3 | 9.1 | 9.3 | 9.1 | 9.9 | 10.0 | 12.1 | 14.5 |
| (-2.9%) | (-4.7%) | (-2.8%) | (-5.1%) | (+3.3%) | (+4.0%) | (+26.5%) | (+51.8%) | |||
| Freeze–thaw | 1 cycle | 2 cycles | 3 cycles | |||||||
| 8.8 | 7.9–9.6 | 9.3 | 8.5 | 8.5 | ||||||
| (+6.0%) | (-2.7%) | (-2.4%) | ||||||||
For plasma samples (Fig. 3 and Table 3), during storage at 4 °C, the PRA concentration increased after 3 days (from 8.3 ng mL−1 h−1 to 10.4 ng mL−1 h−1; +25.2%; p < 0.0001), and then increased by 29.6% after 7 days (from 8.3 ng mL−1 h−1 to 10.8 ng mL−1 h−1; p < 0.0001). During storage at RT, the PRA concentration increased after 3 days (from 9.6 ng mL−1 h−1 to 12.1 ng mL−1 h−1; +26.5%; p < 0.0001), and rapidly increased more than half of the baseline concentration after 7 days (from 9.6 ng mL−1 h−1 to 14.5 ng mL−1 h−1; +51.8%; p < 0.0001). The results of three freeze–thaw cycles for PRA were not significantly different from the baseline concentration (8.8 ng mL−1 h−1), with the change percentage ranging from −2.7% to 6.0% (p > 0.05).
 | ||
| Fig. 3 The short-term stability of PRA plasma samples (n = 4) at 4 °C (A) and room temperature (B). The mean (standard deviation; error bars) concentrations of PRA (y-axis) vs. the storage time (x-axis). *Statistical significance (p < 0.05) vs. baseline concentrations (T0). | ||
Several studies6,24 have evaluated the stability of DRC or PRA in whole blood collected into serum gel or EDTA plasma tubes before centrifugation. Locsei et al.6 proposed that when whole blood was kept at 4 °C for 2 h or at RT within 30 min before centrifugation, the PRA results remained unchanged. They also recommended that −20 °C storage should not exceed 2 weeks, because PRA decreased by 9.4% ± 2.4%. Chakera et al.24 found that DRC collected in EDTA plasma tubes dropped by over 10% after 6 h at RT (percentage change: −10.4%, 95% CI: −17.9% to −2.9%). However, few studies explore the short-term stability of post-centrifugation PRA plasma samples, and this knowledge is critical for EQA program providers and clinical laboratorians to handle EQA samples (plasma-matrixed pools for PRA measurement) throughout the analysis.
In this study, PRA plasma was observed to be stable for one day at 4 °C or RT; if the storage time is prolonged, the PRA results will significantly increase (>10%). This variation is probably attributed to the ex vivo renin catalysis activity or prorenin cryoactivation.6 Prorenin cryoactivation was expected to occur after the cooling stimulation lasting at least 6 h to 24 h,24,29 which is similar to our findings. This study provided more flexibility and convenience to laboratory technicians, especially when handling the EQA plasma materials.
We assessed the influence of freeze–thaw cycles and the results showed that PRA just changed slightly after three freeze–thaw cycles (−2.7% to 6.0%); this also provides confidence in the evaluation of the plasma stability study, since the inevitable freeze–thaw activities might cause little interference. In contrast, Hillebrand et al.30 found that two or three freeze–thaw cycles would increase the PRA concentration significantly (+52.5% vs. baseline, p = 0.0076). These conflicting findings are probably due to different assay principles. The former study adopted an in-house RIA assay based on antigen–antibody binding, with an inter-assay CV of 12%; while the developed LC-MS/MS method showed better specificity and precision (total CV < 10%). Second, variable levels of PRA samples may exhibit changeable responses to prorenin cryoactivation or freeze–thaw stability,6,24 since low concentration (1.2 ± 0.4 ng mL−1 h−1) and high concentration (8.8 ± 0.1 ng mL−1 h−1) plasma were analyzed in the RIA assay and in this established method, respectively.
Therefore, this study may be a piece of the supplementary information for the clinical guideline,4 which suggests that for post-centrifugation plasma samples cannot be tested immediately, and short-term storage of one day at 4 °C or RT is acceptable; this would efficiently work against cryoactivation and avoid multiple freeze–thaw. The residual plasma should be immediately aliquoted and frozen in case any re-testing is required. Moreover, considering the poor stability at −20 °C, a more stringent storage strategy would be preferable (e.g., −70 °C).6,31 More information about the long-term stability at −70 °C is needed.
One limitation of this study is that we did not evaluate the blank subtraction. Previous studies are in favor of neglecting the blanks, and potential benefits include lower sample volume and cheaper labor costs. The small proportion of blank to the generated Ang1 was considered to be quantitatively irrelevant.12 However, a well-designed evaluation of the necessity of blank subtraction was needed in clinical practice, if necessary, in conjunction with its relevance to clinical decision-making. Additionally, we did not perform a detailed interference study because we only obtained the standard materials of Ang1 and Ang2; mass spectrometry analysis could distinguish the two compounds completely (MRM transitions at m/z 433.2 → 647.4 and m/z 524.0 → 263.2 for Ang1 and Ang2, respectively).
Conclusions
A robust, traceable, and reliable designed comparison method based on LC-MS/MS for measuring PRA was established and validated. The recommended incubation strategies might reduce the analytical variability of Ang1 generation, which currently limits the clinical application of this assay. Short-term stability for one day at 4 °C and RT could provide flexibility in clinical practice.Ethical approval
The collection of plasma was approved by the Ethics Committee of Beijing Hospital.Informed consent
This study was approved for the exemption from informed consent.Author contributions
All authors have accepted responsibility for the entire content of this manuscript and approved its submission.Conflicts of interest
The authors state no conflict of interest.Abbreviations
| PRA | Plasma renin activity |
| PA | Primary aldosteronism |
| CLIA | Chemiluminescence immunoassay |
| LC-MS/MS | Liquid chromatography–tandem mass spectrometry |
| CLSI | Clinical Laboratory Standard Institution |
| CV | Coefficient variations |
| Ang1 | Angiotensin 1 |
| RIA | Radioimmunoassay |
| EQA | External Quality Assessment |
| NCCL | National Center for Clinical Laboratories |
| PMSF | Phenylmethanesulfonyl fluoride |
| SBTI | Soybean trypsin inhibitor |
| BSA | Bovine serum albumin |
| EDTA | Ethylenedinitrilotetraacetic acid |
| IS | Internal standard |
| CUR | Curtain gas |
| GS1 | Nebulizer gas |
| GS2 | Auxiliary gas |
| CAD | Collision gas |
| CRMs | Certified reference materials |
| ANOVA | One-way analysis of variance |
| JCTLM | Joint Committee for Traceability in Laboratory Medicine |
| LOQ | Limit of quantification |
| LOD | Limit of detection |
| S/N | Signal-to-noise ratio |
| TCL | Total change limit |
| LDTs | Laboratory-developed tests |
| RT | Room temperature |
Acknowledgements
This study was supported by grants from the Beijing Natural Science Foundation (grant no: 7212087).References
- J. W. Funder, Vitam. Horm., 2019, 109, 285–302 CrossRef CAS PubMed.
- D. A. Calhoun, Annu. Rev. Med., 2013, 64, 233–247 CrossRef CAS PubMed.
- P. Mulatero, M. Stowasser, K. C. Loh, C. E. Fardella, R. D. Gordon and L. Mosso, et al. , J. Clin. Endocrinol. Metab., 2004, 89, 1045–1050 CrossRef CAS PubMed.
- J. W. Funder, R. M. Carey, F. Mantero, M. H. Murad, M. Reincke and H. Shibata, et al. , J. Clin. Endocrinol. Metab., 2016, 101, 1889–1916 CrossRef CAS PubMed.
- K. Schilbach, R. K. Junnila and M. Bidlingmaier, Exp. Clin. Endocrinol. Diabetes, 2019, 127, 84–92 CrossRef CAS PubMed.
- Z. Locsei, K. Racz, A. Patocs, G. L. Kovacs and E. Toldy, Clin. Chim. Acta, 2009, 402, 203–205 CrossRef CAS PubMed.
- L. A. P. Vilela and M. Q. Almeida, Arch. Endocrinol. Metab., 2017, 61, 305–312 CrossRef PubMed.
- A. Morganti, J. Hypertens., 2010, 28, 1307–1312 CrossRef CAS PubMed.
- S. Carter, L. J. Owen, M. N. Kerstens, R. P. Dullaart and B. G. Keevil, Ann. Clin. Biochem., 2012, 49, 570–579 CrossRef CAS PubMed.
- C. E. Bystrom, W. Salameh, R. Reitz and N. J. Clarke, Clin. Chem., 2010, 56, 1561–1569 CrossRef CAS PubMed.
- N. Stoppacher, R. D. Josephs, A. Daireaux, T. Choteau, S. Westwood and R. I. Wielgosz, Rapid Commun. Mass Spectrom., 2015, 29, 1651–1660 CrossRef CAS PubMed.
- J. G. Van Der Gugten and D. T. Holmes, Methods Mol. Biol., 2016, 1378, 243–253 CrossRef CAS PubMed.
- J. E. Sealey, R. D. Gordon and F. Mantero, Trends Endocrinol. Metab., 2005, 16, 86–91 CrossRef CAS PubMed.
- D. J. Campbell, J. Nussberger, M. Stowasser, A. H. Danser, A. Morganti and E. Frandsen, et al. , Clin. Chem., 2009, 55, 867–877 CrossRef CAS PubMed.
- D. L. Chappell, T. McAvoy, B. Weiss, R. Weiner and O. F. Laterza, Bioanalysis, 2012, 4, 2843–2850 CrossRef CAS PubMed.
- Q. Zhang, L. Zhang, H. Lin, Z. Cai, J. Yan and Q. Wang, et al. , Anal. Bioanal. Chem., 2019, 411, 7095–7104 CrossRef CAS PubMed.
- F. Chen, Z. Cheng, Y. Peng, Z. Wang, C. Huang and D. Liu, et al. , J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2021, 1179, 122740 CrossRef CAS PubMed.
- CLSI, C-62A: Liquid chromatography-mass spectrometry methods; approved guideline, Clinical and Laboratory Standards Institute, Wayne, PA, 2014 Search PubMed.
- CLSI, EP07-A2: Interference testing in clinical chemistry; approved guideline, Clinical and Laboratory Standards Institute, Wayne, PA, 2nd edn, 2005 Search PubMed.
- CLSI, EP15-A3: User verification of precision and estimation of bias, approved guideline – 3rd edn, Clinical and Laboratory Standards Institute, Wayne, PA, 2014 Search PubMed.
- J. H. Livesey, M. J. Ellis and M. J. Evans, Clin. Biochem. Rev., 2008, 29, S11–S15 Search PubMed.
- M. Heins, W. Heil and W. Withold, Eur. J. Clin. Chem. Clin. Biochem., 1995, 33, 231–238 CAS.
- C. Oddoze, E. Lombard and H. Portugal, Clin. Biochem., 2012, 45, 464–469 CrossRef CAS PubMed.
- A. J. Chakera, T. J. McDonald, B. A. Knight, B. Vaidya and A. G. Jones, Clin. Med., 2017, 17, 18–21 CrossRef PubMed.
- Z. Liu, Q. Liu, Y. Deng, H. Zhao, J. Zeng and T. Zhang, et al. , Anal. Bioanal. Chem., 2021, 413, 7509–7520 CrossRef CAS PubMed.
- A. G. Camenzind, J. G. van der Gugten, R. Popp, D. T. Holmes and C. H. Borchers, Clin. Proteomics, 2013, 10, 20 CrossRef PubMed.
- J. E. Sealey and J. H. Laragh, Semin. Nucl. Med., 1975, 5, 189–202 CrossRef CAS PubMed.
- V. F. Fredline, E. M. Kovacs, P. J. Taylor and A. G. Johnson, Clin. Chem., 1999, 45, 659–664 CrossRef CAS.
- A. C. Rutledge, A. Johnston, D. Bailey, R. A. Booth, P. Edmond and V. Leung, et al. , Pract. Lab. Med., 2021, 25, e00229 CrossRef CAS PubMed.
- J. J. Hillebrand, A. C. Heijboer and E. Endert, Ann. Clin. Biochem., 2017, 54, 289–292 CrossRef CAS PubMed.
- S. Hepburn, C. Munday, K. Taylor and D. J. Halsall, Clin. Chem. Lab. Med., 2022, 60, 1384–1392 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental Table 1: The analytical recovery experiment of PRA plasma spiked with Ang1 working solution. See DOI: https://doi.org/10.1039/d2ay01646j |
| This journal is © The Royal Society of Chemistry 2023 |