
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonitoring of n-hexane degradation in a plasma reactor by chemical ionization mass spectrometry†
Perla
Trad
 *ab,
Nicole
Blin-Simiand
a,
Pascal
Jeanney
a,
Stéphane
Pasquiers
a,
Joel
Lemaire
c,
Essyllt
Louarn
c,
Hélène
Mestdagh
c and
Michel
Heninger
c
*ab,
Nicole
Blin-Simiand
a,
Pascal
Jeanney
a,
Stéphane
Pasquiers
a,
Joel
Lemaire
c,
Essyllt
Louarn
c,
Hélène
Mestdagh
c and
Michel
Heninger
c
aUniversité Paris-Saclay, CNRS, Laboratoire de Physique des Gaz et des Plasmas, 91405 Orsay, France. E-mail: perla.trad@universite-paris-saclay.fr
bInstitut National de Recherche et de Sécurité, Rue du Morvan, CS60027, 54519 Vandæuvre Cedex, France
cUniversité Paris-Saclay, CNRS, Institut de Chimie Physique, 91405 Orsay, France
First published on 26th October 2023
Abstract
n-Hexane (C6H14) removal and conversion are investigated in a filamentary plasma generated by a pulsed high-voltage Dielectric Barrier Discharge (DBD) at atmospheric pressure and room temperature in a dry N2/O2 (20%) mixture with C6H14. The degradation of n-hexane and the by-product formation are analyzed in real-time using a high-resolution Fourier Transform Ion Cyclotron Resonance (FT-ICR) mass spectrometer coupled with Chemical Ionization (CI). As alkanes are reacting slowly with H3O+ ions, two precursor ions were used: O2+ to follow the n-hexane mixing ratios and H3O+ to follow the mixing ratios of organic by-products. As the CI-FTICR technique can work at high mixing ratios, studies were made between 5 and 200 ppm of n-hexane. Absorption spectroscopy is also used to follow ozone and carbon dioxide molecules. We show that the DBD efficiency increases for lower n-hexane mixing ratios and a large number of by-products are identified, with the major compounds being: formaldehyde, acetaldehyde, propanal, carbon dioxide, and carbon monoxide along with nitrate compounds. Based on the nature of the by-products characterized, a mechanism accounting for their formation is proposed.
1 Introduction
Volatile organic compounds (VOCs) group together a multitude of substances that can be of natural or anthropogenic origin. They are used in industrial processes, including around twenty activity sectors. They have proven toxicity for humans, thus justifying a rigorous classification. An overview of occupational exposures to VOCs in France was published in 2011![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 and is regularly updated.2 This summary work consists in identifying the VOCs most frequently measured between 2003 and 2010 that are subject to an occupational exposure limit value (VLEP). Among these substances, there are nine proven or suspected CMR (Carcinogenic, Mutagenic, and Reprotoxic) agents according to the European classification and the International Center for Cancer Research (IARC). The agents listed are toluene, styrene, formaldehyde, ethylbenzene, dichloromethane, n-hexane, tetrachloroethylene, trichloroethylene, and benzene.
1 and is regularly updated.2 This summary work consists in identifying the VOCs most frequently measured between 2003 and 2010 that are subject to an occupational exposure limit value (VLEP). Among these substances, there are nine proven or suspected CMR (Carcinogenic, Mutagenic, and Reprotoxic) agents according to the European classification and the International Center for Cancer Research (IARC). The agents listed are toluene, styrene, formaldehyde, ethylbenzene, dichloromethane, n-hexane, tetrachloroethylene, trichloroethylene, and benzene.
Given the high volatility of most VOCs, workstations using VOCs are often sources of operator exposure. This can lead, if necessary, to the implementation of solutions that limit the risk of exposure or even eliminate it. While activated carbon adsorption systems are still prevalent methods to trap VOCs, air purification devices using advanced oxidation techniques have existed for several years in different sectors of activity.
Non-thermal plasmas are used in such oxidation techniques.3,4 A non-thermal plasma is not in thermodynamic equilibrium since the electron temperature is much higher than the temperature of heavier species, which may be close to room temperature. High-velocity electrons thus react on stable molecules in the gas. This entails the formation of reactive species like atomic oxygen O, hydroxyl radical OH, and metastable nitrogen species N2*. In the presence of organic molecules in the gaseous mixture, removal processes initiated by these reactive species lead to the total or partial oxidation of the polluting compounds. Since the beginning of the 1990s, a great amount of work has been performed on the conversion of VOCs by non-thermal plasmas produced in mixtures of atmospheric gases N2/O2/H2O.
Several studies have been published concerning VOCs removal by these plasmas or their coupling with different types of catalysts, they concern various types of molecules: hydrocarbons,5,6 carbonyl compounds,7,8 and alcohols.9 Multiple ways of producing plasma with pulsed electrical discharge have been reported,10–19 such as Dielectric Barrier Discharge (DBD) and packed-bed plasma reactors.
The pollutant investigated in this study, n-hexane, a highly toxic VOC, is found in different types of industries where it is heavily used as an industrial solvent. Due to n-hexane's long carbon chain, its treatment generates numerous by-products from different chemical families, whose formation mechanism is not yet well established.
Studies concerning products issued from controlled oxidation of n-hexane were published for temperatures between 450–1000 K at 1 atm.20,21 Experiments in a jet-stirred reactor in the temperature range of 530–1160 K at 10 atm allow for the identification of species and measure their mixing ratio as functions of temperature. The results were used to validate a chemical kinetic model for the n-hexane oxidation mechanism.22
For the studies concerning n-hexane degradation with different plasma reactors,23–27 none of them uses PTR-MS instruments (Proton Transfer Reaction Mass Spectrometry) for gas analysis. PTR-MS use proton transfer from H3O+ ions to ionize specifically the VOCs present which are then detected by mass spectrometry. It is implemented with different kinds of mass spectrometers such as quadrupole,28 time of flight29 or FTICR.30 It is a very sensitive technique for the real time analysis of VOCs and has applications in many areas: atmospheric research, food analysis, heath monitoring.31,32
Only in a few cases PTR-MS instruments have been applied to the determination of plasma degradation products: results are available for acetone,8,33 toluene,3 2-heptanone,34 ethyl acetate.35 The precursor ions used in all these studies are H3O+ ions, since they are quite efficient in detecting the target VOCs and most of their degradation products, and only in the case of acetone, O2+ ions were also used to detect alkane degradation products. In the case of n-hexane a difficulty is that H3O+ ions do not react or react very slowly with n-hexane. In general the study of alkanes by mass spectrometry coupled to chemical ionization in PTR-MS instruments is difficult since alkanes do not give stable protonation products with the H3O+ ions, and the rate coefficients are not known.
To solve this problem, we have used two different ionic precursors in the CI-FTICR analyzer: O2+ for the detection of n-hexane (and some of the by-products) and H3O+ for the detection of most degradation products. High mixing ratio such as the one used for n-hexane are often a problem PTR-MS instruments: those based on chemical ionization in a drift tube operated at pressures close to 1 mbar are very sensitive and able to detect VOCs with mixing ratio <1 ppt but in order to deal with high mixing ratio a dilution in air in necessary. This is not the case with CI-FTICR instrument in which the reaction time can be adjusted resulting in a range of accessible mixing ratio from ∼10 ppb up to a few %.
Therefore studying n-hexane plasma degradation by CI-FTICR is interesting for trace analysis in a complex mixture, allowing (i) to validate the consistency of the different analytical methods used, (ii) to understand the physicochemical processes involved.
In this study, we present the removal of n-hexane by a non-thermal plasma reactor, in order to evaluate its efficiency, while analyzing the treated effluents to quantify the by-products that can be issued from the degradation of the molecule.
2 Materials and methods
2.1 Plasma discharge reactor
A scheme of the experimental set-up, similar to that already described for the treatment of toluene,3 is given in Fig. 1. The plasma reactor consists of an alumina tube (internal diameter 10 mm, thickness 2.5 mm, permittivity εr = 9.05) functioning as a dielectric barrier. On its exterior is an electrically grounded copper tape (5 cm length). At the center of the tube, a tungsten rod, with a diameter of 1 mm, is connected to a high-voltage power supply. These elements make it possible to generate a DBD in a cylindrical coaxial geometry with a reactor volume of 3.9 cm3. The plasma is created at atmospheric pressure and at room temperature in dry synthetic air (20% oxygen). The n-hexane-containing gas mixture was prepared and introduced into the reactor using a GasMix™ Zephyr (AlyTech, Juvisy, France) gas diluter from a calibrated air/n-hexane standard mixture (500 ppm, Crystal mixture Air Liquide) and dry synthetic air, for a total volumetric flow rate of 0.5 L min−1 NTP. The resulting mixing ratio of n-hexane in the gas before treatment varies from 5 ppm to 200 ppm. | ||
| Fig. 1 Schematic drawing of the experimental set-up. | ||
2.2 Electrical characterization
For most of the results discussed in this paper, the DBD was driven by HV pulses at a repetition frequency, f = 500 Hz. At this repetition frequency and flow rate, the gas will be treated with about 234 HV-pulses before exiting the reactor. The applied voltage (peak value measured on a purely capacitive load, without plasma) was chosen in the range between 17 kV and 24 kV.The electrical energy deposited in the plasma during each HV-pulse, Epulse, was determined through measurements of the voltage, V(t), and current, I(t), and integration of the instantaneous electrical power over the duration of one pulse:
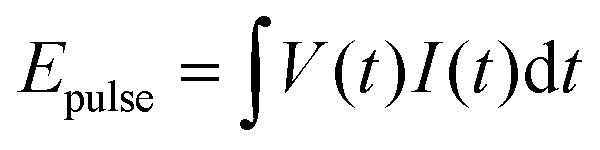 | (1) |
This method allowed real-time energy monitoring; for each experiment, Epulse was systematically followed throughout the discharge operation. Epm, the mean energy values were calculated over several thousand pulses. Current and voltage waveforms were measured by means of electrical probes (Lecroy PPE, TM Research Products SBNC-5-5) connected to a fast digital oscilloscope (Lecroy 204 MXi-A, 2.0 GHz, 10 GS s−1, New York US).
The energy deposited into the plasma per unit of volume, i.e. the specific energy, SED is defined by the following:
 | (2) |
Plotted in Fig. 2 are examples of current and voltage temporal evolutions measured for the gas mixture containing 100 ppm of n-hexane, and other parameters indicated in the figure caption.
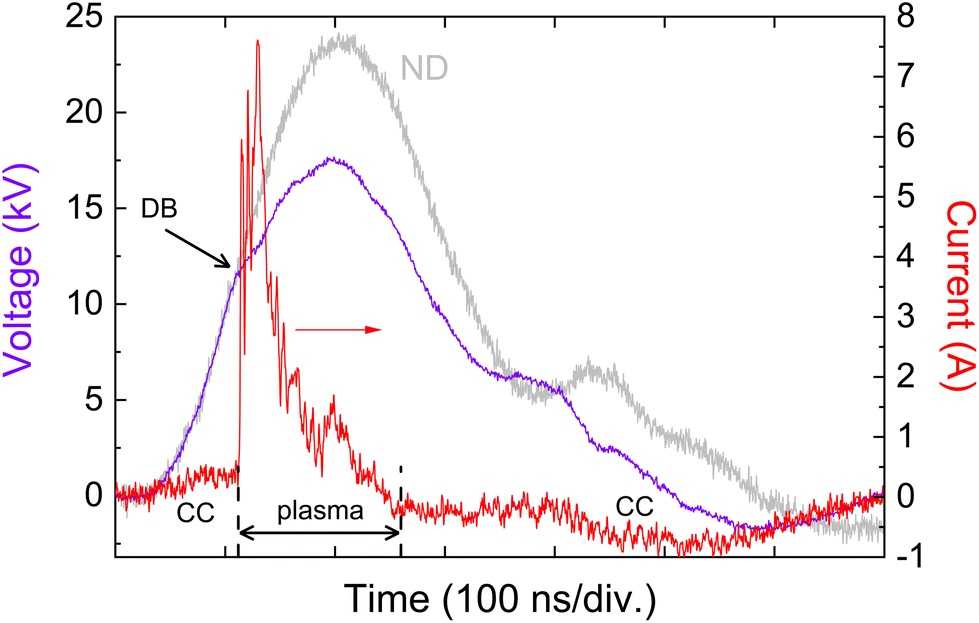 | ||
| Fig. 2 Typical temporal evolutions of current and voltage. HV-pulse repetition frequency: 500 Hz, applied voltage: 23.6 kV (ND: no discharge, voltage plot in grey line), gas flow rate: 0.5 L mn−1 NTP, n-hexane mixing ratio: 100 ppm. DB: discharge breakdown, CC: capacitive current. | ||
2.3. Analytical methods
It allows absolute quantification of the compounds identified based on the relative intensities of the ions and the CI reaction rate coefficient. The high mass resolution allows good identification of molecular formulas, making it possible to separate quasi-isobaric compounds and follow their mixing ratio during the experiment.
Chemical ionization is based on the use of a selective ion-molecule reaction to ionize and detect the compounds of interest. Different precursor ions may be used in positive mode such as H3O+,37 CF3+,38 C6H4F2–H+39 or negative mode, such as HO−, O−.40 For this study two precursor ions were used, O2+ and H3O+. With H3O+ precursor the main CI reaction is protonation of the analyte A leading to AH+ ion, provided its proton affinity PA(A) is higher than PA(H2O) = 691 kJ mol−1. Quite differently, O2+ reacts by initial charge transfer giving A+, generally followed by fragmentation. The energy of the trapped ions remains very low, it is acquired solely from the exothermicity of the CI reaction, which may lead to the fragmentation of the ions produced. This charge transfer is fast with most VOCs.
Thus, H3O+ is well suited for the detection of n-hexane oxidation products in the discharge effluents since these products are oxygenated or unsaturated molecules with relatively high PAs. Towards alkanes such as n-hexane, H3O+ has a low reactivity since PA(n-hexane) = 676 kJ mol−1 < PA(H2O);41 H3O+ is not appropriate for n-hexane quantification.
Therefore, the O2+ ion was used to measure the degradation rates of n-hexane as a function of the deposited energy and initial n-hexane mixing ratio, while H3O+ allowed analysis of its partial oxidation products in the plasma. The reaction of O2+ ion with n-hexane is known to be fast with many fragmentation pathways. A slow reaction between the H3O+ ion and n-hexane was observed. Accordingly, we have started this study by re-measuring the rate coefficients and branching ratios of the reactions of O2+ and H3O+ with n-hexane in our experimental conditions.
In the case of a complex mixture such as the DBD effluents under study, interpretation of the O2+ results is made difficult by the large numbers of fragmentation pathways. Moreover, the rate coefficients and branching ratios of the reactions of O2+ are not well known for all VOCs of interest. For these different reasons, the O2+ ion will be mainly used to quantify n-hexane and products that cannot be detected with the H3O+ ion.
In consequence, n-hexane mixing ratio is measured by CI-FTICR-MS using O2+ as precursor ion for the mixing ratios in the range of (5 ppm–200 ppm) and by Micro GC for mixing ratios in the range of (50 ppm–150 ppm).
VOCs by-product formation is followed by CI-FTICR-MS using H3O+ as precursor ion. The mixing ratios are calculated using the experimental rate coefficients, when available, or using the capture rate coefficient calculated from Su and Chesnavich formula.42 NOx and C2H4 are monitored using O2+ precursor ion.
If rate coefficients are known, absolute quantification of detected molecules is possible.
The mixing ratio of the molecules detected with the H3O+ precursor in the mass spectrometer30 when no fragmentation occurs on the AH+ ion, is calculated with the following equation:
 | (3) |
Firstly, the discharge is started in synthetic air at a given SED. At this step, the chemical noise is measured, i.e. the by-products that could be produced by residual pollution in the tubes, this corresponds to measurements with 0 ppm of n-hexane. The chemical noise value is then subtracted from any result obtained with n-hexane to eliminate the effect of pollution in our system.
Secondly, n-hexane is introduced into the discharge at a certain mixing ratio, and the generated by-products are measured.
These two steps are repeated for several mixing ratios of n-hexane while keeping the discharge running and all operating conditions constant.
Lastly, the interior walls of the tubes are cleaned, by running a discharge in air.
3 Results and discussion
3.1 Rate coefficient measurements
To quantify the n-hexane mixing ratio, the rate coefficient of the reaction of O2+ with n-hexane was measured in our experimental conditions. The reaction of H3O+ with n-hexane, which is known to be very slow, was also studied. The rate coefficients were measured in the absence of discharge and under chemical ionization conditions using the gas cylinder of 500 ppm n-hexane in an air matrix and diluting it with synthetic air in the 5–200 ppm range.The main reaction channels are the formation of the fragments ions C4H9+(6) and C4H8+(7), then charge transfer to C6H14+(4). These results are similar to those reported by Francis et al.44 given in parentheses for a CI pressure of 0.25 Torr. Since the CI reaction occurs at lower pressure (ca. 10–5 Torr) in our instrument, we expect to get a larger fragmentation extent. The agreement with Francis's results is thus quite good. The somewhat lower fragmentation in the flow tube experiment could be explained by some collisional relaxation at the much higher pressure (0.25 Torr) used.
| O2+ + C6H14 → C6H14+ + O2 11% (20%) | (4) |
| →C5H11+ + O2 + CH3 3% (5%) | (5) |
| →C4H9+ + O2 + C2H5 37% (35%) | (6) |
| →C4H8+ + O2 + C2H6 32% (25%) | (7) |
| →C3H7+ + O2 + C3H7 8% (5%) | (8) |
| →C3H6+ + O2 + C3H8 8% (10%) | (9) |
Three minor channels with less than 1% were also observed: C4H7+, C6H13+, C5H1O+.
The total rate coefficient measured is 1.9 × 10−9 molecule−1 cm3 s−1 in good agreement with previous experimental values: 1.7–1.9 × 10−9 molecule−1 cm3 s−1 (ref. 44) or with the capture rate coefficient, 1.8 × 10−9 molecule−1 cm3 s−1.42
We observe a linear evolution of the products as a function of injected n-hexane with correlation coefficients of 0.99 for all products.
| H3O+ + C6H14 → C3H7+ + H2O + C3H8 24% | (10) |
| →C4H9+ + H2O + C2H6 44% | (11) |
| →C5H11+ + H2O + CH4 3% | (12) |
| →C6H13+ + H2O + H2 29% | (13) |
 | ||
| Fig. 3 Intensities of the products issued from H3O+ reaction with n-hexane as a function of n-hexane mixing ratio. | ||
Arnold et al.45 studied the reactivity of H3O+ ion with C2–C12 alkanes. For n-hexane they measured an overall rate coefficient at 300 K of 3 × 10−11 molecule−1 cm3 s−1 with two main products: C6H14H3O+ (association product 63%) and C6H13+ ion (20%) and the formation of minor products C4H9+ (4%), C4H9OH2+ (3%) and C3H7OH2+ (10%).
Proton transfer reaction of H3O+ ions with n-hexane is endothermic by 15 kJ mol−1 and is expected to be very slow. The pressure conditions in our experiment are also much lower than 10−5 Torr compared to those used in SIFT techniques (∼0.45 Torr) therefore the association product cannot be observed.
The value of the rate coefficient measured here is inferior to the value measured by Arnold as the association reaction is not possible.
3.2 n-Hexane degradation
The mixing ratios of n-hexane are calculated from the measurement of the C6H14+ ion intensities using the rate coefficient and the branching ratio measured in this study. Fig. 4 shows the evolution of the total removal efficiency, defined as the ratio of removed mixing ratio to injected mixing ratio (reactor inlet), as a function of the mixing ratio of n-hexane injected.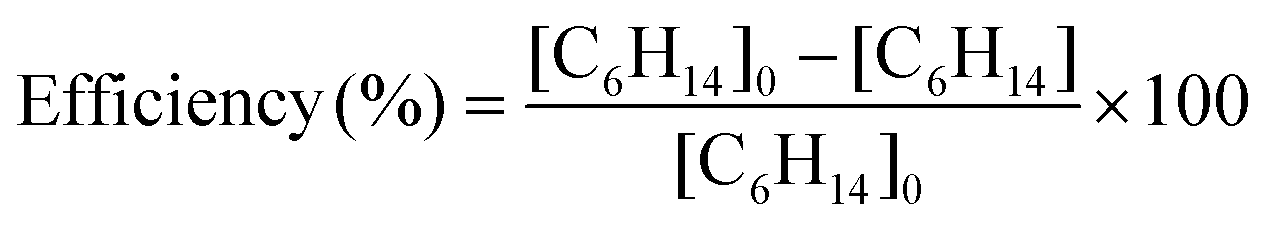 | (14) |
 | ||
| Fig. 4 Efficiency of the degradation of n-hexane as a function of the mixing ratio of n-hexane injected (SED = 265 J L−1, applied voltage: 24.3 kV, HV-pulse repetition frequency: 500 Hz, gas flow rate = 0.5 L min−1). | ||
The degradation is more efficient for the low injected mixing ratios. This result is in good agreement with previous studies concerning plasma treatment in air of various molecules.46,47 The results obtained with the two techniques are in good agreement in the (50 ppm–200 ppm) range as shown in Fig. 4.
3.3 By-products
The treatment of n-hexane by the plasma is initiated by an oxidation reaction with atomic oxygen and hydroxyl radical, leading to radical formation on one of the carbon atoms of n-hexane. This radical rapidly reacts in different ways with the components of the discharge mixture. This leads to the production of several VOCs alongside CO, H2O, CO2, and H2. Ozone is also generated in atmospheric plasmas; however, it is not an oxidation by-product (ESI: Fig. 4†). | ||
| Fig. 5 Measurement of CO2 mixing ratio (black) and CO mixing ratio (red) at the exit of the DBD for mixing ratios of n-hexane in air up to 200 ppm (SED: 225 J L−1, applied voltage: 23.4 kV, HV-pulse repetition frequency: 500 Hz, gas flow rate: 0.5 L min−1). Measurements are obtained using different detectors: CO by gas chromatography and CO2 by IR detector. | ||
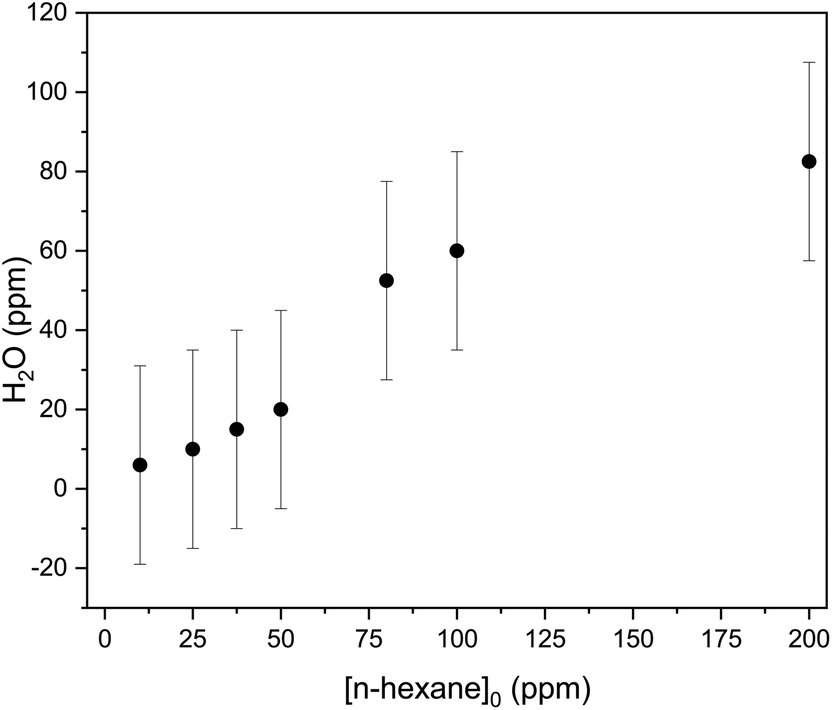 | ||
| Fig. 6 Measurement of H2O mixing ratio at the exit of the DBD for mixing ratios of n-hexane in air up to 200 ppm (SED: 225 J L−1, applied voltage: 23.4 kV, HV-pulse repetition frequency: 500 Hz, gas flow rate: 0.5 L min−1). | ||
As shown in Fig. 6 H2O mixing ratio steadily increases from [n-hexane] = 10 ppm to [n-hexane] = 200 ppm.
The experimental results are to be compared with the maximal possible CO2 mixing ratio, i.e. the value [CO2]max that would be obtained if all of the n-hexane consumed in the reactor was totally converted to CO2 and H2O. [CO2]max is equal to 6 times the mixing ratio of n-hexane degraded in the plasma reactor. For a 50 ppm injected n-hexane mixing ratio, the mixing ratio of removed n-hexane is known from the data reported in Fig. 4 and is equal to 25 ppm. Therefore [CO2]max is equal to 150 ppm. This value is higher than the CO2 mixing ratio measured experimentally at the discharge exit, with [CO2]exp = 29 ppm, as shown in Fig. 5. Taking into account the chemical noise measured in the absence of n-hexane, we obtain: [CO2]exp = 16% [CO2]max. Only 16% of the carbon in the treated n-hexane was converted into CO2, this indicates that the remaining carbon was converted into intermediate by-products.
| [n-hexane]0 (ppm) | 40 | 50 | 80 | 100 | 200 |
| H2 (ppm) | 0.71 | 1.16 | 1.61 | 1.88 | 4.48 |
 | ||
| Fig. 7 Mass spectra obtained for the O2+ precursor ion for an n-hexane mixing ratio of 100 ppm, with C6H4F2+ being used as an internal mass calibrant (m/z 114.028). | ||
 | ||
| Fig. 8 Mass spectra obtained for the H3O+ precursor ion for an n-hexane mixing ratio of 100 ppm, with C6H4F2+ being used as an internal mass calibrant (m/z 114.028). | ||
With O2+ precursor ion, the main ions detected are those of the reaction of the O2+ ion with n-hexane: C4H8+, C4H9+, C6H14+ (since this reaction is exothermic). O2+ is mainly used to measure the evolution of the n-hexane mixing ratio with the discharge conditions. Small organic compounds not reacting with H3O+ are also detected like ethylene, NO and NO2.
As H3O+ precursor ion reacts very slowly with n-hexane, the main ions detected are the by-products of the discharge. H2 is a by-product that is not detected on the BTrap, but is detected with gas-chromatography at very low concentrations.
• Aldehydes: The major compound detected is formaldehyde. The chemical noise for the formaldehyde is quite high, about 50 ppm, probably due to polymerization leading to solid formaldehyde polymer dust deposited in the connection tubings. Acetaldehydeis the second most abundant by-product. Propanal or acetone, that cannot be distinguished, is also detected. Hexanal and/or hexanones are detected in mixing ratios lower than 500 ppb for a mixing ratio of n-hexane of 50 ppm injected in the reactor.
• Small oxygenated compounds: methanol, ketene, acetic acid, hexanal, or hexanones are detected in mixing ratios lower than 500 ppb for the same injected hexane mixing ratio.
• Alkenes: propene, hexene and hexadiene C6H1O are detected. The corresponding detected ions (protonated alkenes) may also be due to propanol, hexanol, or hexenol via dehydration accompanying alcohol protonation by H3O+.
• Nitrite and nitrate compounds: nitrous acid, nitromethane, nitric acid, methyl nitrate, ethyl nitrate. Peroxyacetyl nitrate (PAN, C2H3NO5 leading to m/z 122.009 ion) was also measured. In addition to these nitrogen-containing compounds, a small quantity of hydrogen cyanide is detected. The generation of HCN under our experimental conditions cannot be explained by oxidation but rather reactions between n-hexane and nitrogen metastable states like in the case of H2.48
Fig. 9 shows the variation of the mixing ratio of the main by-products detected with H3O+ precursor for different n-hexane mixing ratios injected in the reactor. The by-products show a quite linear evolution as a function of n-hexane mixing ratio.
 | ||
| Fig. 9 Mixing ratios of the main by-products detected as a function of the mixing ratio of n-hexane injected. | ||
The main by-products: formaldehyde, acetaldehyde, and propanal were also detected with the O2+ precursor ion, but they are more difficult to quantify because of the numerous fragmentations. More interestingly, ethylene which is not detectable by H3O+, was detected at low mixing ratios: 0.76 ppm for 50 ppm and 1.65 ppm for 100 ppm of n-hexane injected, respectively.
Since only a small proportion of carbon from consumed n-hexane appears as CO2 after degradation and we have measured the mixing ratios of numerous carbon-containing by-products, it is interesting to estimate the global “elemental carbon mixing ratio” (C)exp in the measured VOC by-products. (C)exp is expressed in ppmC as:
 | (15) |
According to the data reported in Fig. 9, we find for a 50 ppm injected n-hexane mixing ratio: (C)exp = 43.7 ppmC. This value is about twice (CO2)exp, and 27.5% of (CO2)max.
According to gas chromatography measurements in the same experimental conditions, for a 50 ppm injected n-hexane mixing ratio, we find a mixing ratio of CO equal to 40 ppm.
Finally, the summed experimental mixing ratios of CO2, CO, and carbon in VOC products account for ca. 108 ppmC, representing 66–67% of the degraded n-hexane carbon. The undetected carbon may be in the form of soot.49
| C6H14 + O → C6H13˙ + OH˙ |
| C6H14 + OH˙ → C6H13˙ + H2O |
The molecule is symmetrical, three alkyl radicals are therefore possible depending on the site of H abstraction: C1, C2, or C3. A scheme for the first reaction steps is given in Fig. 10. Each alkyl radical issued from these initiation reactions can react in different ways: oxidation, C–C cleavage, H abstraction or NO2 addition, NO2 being produced in plasma of N2/O2 mixture.
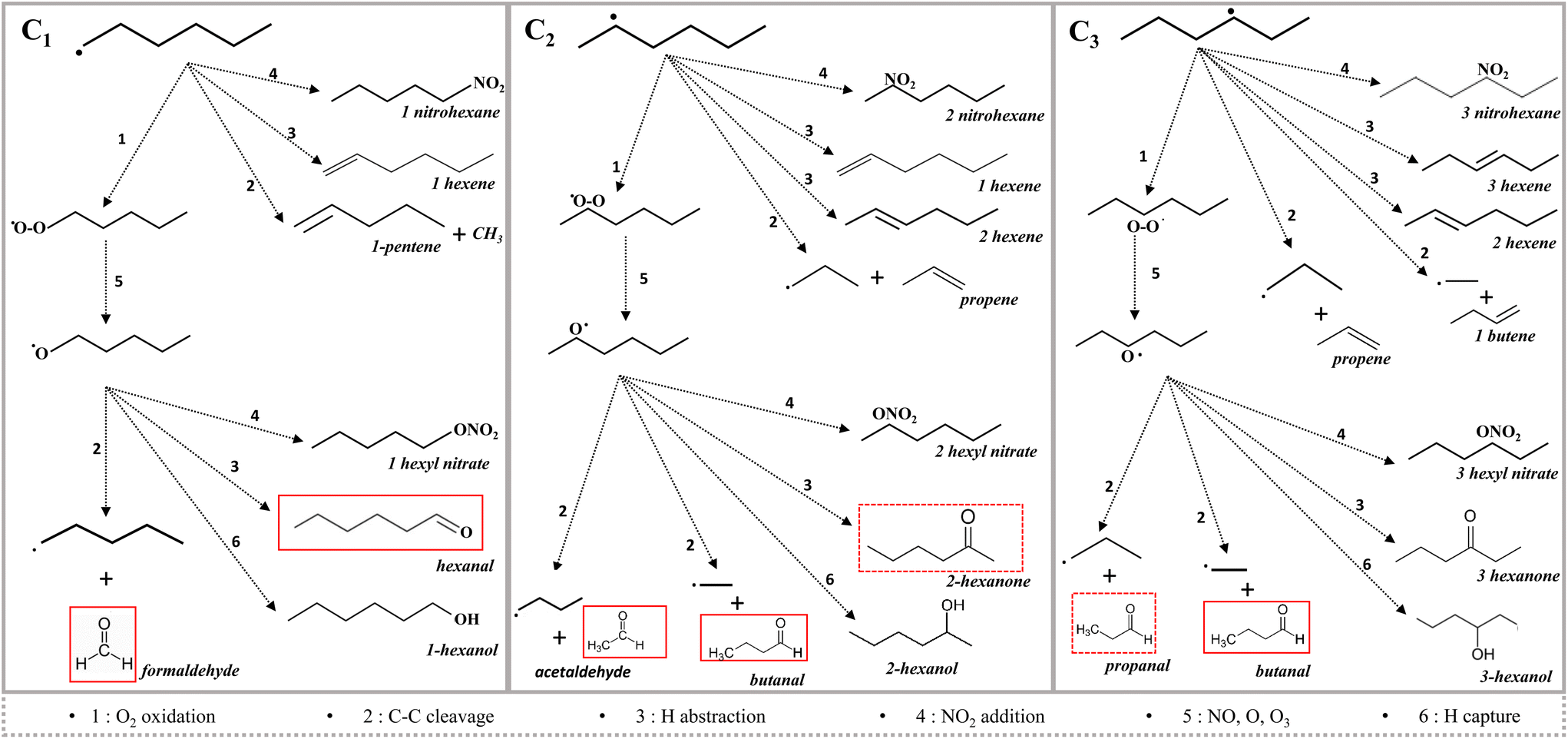 | ||
| Fig. 10 Proposed mechanism for by-product formation that shows the three main reaction channels for the initiation reactions. | ||
The abstraction of a hydrogen atom in α of the different hexyl radicals may lead to the formation of different isomers: 1-, 2- and 3-hexene C6H12. Protonated hexene C6H13+ ion is observed by the BTrap.
C–C cleavage leads to a shorter alkene (1-pentene, propene or 1-butene) and a shorter alkyl radical (methyl, ethyl, 1-propyl). Those alkyl radicals being very reactive, they enter into the same reaction process as that of hexyl, leading to shorter molecules.
The process that best explains the identified by-products is that of oxidation by the addition of O2 on each hexyl radical, leading to three isomers of peroxy radicals C6H13O2. These radicals lead rapidly to alkoxy radicals C6H13O by reactions with NO, O or O3.
Alkoxy radicals, RO, are reactive species, which may lead to carbonyl compounds either by H abstraction on the radical carbon atom or by C–C cleavage in β position yielding an aldehyde and a shorter radical. In this way 1-hexoxy radical leads to formaldehyde, CH2O, and 1-pentyl radical. Two possibilities exist for 2-hexoxy radical, leading to acetaldehyde and 1-butyl radical or butanal with ethyl radical. Similarly, 3-hexoxy radical yields propanal and butanal, along with 1-propyl and1-ethyl radicals respectively. As shown in Fig. 10, the operation of this mechanism on the three hexoxy isomers allows to explain the formation of aldehydes with 1 to 6 carbon atoms, along with C1–C5 radicals which in turn undergo O2 addition and so forth.
Aldehydes are actually the major by-products characterized, with formaldehyde representing ca. 67% of the total by-products. The presence of formaldehyde is consistent with the proposed mechanism since all the C–C cleavage chains all end with a CH3 radical leading to CH3O and CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.
O.
Alkyl or alkoxy radicals can also react by addition with NO2 to give RNO2 and RONO2 respectively. While CH3NO2 is detected in low amount, CH3ONO2 is quite significant and C2H5ONO2 is detected. Larger nitrites or nitrates are not detected, either due to their low mixing ratio or due to fragmentation of the protonated species. Interestingly, peroxyacetyl nitrate (PAN) corresponding to C2H4NO5+ ion, was detected at m/z 122 in a similar amount as methyl nitrate. The formation of PAN can be explained by NO2 association with CH3C(O)O–O radical, resulting from O2 association with CH3CO. The latter radical might be formed by H abstraction from acetaldehyde, in analogy with the atmospheric process of PAN formation.
In the same manner, the formation of alcohols by H from hexoxy or smaller radicals by H abstraction on another species is not observed except for methanol, but protonation of many alcohols by H3O+ is known to be accompanied by dehydration and/or fragmentation. In this case, the resulting ions are identical to those issued from n-hexane reaction with H3O+.
This mechanism does not include the reactivity of nitrogen metastable states, as observed for other molecules,50 that lead to the formation of HCN and H2 in very small amounts.
4 Conclusions
In this study, we present the results obtained for the degradation of n-hexane at low mixing ratios in a DBD reactor.To follow the evolution of the multitude of molecules at the exit of the plasma, different analytical methods were used. For the chemical ionization measurements, two precursor ions were used: O2+ to follow mainly n-hexane and H3O+ to follow organic by-products.
The rate coefficients of the ion precursors with n-hexane were remeasured in our experimental conditions for a better quantification. The association of the two precursors has allowed to quantify both n-hexane (O2+) and most of its degradation by-products (H3O+) in real-time and simultaneously on the same sample. O2+ precursor also allows the detection of additional degradation products such as nitrogen oxides or ethylene. Moreover, comparing the O2+ and H3O+ results supports the consistency of product identification.
A satisfying agreement between Micro GC and mass spectrometry measurements of n-hexane mixing ratios was observed, confirming that the efficiency of the degradation is higher for low mixing ratios injected.
Only a small proportion of carbon from treated n-hexane appears as CO2 after degradation. The remaining consists of carbon monoxide, carbon-containing by-products, and solid carbonaceous particles (soots), which may be deposited in the reactor or driven by the gas flow. Aldehydes are the main by-products detected with small oxygenated compounds, alkenes, nitrite, and nitrate compounds.
Understanding the mechanisms of degradation becomes necessary with the presence of hazardous compounds in the treated effluent, and a simplified mechanism of by-product formation is proposed. The nature and distribution of the main by-products suggest the hypothesis that oxidation beginning with O2 addition on a hydrocarbon radical is its most probable reaction in air atmosphere.
The presence of hazardous compounds such as ozone and nitrogen oxide by-products formed are undesirable in the context of industrial applications for depollution. This can be remedied by the addition of a complementary oxidation technique to the plasma, such as catalysis, which is proven to have high removal efficiency and low by-product formation.
Author contributions
Perla Trad: Conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing; Nicole Blin-Simiand: Conceptualization, funding acquisition, supervision, writing – original draft, writing – review & editing; Pascal Jeanney: Conceptualization, resources, software; Stéphane Pasquiers: Writing – original draft, writing – review & Editing; Joel Lemaire: Writing – review & editing; Essylt Louarn: Writing – review & editing; Hélène Mestdagh: Writing – review & editing; Michel Heninger: Conceptualization, data curation, formal analysis, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Institut National de Recherche et de Sécurité (INRS) [Grant Number 170429], and by Région Ile-de-France (research project n°16016327 Diagplas - Sésame 2016).References
- N. Bertrand and F. Clerc, Hyg. Secur. Trav., 2011, 4, 31–44 Search PubMed.
- A. Emili and G. Mater, Hyg. Secur. Trav., 2018, 1, 72–74 Search PubMed.
- S. Pasquiers, M. Heninger, N. Blin-Simiand, J. Lemaire, G. Bauville, B. Bournonville, E. Louarn, F. Jorand and H. Mestdagh, J. Phys. D: Appl. Phys., 2018, 51, 425201 CrossRef.
- S. Li, X. Dang, X. Yu, G. Abbas, Q. Zhang and L. Cao, Chem. Eng. J., 2020, 388, 124275 CrossRef CAS.
- R. Cimerman, D. Racková and K. Hensel, J. Phys. D: Appl. Phys., 2018, 51, 274003 CrossRef.
- T. Zhu, J. Li, Y.-q. Jin, Y. Liang and G. Ma, Int. J. Environ. Sci. Technol., 2008, 5, 375–384 CrossRef CAS.
- C. Klett, X. Duten, S. Tieng, S. Touchard, P. Jestin, K. Hassouni and A. Vega-González, J. Hazard. Mater., 2014, 279, 356–364 CrossRef CAS PubMed.
- S. Thomas, N. Blin-Simiand, M. Héninger, P. Jeanney, J. Lemaire, L. Magne, H. Mestdagh, S. Pasquiers and E. Louarn, J. Am. Soc. Mass Spectrom., 2020, 31, 1579–1586 CrossRef CAS PubMed.
- T. Sato, M. Kambe and H. Nishiyama, JSME Int. J., Ser. B, 2005, 48, 432–439 CrossRef CAS.
- K. L. L. Vercammen, A. A. Berezin, F. Lox and J.-S. Chang, J. Adv. Oxid. Technol., 1997, 2, 312–329 CAS.
- L. A. Rosocha and R. A. Korzekwa, J. Adv. Oxid. Technol., 1999, 4, 247–264 CAS.
- R. Hackam and H. Aklyama, IEEE Trans. Dielectr. Electr. Insul., 2000, 7, 654–683 CrossRef CAS.
- H.-H. Kim, Plasma Processes Polym., 2004, 1, 91–110 CrossRef CAS.
- J. Van Durme, J. Dewulf, C. Leys and H. Van Langenhove, Appl. Catal., B, 2008, 78, 324–333 CrossRef CAS.
- H. L. Chen, H. M. Lee, S. H. Chen, M. B. Chang, S. J. Yu and S. N. Li, Environ. Sci. Technol., 2009, 43, 2216–2227 CrossRef CAS PubMed.
- A. M. Vandenbroucke, R. Morent, N. De Geyter and C. Leys, J. Hazard. Mater., 2011, 195, 30–54 CrossRef CAS PubMed.
- G. Xiao, W. Xu, R. Wu, M. Ni, C. Du, X. Gao, Z. Luo and K. Cen, Plasma Chem. Plasma Process., 2014, 34, 1033–1065 CrossRef CAS.
- F. Thévenet, L. Sivachandiran, O. Guaitella, C. Barakat and A. Rousseau, J. Phys. D: Appl. Phys., 2014, 47, 224011 CrossRef.
- X. Feng, H. Liu, C. He, Z. Shen and T. Wang, Catal. Sci. Technol., 2018, 8, 936–954 RSC.
- R. Mével, K. Chatelain, P. Boettcher, G. Dayma and J. Shepherd, Fuel, 2014, 126, 282–293 CrossRef.
- Z. Wang, O. Herbinet, Z. Cheng, B. Husson, R. Fournet, F. Qi and F. Battin-Leclerc, J. Phys. Chem. A, 2014, 118, 5573–5594 CrossRef CAS PubMed.
- K. Zhang, C. Banyon, C. Togbé, P. Dagaut, J. Bugler and H. J. Curran, Combust. Flame, 2015, 162, 4194–4207 CrossRef CAS.
- S. Kudryashov, G. Shchegoleva, E. Sirotkina and A. Y. Ryabov, High Energy Chem., 2000, 34, 112–115 CrossRef CAS.
- E. Marotta, A. Callea, X. Ren, M. Rea and C. Paradisi, et al. , Int. J. Plasma Environ., 2007, 1, 39–45 Search PubMed.
- E. Marotta, A. Callea, M. Rea and C. Paradisi, Environ. Sci. Technol., 2007, 41, 5862–5868 CrossRef CAS PubMed.
- E. Marotta, A. Callea, X. Ren, M. Rea and C. Paradisi, Plasma Processes Polym., 2008, 5, 146–154 CrossRef CAS.
- Q. Jin, B. Jiang, J. Han and S. Yao, Chem. Eng. J., 2016, 286, 300–310 CrossRef CAS.
- D. Smith, P. Španěl, N. Demarais, V. S. Langford and M. J. McEwan, Mass Spectrom. Rev., 2023, e21835 CrossRef PubMed.
- F. Piel, M. Müller, K. Winkler, J. Skytte af Sätra and A. Wisthaler, Atmos. Meas. Tech., 2021, 14, 1355–1363 CrossRef CAS.
- J. Lemaire, S. Thomas, A. Lopes, E. Louarn, H. Mestdagh, H. Latappy, J. Leprovost and M. Heninger, Sensors, 2018, 18, 1415 CrossRef PubMed.
- W. Lindinger, A. Hansel and A. Jordan, Int. J. Mass Spectrom. Ion Processes, 1998, 173, 191–241 CrossRef CAS.
- B. Yuan, A. R. Koss, C. Warneke, M. Coggon, K. Sekimoto and J. A. de Gouw, Chem. Rev., 2017, 117, 13187–13229 CrossRef CAS PubMed.
- S. Thomas, N. Blin-Simiand, M. Héninger, P. Jeanney, J. Lemaire, L. Magne, H. Mestdagh, S. Pasquiers and E. Louarn, Phys. Chem. Chem. Phys., 2022, 24, 20553–20564 RSC.
- A. S. Chiper, N. Blin-Simiand, M. Heninger, H. Mestdagh, P. Boissel, F. Jorand, J. Lemaire, J. Leprovost, S. Pasquiers and G. Popa, et al. , J. Phys. Chem. A, 2010, 114, 397–407 CrossRef CAS PubMed.
- T. Guo, G. Cheng, G. Tan, L. Xu, Z. Huang, P. Cheng and Z. Zhou, Chemosphere, 2021, 264, 128430 CrossRef CAS PubMed.
- M. Heninger, H. Mestdagh, E. Louarn, G. Mauclaire, P. Boissel, J. Leprovost, E. Bauchard, S. Thomas and J. Lemaire, Anal. Chem., 2018, 90, 7517–7525 CrossRef CAS PubMed.
- A. M. Ellis and C. A. Mayhew, in Chemical Ionization: Chemistry, Thermodynamics and Kinetics, John Wiley & Sons, Ltd, 2014, ch. 2, pp. 25–48 Search PubMed.
- C. Dehon, J. Lemaire, M. Heninger, A. Chaput and H. Mestdagh, Int. J. Mass Spectrom., 2011, 299, 113–119 CrossRef CAS.
- H. Latappy, J. Lemaire, M. Heninger, E. Louarn, E. Bauchard and H. Mestdagh, Int. J. Mass Spectrom., 2016, 405, 13–23 CrossRef CAS.
- C. Le Vot, J. Lemaire, P. Pernot, M. Heninger, H. Mestdagh and E. Louarn, J. Mass Spectrom., 2018, 53, 336–352 CrossRef CAS PubMed.
- T. Wróblewski, L. Ziemczonek, K. Szerement and G. P. Karwasz, Czech J. Phys., 2006, 56, B1110–B1115 CrossRef.
- T. Su and W. J. Chesnavich, J. Chem. Phys., 1982, 76, 5183–5185 CrossRef CAS.
- W. E. Wallace, NIST chemistry webbook, NIST standard reference database, 2018, p. 20899 Search PubMed.
- G. J. Francis, P. F. Wilson, D. B. Milligan, V. S. Langford and M. J. McEwan, Int. J. Mass Spectrom., 2007, 268, 38–46 CrossRef CAS.
- S. T. Arnold, A. Viggiano and R. A. Morris, J. Phys. Chem. A, 1998, 102, 8881–8887 CrossRef CAS.
- N. Blin-Simiand, S. Pasquiers and L. Magne, J. Phys. D: Appl. Phys., 2016, 49, 195202 CrossRef.
- M. Schiavon, V. Torretta, A. Casazza and M. Ragazzi, Water, Air, Soil Pollut., 2017, 228, 1–20 CrossRef CAS.
- S. Pasquiers, W. Faider, N. Blin-Simiand, L. Magne, P. Jeanney and F. Jorand, Databases, 2012, 4, 19–21 Search PubMed.
- Y.-S. Son, J. Kim, I.-Y. Choi and J.-C. Kim, Environ. Technol., 2021, 42, 2067–2076 CrossRef CAS PubMed.
- S. Pasquiers, N. Blin-Simiand and L. Magne, Plasma Phys. Controlled Fusion, 2013, 55, 124023 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an01617j |
| This journal is © The Royal Society of Chemistry 2023 |