
Multivariate curve Resolution-Alternating least squares coupled with Raman microspectroscopy: new insights into the kinetic response of primary oral squamous carcinoma cells to cisplatin
Valentina
Notarstefano
 *a,
Alessia
Belloni
a,
Paolo
Mariani
a,
Giulia
Orilisi
b,
Giovanna
Orsini
b,
Elisabetta
Giorgini
*a and
Hugh J.
Byrne
c
*a,
Alessia
Belloni
a,
Paolo
Mariani
a,
Giulia
Orilisi
b,
Giovanna
Orsini
b,
Elisabetta
Giorgini
*a and
Hugh J.
Byrne
c
aDepartment of Life and Environmental Sciences, Università Politecnica delle Marche, Via Brecce Bianche, 60131 Ancona, Italy. E-mail: v.notarstefano@univpm.it; e.giorgini@univpm.it
bDepartment of Clinical Sciences and Stomatology, Università Politecnica delle Marche, Via Brecce Bianche, 60126 Ancona, Italy
cFOCAS Research Institute, Technological University Dublin, City Campus, Dublin, Ireland
First published on 2nd August 2023
Abstract
Raman MicroSpectroscopy (RMS) is a powerful label-free tool to probe the effects of drugs at a cellular/subcellular level. It is important, however, to be able to extract relevant biochemical and kinetic spectroscopic signatures of the specific cellular responses. In the present study, a combination of Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS) and Principal Component Analysis (PCA) is used to analyse the RMS data for the example of exposure of primary Oral Squamous Carcinoma Cells (OSCC) to the chemotherapeutic agent cisplatin. Dosing regimens were established by cytotoxicity assays, and the effects of the drug on cellular spectral profiles were monitored from 16 to 72 hours post-exposure using an apoptosis assay, to establish the relative populations of viable (V), early (EA) and late apoptotic/dead (LA/D) cells after the drug treatment. Based on a kinetic model of the progression from V > EA > D, MCR-ALS regression analysis of the RMS responses was able to extract spectral profiles associated with each stage of the cellular responses, enabling a quantitative comparison of the response rates for the respective drug treatments. Moreover, PCA was used to compare the spectral profiles of the viable cells exposed to the drug. Spectral differences were highlighted in the early stages (16 hours exposure), indicative of the initial cellular response to the drug treatment, and also in the late stages (48–72 hours exposure), representing the cell death pathway. The study demonstrates that RMS coupled with multivariate analysis can be used to quantitatively monitor the progression of cellular responses to different drugs, towards future applications for label-free, in vitro, pre-clinical screening.
Introduction
Raman MicroSpectroscopy (RMS) is a vibrational spectroscopy widely and effectively applied in life sciences to examine the biomolecular structure and composition of cells and tissues.1–4 The analysis of the position, intensity, and width of Raman peaks reveals the most relevant biological molecules (such as lipids, proteins, nucleic acids, and sugars) within the investigated samples by their molecular fingerprints. RMS provides very informative data related to biological processes, affording this technique many research applications, including cell and tissue diagnosis5–7 and evaluation of drug toxicity and mode of action.8–11 In particular, as regards the research area of drug development, the possible application of RMS as a pre-clinical screening tool is quite appealing: in contrast to other routine assays, which can only probe one target at a time, RMS provides relevant information on all the most important biomolecules within a single measurement, simultaneously, and in a label-free manner.12 In this sense, thorough analysis of the spectral data can reveal specific modifications in the biomolecular composition of treated cells, which can potentially elucidate new aspects of action of the drug and the cellular response.12–15Cisplatin is a chemotherapeutic drug with well-established clinical applications. Widely employed for the treatment of several solid tumours, such as gastric cancer, Non-Small Cell Lung Cancer (NSCLC), and Oral Squamous Cells Carcinoma (OSCC),16 cisplatin (cisPt) is known to form adducts with the N7 atom of DNA purine nucleobases, subsequently producing inter- and intra-strand DNA cross-linkages.16,17 As a consequence of this binding, DNA becomes misfolded and degraded, which triggers several cellular effects, like the arrest of the cell cycle at the G2/M checkpoint and the initiation of the apoptosis cascade.15,18,19
The effects of cisplatin, as well as the drugs 5-fluorouracil, and 5-azacytidine, on OSCC primary tumoral cells have already been explored using Fourier Transform InfraRed Microspectroscopy (FTIRM).17,20,21 In these studies, analyses of the cell populations were made, as the limited spatial resolution FTIRM makes subcellular analysis more difficult. In the current work, the higher spectral and spatial resolutions of Raman MicroSpectroscopy are employed to specifically target the cell nuclei, and the kinetics of the spectroscopic responses of the cell are analysed to explore in greater detail the biomolecular modifications induced by cisPt in vitro on OSCC primary cells, with the aim of elucidating subtle and specific spectral changes characteristic of the particular aspects of the cellular response. Towards this aim, various multivariate analysis techniques, notably Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS) regression and Principal Components Analysis (PCA), have been employed. MCR-ALS was developed to describe the evolution of mixtures of multiple concentration profiles in chemical analyses.22,23 It is attracting increasing attention for addressing various biological questions, including extracting biochemical and kinetic information of drug uptake24 and time- and drug-dependent cellular processes, in vitro, such that the evolution of distinct, characteristic spectra of constituent components or states of the system can be identified and monitored.25,26 In the present work, MCR-ALS will be employed to deconvolute the Raman spectral profiles acquired from OSCCs at different timepoints, and correlate the kinetic progression of the cell population from viable to early and late apoptotic/dead, with the action of cisPt. PCA, on the other hand, is a multivariate data analysis technique which seeks to reduce the dimensionality of the dataset by describing the maximum variance by a series of principal components of decreasing order. It is often used to identify clustering of subsets of similar spectra within a dataset,27 and in this work is used to further explore the spectral profiles of the cell populations at different kinetic timepoints. The spectroscopic analysis has been guided by a preliminary cytotoxicity screening to establish the appropriate dosing regimens and to monitor the progression from viable to early and then late apoptotic cell populations, after drug exposure.
This study aimed to demonstrate the ability of RMS not only to show the effects of a treatment but also quantitatively monitor the kinetics of the cellular responses, potentially paving the way to the application of this vibrational spectroscopic technique as a pre-clinical screening tool in drug development research. Moreover, applied to primary cells, this approach may lead the way to quantification of the efficacy of the drug directly on patient cells, potentially the basis for applications in Companion Diagnostics.
Experimental
The present study was agreed by the Ethics Committee of Azienda Ospedaliero Universitaria – Ospedali Riuniti Ancona and was executed in full agreement with ethical principles for experimental procedures involving humans, comprising The Code of Ethics of the World Medical Association (Declaration of Helsinki) and signature of necessary informed consent. Cisplatin was kindly provided by Azienda Ospedaliero Universitaria – Ospedali Riuniti Ancona (TEVA, Italy). The drug was diluted to the final concentrations with 0.9% NaCl.Culture of primary OSCC cells
Following a previously validated protocol, primary OSCC cells (hereafter OSCCs) were isolated.17,20,21 Briefly, lingual biopsy samples diagnosed for poorly differentiated OSCC (tumour grade 3, histologically assessed) were collected from 3 patients (males; mean age 63.0 ± 1.1 years). Each sample was carefully fragmented and cultured in a humidified atmosphere (37 °C, 5% CO2), using Dulbecco's modified Eagle's medium (DMEM F-12), supplemented with foetal bovine serum (10% FBS) and 1% penicillin–streptomycin solution. The medium was changed every 24 h; once 90% of confluence was reached, primary OSCCs were trypsinised, counted by a haemocytometer, centrifuged (1200 rpm for 5 min), and pooled together, for analysis.Apoptosis assessment by annexin V/PI staining
The concentration of cisplatin which reduced the viability of the pooled OSCCs by 50% (IC50) was calculated employing the colorimetric MTT (3-(4,5-dimethylthiazol-2yl)-diphenyltetrazolium bromide) assay.17,20 Based on these calculations, cisplatin was used at a concentration of 7 μg mL−1 (IC50 at 24 h).The apoptotic index of OSCCs, after treatments with cisplatin as described in the following section, was assessed by the Tali™ Apoptosis assay kit – Annexin V Alexa Fluor® 488 and propidium iodide (Invitrogen, Milan, Italy). Cells were seeded into 6-well plates (2 × 105 cells per well) in 3 mL DMEM F-12 with 10% FBS and incubated overnight in a humidified atmosphere (37 °C, 5% CO2). Then, cisplatin, at the calculated IC50 concentration (7 μg mL−1), was added, and the cells incubated for 16, 24, 36, 48, and 72 h in a humidified atmosphere (37 °C, 5% CO2). For control groups, additional cell aliquots were cultured with no chemotherapeutic treatment for 16, 24, 36, 48, and 72 h. All the experiments were performed in triplicate. For each experimental time of interest, cell samples were washed with cold PBS (pH 7.4), centrifuged, resuspended in binding buffer (BB; 100 μL), and incubated in 5 μL of Tali™ Annexin V Alexa Fluor® 488 (20 min, room temperature, dark conditions). After an additional cycle of centrifugation (1000 rpm, 5 min), cells were then resuspended and incubated for 5 min, at room temperature, in the dark, in a mixture of BB (100 μL) Tali™ Propidium Iodide (PI) (5 μL), for being finally analysed with flow cytometer (Tali® image-based cytometer). After staining, live cells were characterised by absent or low fluorescence, early apoptotic cells were green fluorescent (positive to Annexin V and negative to PI), and late apoptotic cells were red/green fluorescent (positive to both dyes).17 The fluorescence histograms of viable (V), early apoptotic (EA), and late apoptotic and dead (LA/D) cells were compared with those of control cells, to assess their percentages within the cell populations.
OSCCs in vitro chemotherapy treatments
OSCCs were seeded in 6-well plates (2 × 105 cells per well) in DMEM F-12 with 10% FBS and incubated at 37 °C in a humidified atmosphere (37 °C, 5% CO2). Cells were then: (a) treated with cisplatin (7 μg mL−1) for 16, 24 and 48 hours (cisPt-16, cisPt-24, and cisPt-48 groups) and (b) normally cultured without any chemotherapy drug for 16, 24, 36, 48 and 72 hours (Ctrl-16, Ctrl-24, Ctrl-36, Ctrl-48, Ctrl-72 groups). The choice of the maximum time point selected was based on avoiding maximum confluence and the consequent cell detachment. At each selected time point, the culture medium was collected, OSCCs were harvested using 0.5% trypsin solution with 0.2% EDTA and centrifuged at 1200 rpm for 5 min. To eliminate FBS residues, pellets were washed twice with 100 μL DMEM F-12, centrifuged again at 1200 rpm for 5 min and fixed in 4% paraformaldehyde (PFA) for 15 min; after fixation, cells were washed twice in physiological solution, and then stored in PBS at 4 °C. This procedure has been validated to maintain the biochemical profile of cells as close as possible to that of live cells28 and preserve them from biological damage during storage and shipping.6,29RMS measurements
RMS measurements were performed at the FOCAS Research Institute, Technological University Dublin (Ireland). A Horiba Jobin-Yvon LabRAMHR800 spectrometer was exploited, using a 532 nm diode laser as a source (∼50 mW laser power at the sample). All measurements were performed by using a ×100 objective (Olympus, numerical aperture 1, spot size ∼1 μm). The 520.7 cm−1 line of silicon was employed for calibration prior to spectral acquisition. Measurements were performed by using a 600-lines per mm grating and 100 μm confocal pinhole. A Peltier-cooled CCD detector (16-bit dynamic range) was employed for spectra dispersion. The spectral range from 400 to 1800 cm−1 was selected.For Raman measurement, after in vitro chemotherapy treatments as described in the previous section, cells were deposited onto glass slides and allowed to dry. Just before RMS measurement, cells were washed with physiological solution to remove the phosphate buffer, and spectra were acquired for 3 × 10 seconds at each spot. For each sample, a single point spectrum was acquired from the centre of each of ∼50 individual cells seeded on the glass slide; spectra displayed homogeneous profiles. It is noteworthy that working with detached cells provides Raman spectra predominantly reflecting the molecular composition of cellular nuclei;30–32 in this specific study, the present procedure was justified by the modes of action of the three selected drugs, which mainly act at a nuclear level. The spot size of size ∼1 μm means that only a small portion of the nuclear material is sampled per cell, and the variability of measurements will be influenced by the inhomogeneity of the nuclear material. Raman spectra underwent 9-point smoothing, baseline correction (polynomial method, 2 iterations; OPUS 7.5 software, Bruker Optics, Ettlingen, Germany), and vector normalisation prior to multivariate analyses. The contribution of glass to the spectra was removed by the extended multivariate signal correction algorithm (EMSC).27,33
Multivariate analysis of RMS data
Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS) was performed to resolve multiple component responses of unresolved mixtures, as explained in detail by de Juan and colleagues.34 Briefly, MCR-ALS can be employed for the analysis of the kinetics of a process and to identify the spectroscopic signature of each component in the process. In particular, in the present work, MCR-ALS was exploited to develop a kinetic model of the evolution of the cell populations, from viable to early and late apoptotic/dead, as a result of the action of cisPt, by decomposing the Raman spectral profiles acquired on OSCCs. The MCR-ALS 2.0 Graphical User Interface 2.0 was employed for all analysis presented in this study.35,36 It is freely available from the Multivariate Curve Resolution Homepage (https://mcrals.wordpress.com/). The GUI was run in Matlab 2022a (Mathworks). Singular Value Decomposition was used to initially identify the number of components as 3. Initial estimates of the components were then performed using Evolving Factor Analysis. The fitting procedure was subjected to non-negativity constraints in both the concentration and spectral domains. Kinetic constraints of A > B > C were applied to the concentration domain.Principal Components Analysis (PCA) was then employed as an unsupervised multivariate approach to analyse spectral data of OSCCs, comparing the evolution of spectral profiles as a function of time for each drug and for all drugs at different time points (OriginPro 2018b software, OriginLab Corporation, Northampton, Massachusetts).37
Results and discussion
This study presents the results of the in vitro RMS analysis of primary OSCC cells treated with cisplatin (7 μg mL−1) for 16, 24, and 48 hours (cisPt-16, cisPt-24, and cisPt-48 groups. The vibrational spectroscopic assessment of the effects of cisPt on OSCC primary tumoral cells and cancer stem cells, already carried out by means of FTIRM,17,20,21 provided clear spectral signals which characterised a time-dependent, drug-specific cellular response, and offered additional information to establish the spectral signature typical of the different modes of action of the three compounds. Here, a higher spectral and spatial resolution approach coupling RMS with MCR-ALS and PCA was exploited to monitor the evolution of the cell population as a result of the action of cisPt, from viable to early and late apoptotic/dead.The percentages of viable (V), early apoptotic (EA) and late apoptotic/dead (LA/D) primary OSCCs were assessed after 16, 24, 36, 48 and 72 hours of treatment with cisPt (7 μg mL−1), by using image-based cytometry (Fig. 1). The stability of the Ctrl group is confirmed by the percentage of viable cells higher than 80% at 72 h (Fig. 1A). Conversely, in the case of the same cells treated with the chemotherapic drug, the results suggest the triggering of apoptotic processes, which induce death of the cell population. In more detail, considerable levels of early apoptosis were evident at 16 h (∼30% of cell population), which persisted at 24 h (∼35% of cell population), while late apoptotic/dead cells became predominant at 36, 48, and 72 h (∼80%, ∼91%, and ∼97%, respectively) (Fig. 1B). The kinetics of the population distributions displayed in Fig. 1C for cisPt, indicates that the process can be viewed as a progression, or adverse outcome pathway,38 from viable, to early, to late apoptotic/dead. Such a progression can be modelled with a simple system of ordinary differential rate equations (eqn (1)–(3)), with rates k1, to describe the progression from viable (V) to early apoptotic (EA), and k2 to describe the progression of early to late apoptotic (LA).
| dV/dt = −k1V | (1) |
| dEA/dt = k1V − k2EA | (2) |
| dLA/dt = k2EA | (3) |
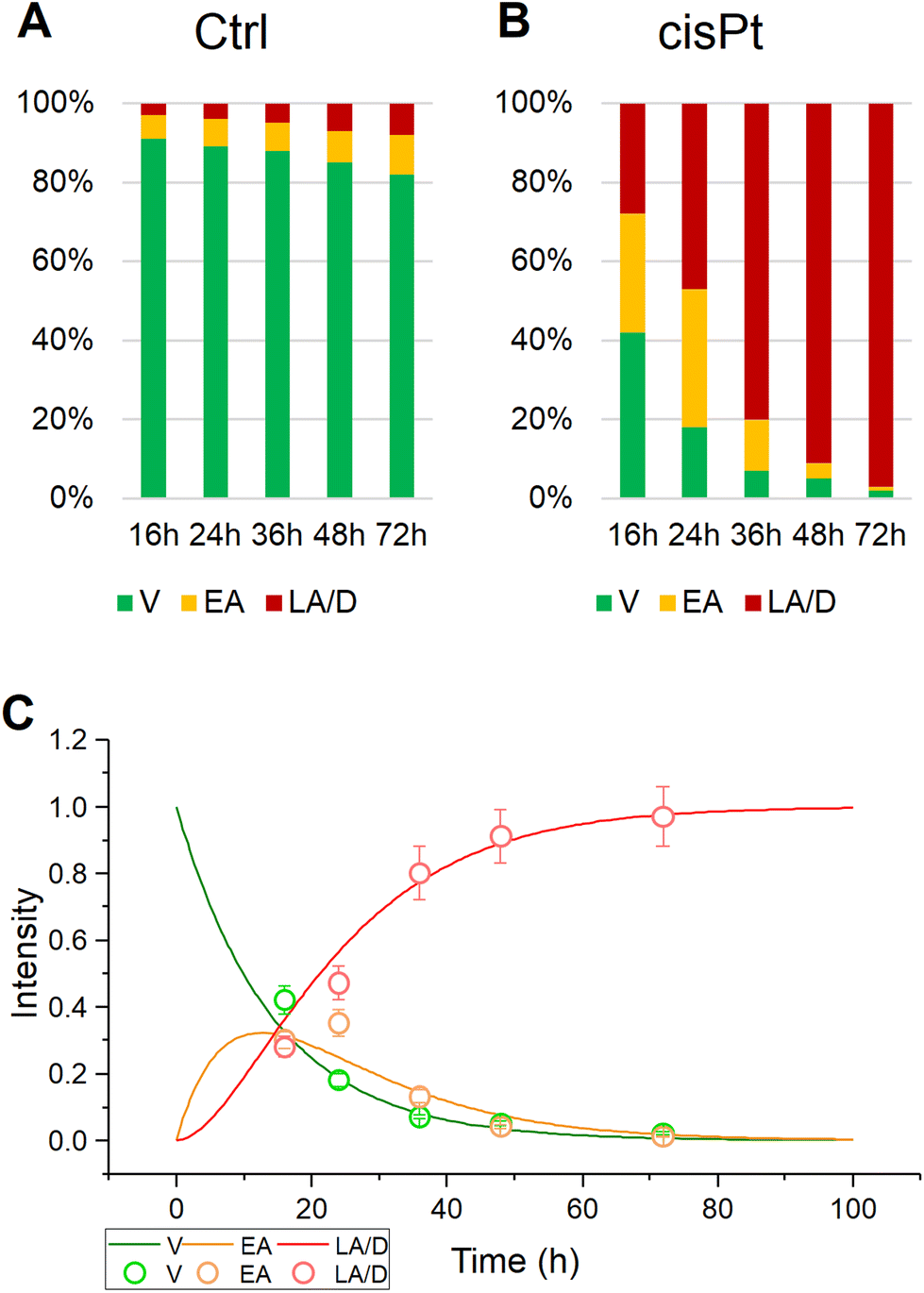 | ||
| Fig. 1 Percentages of viable (V), early apoptotic (EA) and late apoptotic/dead (LA/D) primary OSCC cells treated as follows: (A) no treatment (Ctrl); (B) treated for 16, 24, 36, 48, and 72 h with 7 μg mL−1 cisplatin (cisPt); (C) kinetic progression from viable, to early, to late apoptotic state for cisPt. The standard deviation of the measurements is indicated by the error bars in (C). | ||
Table 1 details the results of such modelling of the progression for the control and cisPt exposed calls. Using the inverse value of IC50 as a measure of drug toxicity/efficacy,39 it can be seen that cisplatin has a high rate of progression compared to control, both from viable to early (k1) and from early to late apoptotic (k2) cell populations, consistent with its high degree of toxicity.
| Drug | Ctrl | cisPt |
|---|---|---|
| Inverse IC50 (mL μg−1) | 0.143 (24 h) | |
| k 1 (h−1) | 0.003 | 0.075 |
| k 2 (h−1) | 0.020 | 0.095 |
In Fig. 2, the average Raman spectra of all the experimental groups are shown in the 400–1800 cm−1 spectral range (average of ∼110–135 spectra); the most relevant Raman peaks are labelled in the spectra, and listed in Table 2, together with the related vibrational modes and biological assignments.
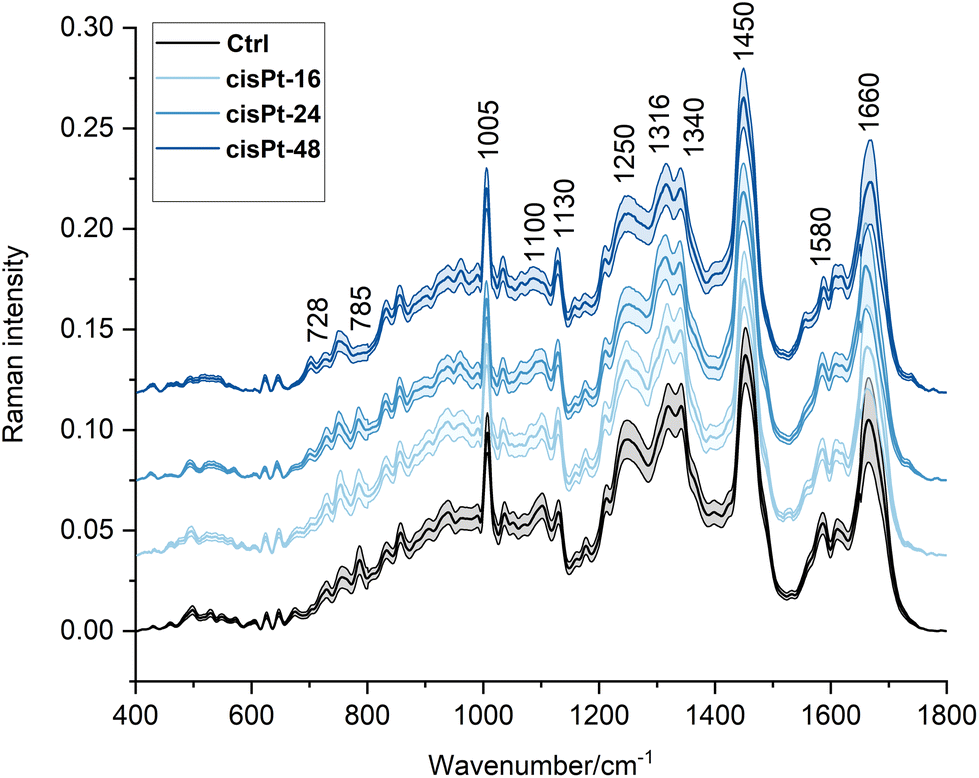 | ||
| Fig. 2 Average RMS spectra of cisPt exposed cells in the 400–1800 cm−1 spectral range. For better viewing, spectra have been offset along the y-axis. An average Ctrl spectrum is displayed for comparison in each case. | ||
| Wavenumber (cm−1) | Raman assignments19,21,40–43 |
|---|---|
| ∼624 | C–C twisting mode of phenylalanine |
| ∼644 | C–C twisting mode of tyrosine |
| ∼670 | Vibration of thymine and guanine from DNA and RNA |
| ∼728 | O–P–O stretching of B-form DNA |
| ∼752 | Ring breathing of tryptophan |
| ∼785 | O–P–O stretching of DNA backbone |
| ∼830 | O–P–O asymmetric stretching of B-form DNA |
| ∼855 | Ring breathing of proline and tyrosine |
| ∼1005 | Breathing vibration of phenylalanine |
| ∼1034 | C–H bending vibration of phenylalanine |
| ∼1095 | Symmetric PO2− stretching vibration of DNA backbone |
| ∼1120 | C–O band of ribose |
| ∼1160 | C–C/C–N stretching of proteins |
| ∼1180 | C–N stretching of cytosine and guanine |
| ∼1208 | Stretching of phenylalanine, tyrosine, and tryptophan |
| ∼1245 | Amide III |
| ∼1316 | CH3CH2 twisting mode of lipids |
| ∼1340 | Ring breathing mode of adenine and guanine |
| ∼1450 | CH2 bending of lipids |
| ∼1580 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bending of phenylalanine/adenine and guanine C bending of phenylalanine/adenine and guanine |
| ∼1610 | CC stretching mode of tyrosine and tryptophan |
| ∼1660 | Amide I |
The homogeneity and stability of control groups were tested by means of PCA, using all RMS spectra of Ctrl-16, Ctrl-24, Ctrl-36, Ctrl-48, and Ctrl-72 cell populations; no segregation was found along PC1 (44.3% explained variance) or PC2 (19.9% explained variance; graph not shown).
Based on the kinetic analysis previously described, MCR-ALS, using kinetic constraints to model the progression of V → EA → LA, in a Hard-Soft modelling approach, was explored to examine the evolution of the spectral responses. For cisPt exposure, three component spectra were identified, as shown in Fig. 3A, which had an MCR-ALS optimised kinetic evolution, as shown in Fig. 3B. The optimised rates of k1 and k2, respectively equal to 0.0346 and 0.0335 h−1, are in reasonable agreement with the values of the simulated apoptosis response listed in Table 1. Fig. 3B also shows the full kinetic evolution of the relative contributions of the V, EA and LA spectral profiles to the Raman spectrum, calculated according to eqn (1)–(3), indicating that the model can quantitatively predict the efficacy of the drug in eliciting the cellular response. Given that the predicted contributions of each component spectrum match the overall fit at each timepoint, the combination spectrum can be predicted at any time, or the ratio of V![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EA:LA can be predicted from a spectrum at any time.
:EA:LA can be predicted from a spectrum at any time.
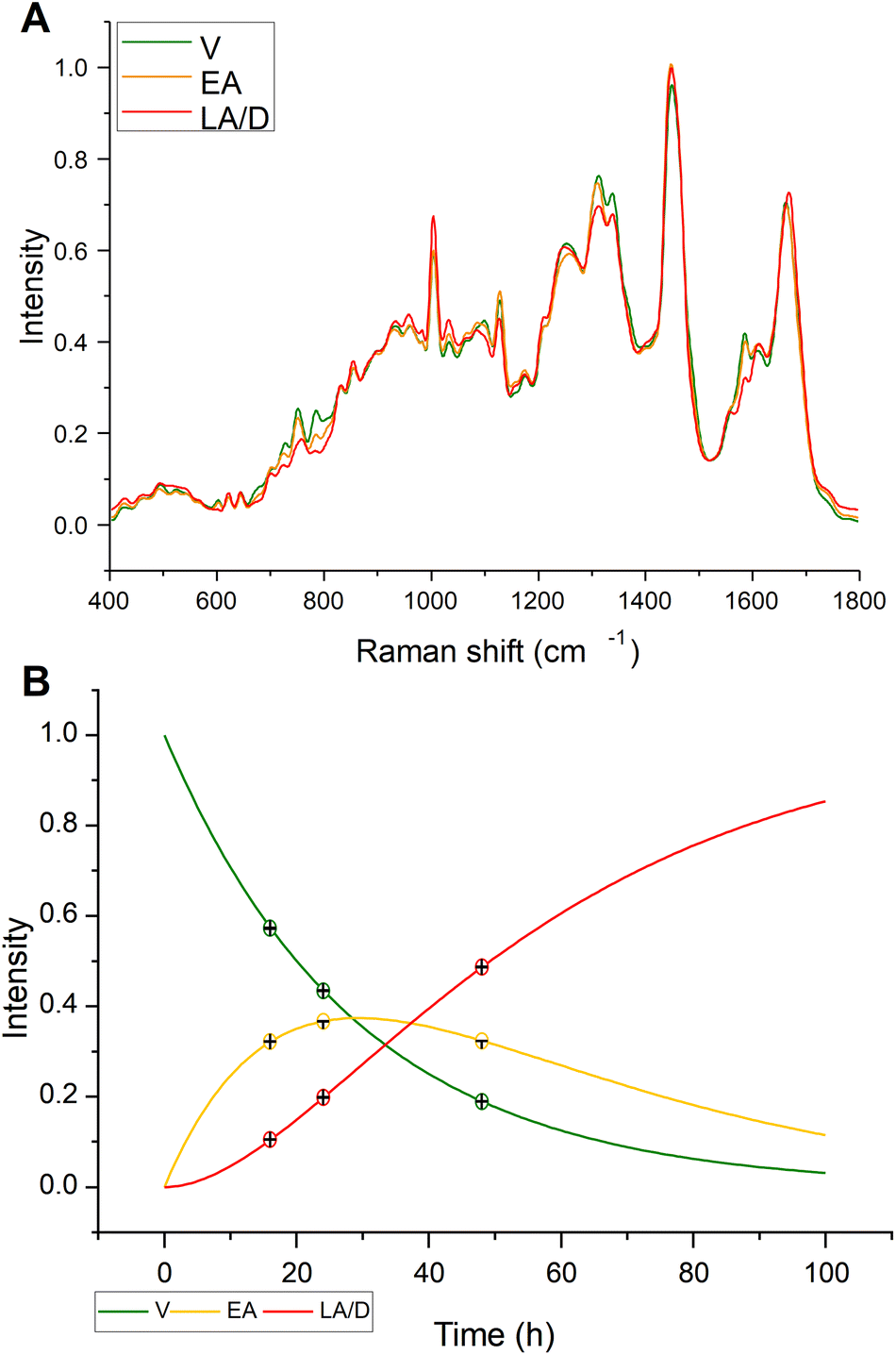 | ||
| Fig. 3 (A) MCR-ALS component spectra and (B) kinetic evolution for cisPt treated cell populations. | ||
The MCR-ALS component spectra were compared to assess the progression of cisPt action and the cellular responses (Fig. 4). Several spectral features changed between Ctrl cells and the MCR-ALS component spectrum representing viable cells after cisPt treatment. The difference spectrum obtained by subtracting the mean spectrum of Ctrl from the spectrum representing viable cells indicates a decrease of the protein related peaks centred at ∼1005 cm−1, representing phenylalanine, and ∼1250 cm−1, representing Amide III, and of the peaks at ∼1095 cm−1 and ∼1340 cm−1, related to DNA, together with an increase of the peaks centred at ∼750 cm−1, representing tryptophan, and ∼1120 cm−1, representing RNA. The RMS signature of the early stage interaction of the drug with the cells should be contained in these initial cellular response. However, as the first timepoint was taken 16 h after the initial interaction, any signatures of the drug interaction are mixed with the cellular response. The comparison of the MCR-ALS component spectra of viable and early apoptotic cells indicates a less profound spectrum evolution. The difference spectrum highlighted a decrease of the peaks centred at ∼784 cm−1, representing DNA backbone, ∼1250 cm−1, representing amide III, and ∼1340 cm−1, representing adenine and guanine, together with an increase of the peak centred at 1450 cm−1, representing lipids. Finally, the difference spectrum, obtained by subtracting the MCR-ALS component spectrum of LA/D cells from the one of EA cells, after cisPt treatment, showed a decrease of the peaks centred at ∼750 cm−1, representing tryptophan, ∼784 cm−1, representing DNA backbone, ∼1120 cm−1, representing RNA, and ∼1580 cm−1, representing adenine and guanine, together with an increase of the peak centred at ∼1005 cm−1, representing phenylalanine.
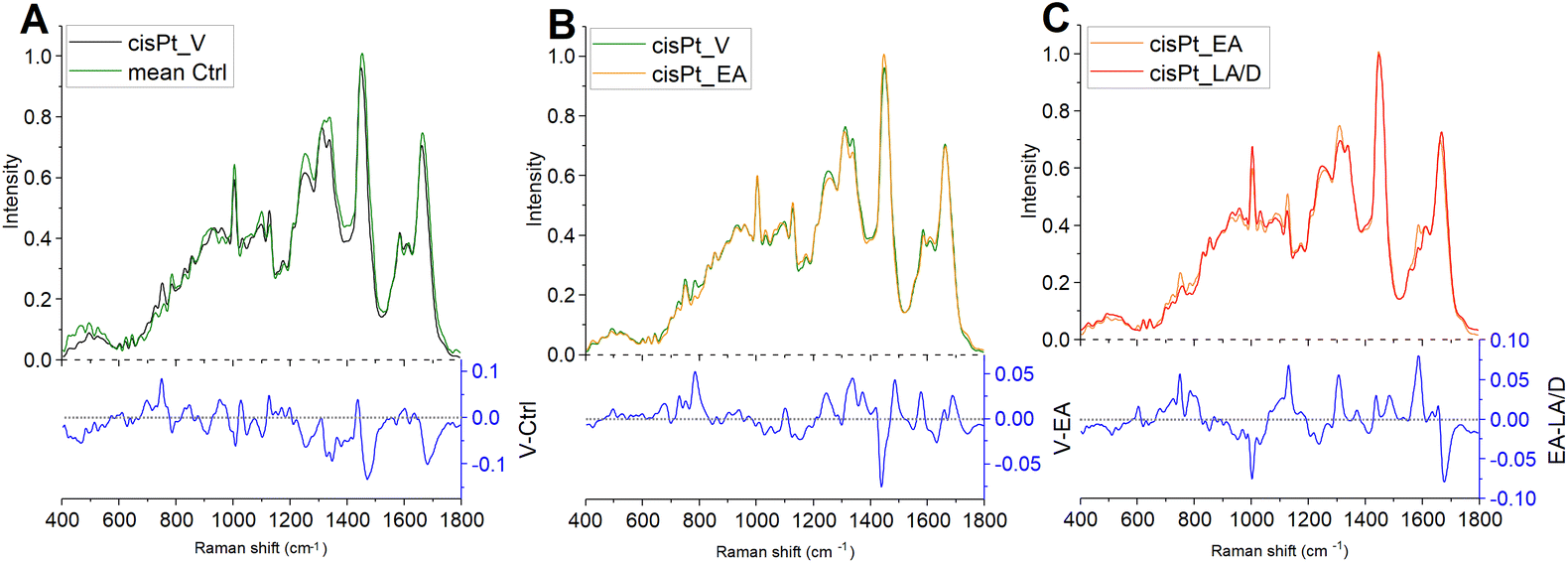 | ||
| Fig. 4 Difference spectra obtained by subtracting: (A) the Ctrl mean spectrum to the MCR-ALS component of viable cells after the cisPt treatment (V-Ctrl); (B) the MCR-ALS component of early apoptotic cells to the one of viable cells (V-EA), and (C) the MCR-ALS component of late apoptotic/dead cells to the one of early apoptotic cells (EA-LA/D). | ||
The comparison of MCR-ALS component spectra elucidated some traits of the known cellular responses to cisPt, which primarily induces inter- and intra-strand DNA cross-linkages by forming adducts with the N7 atom of the purine nucleobases of DNA.44 In particular, consistent with its known and well-described activity, significant alterations were found in DNA structure, as shown by the decrease of the peak at 1095 cm−1 in early apoptotic treated cells and of the peak at 785 cm−1, decreasing in early apoptotic and, progressively, in late apoptotic/dead cells. This result suggests a profound loss in the integrity of DNA backbone, probably due to an apoptosis-related general fragmentation and rearrangement of DNA.15,17,21 The impact of the cisPt treatment on DNA integrity was also evidenced by a significant decrease of adenine and guanine content in treated cells, represented by the peaks at 1340 cm−1 and 1580 cm−1. Finally, the increase of the peak at 1450 cm−1, representative of lipids, evidenced in early apoptotic cells with respect to viable ones, may be indicative of the known apoptosis-related accumulation of lipid droplets within the cytoplasm, as reported elsewhere.10,17,21,44
The progression from viable, through early to late apoptotic states of cisPt exposed cells, was further analysed by comparison with the results of a Principal Component Analysis of the spectra, at the different stages (Fig. 5). Notably, similar features highlighted by the differences of the MCR-ALS component spectra were found in the respective PC loadings which are primarily responsible for the differentiation. In particular, both the PC1 loading of the PCA comparing Ctrl and cisPt-16 groups (Fig. 5D), and the PC2 loading of the comparison between cisPt-24 and cisPt-48 groups (Fig. 5F), compare well with the respective difference spectra of the MCR-ALS components, (V-Ctrl – Fig. 4A) and (LA/D-EA – Fig. 4C).
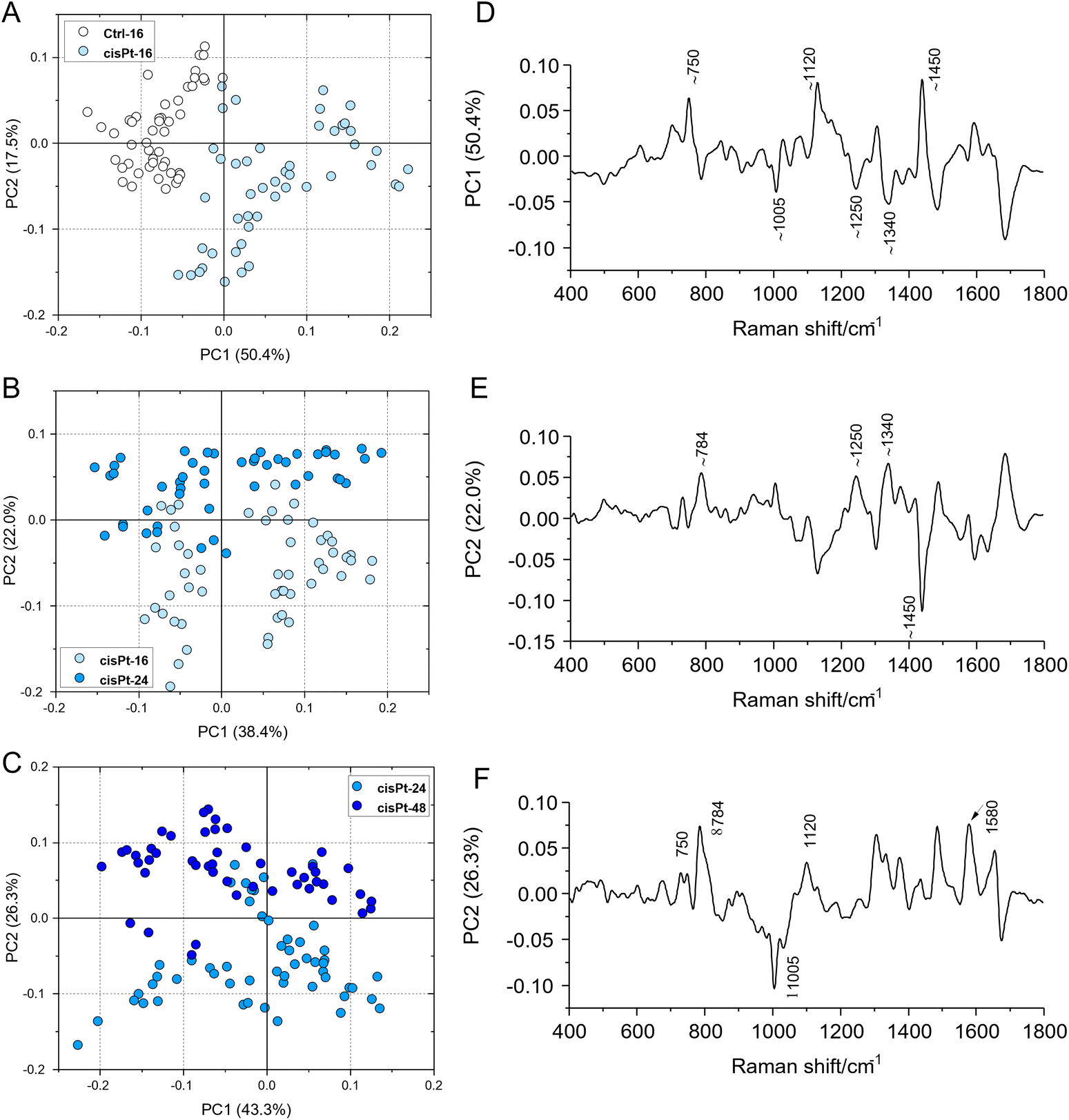 | ||
| Fig. 5 PCA scores plots and corresponding loading spectra calculated on Raman spectra in the 400–1800 cm−1 range of the following experimental groups: (A and D) Ctrl/cisPt-16; (B and E) cisPt-16/cisPt-24; (C and F) cisPt-24/cisPt-48. | ||
The comparison of the PC2 loading of cisPt-16 and cisPt-24 (Fig. 5E) and the difference of the spectra of the MCR-ALS components representing V and EA (Fig. 4B), is perhaps less convincing, although there are clear similarities.
Therefore, although the cisPt-16 group represents only 42% viable, the cisPt-24 group 35% early apoptotic and cisPt-48 91% late apoptotic cells, PCA provides a good indication of the biochemical evolution from V → EA → LA/D cells.
Conclusions
To date, chemotherapy remains one of the main strategies to defeat cancer, and several molecules are routinely used both individually and in mixture in the clinical practice. Nevertheless, contrary to the routine of the past years, the choice of a chemotherapy drug depends not only on its mechanisms of action, but also on the personal response of the patient. In fact, the demand for personalised therapies is increasing, and hence, deepening the knowledge of the activity of the clinically employed molecules is mandatory. Cisplatin is a well-known chemotherapic drug, routinely used in many types of cancer, and in particular for the treatment of the oral squamous cell carcinoma, and a complete understanding of its mechanism of action could be helpful for a more appropriate use of these molecules. Among all the analytical techniques, RMS has been demonstrated to have the ability to provide reliable information on the interaction between drugs and cells, paving the way to the application of this vibrational spectroscopic technique as a pre-clinical screening tool in drug development research.In the present work, RMS was employed to assess the biomolecular modifications induced by cisplatin, and in particular to demonstrate the ability to track the cellular responses from the early stages at which the cells are still identified as viable, through the early and late stages of apoptosis. The effects of these drugs were previously assessed by means of FTIRM, which identified a time-dependent, drug-specific cellular response. Pursuing this approach further, and considering the spatial resolution achievable by RMS, which makes it suitable to target subcellular regions, it was possible to focus specifically on changes occurring in the nuclear regions of the analysed cells, which are the target sites of the drugs. In terms of the potential applications of these techniques, first would be being able to identify signatures of the mode of action of drugs, even in reference cell lines, such that new drugs could be examined (pre-clinical screening) to easily establish their mode of action. If the method can be further developed to quantify the efficacy of the drug, known drugs could be tested on patient cells (Companion Diagnostics).
Raman spectra exhibited signatures of the progression of the cell death process, from viable, to early apoptotic, late apoptotic/dead cells. Although preliminary, the study explored the application of MCR-ALS regression to build kinetic models, and use the constraints of the model to data mine relevant spectral profiles, and rates, which can be better employed to characterise the efficacy of the drug, and the response of a specific patient to that drug. The MCR-ALS result enables the prediction the ratio of V, EA, and LA/D cells in the cell populations from a Raman spectrum. Potentially, the most significant and novel aspect of the present work is being able to quantify the rates of cellular response, which could be used to quantify and compare the efficacy of different drugs, as well as the individual response of patients.
Ultimately, the study supports the ability of RMS to shed new light on the mode of action of different types of drugs, possibly paving the way to its application as a pre-clinical screening tool in drug development research.
Author contributions
Conceptualization, data curation, and writing – original draft preparation: V. N., E. G., H. J. B. Formal analysis and methodology: V. N., H. J. B. Investigation: A. B., G. O., G. O. Resources: G. O., G. O. Writing – review and editing:V. N., E. G., H. J. B., A. B. Supervision: E. G., H. J. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors V. N. and P. M. acknowledge for financial support the European Union – Next Generation EU. Project Code: ECS00000041; Project Title: Innovation, digitalisation and sustainability for the diffused economy in Central Italy – VITALITY.References
- V. Notarstefano, G. Gioacchini, E. Giorgini, N. Montik, A. Ciavattini, A. R. Polidori, F. A. Candela, L. Vaccari, M. Cignitti and O. Carnevali, Int. J. Mater. Sci., 2020, 21, 7124 CAS.
- H. J. Butler, L. Ashton, B. Bird, G. Cinque, K. Curtis, J. Dorney, K. Esmonde-White, N. J. Fullwood, B. Gardner, P. L. Martin-Hirsch, M. J. Walsh, M. R. McAinsh, N. Stone and F. L. Martin, Nat. Protoc., 2016, 11, 664–687 CrossRef CAS PubMed.
- H. J. Butler, J. M. Cameron, C. A. Jenkins, G. Hithell, S. Hume, N. T. Hunt and M. J. Baker, Clin. Spectrosc., 2019, 1, 100003 CrossRef.
- G. Clemens, J. R. Hands, K. M. Dorling and M. J. Baker, Analyst, 2014, 139, 4411–4444 RSC.
- G. Orilisi, R. Monterubbianesi, V. Notarstefano, V. Tosco, F. Vitiello, G. Giuliani, A. Putignano and G. Orsini, Spectrochim. Acta, Part A, 2021, 260, 119966 CrossRef CAS PubMed.
- V. Notarstefano, G. Gioacchini, H. J. Byrne, C. Zacà, E. Sereni, L. Vaccari, A. Borini, O. Carnevali and E. Giorgini, Spectrochim. Acta, Part A, 2019, 212, 206–214 CrossRef CAS PubMed.
- M. Paraskevaidi, B. J. Matthew, B. J. Holly, B. J. Hugh, C. P. V. Thulya, C. Loren, C. StJohn, G. Peter, G. Callum, K. G. Sergei, K. Kamila, K. Maria, L. M. G. Kássio, M.-H. L. Pierre, P. Evangelos, P. Savithri, A. A. John, S. Alexandra, S. Marfran, S.-S. Josep, T. Gunjan, W. Michael and W. Bayden, Appl. Spectrosc. Rev., 2021, 56, 804–868 CrossRef CAS.
- H. J. Byrne, F. Bonnier, A. Casey, M. Maher, J. McIntyre, E. Efeoglu and Z. Farhane, Appl. Opt., 2018, 57, E11 CrossRef CAS PubMed.
- H. J. Byrne, F. Bonnier, E. Efeoglu, C. Moore and J. McIntyre, Front. Bioeng. Biotechnol., 2020, 8, 544311 CrossRef PubMed.
- V. Notarstefano, S. Sabbatini, M. Sabbatini, A. Arrais, A. Belloni, C. Pro, L. Vaccari, D. Osella and E. Giorgini, Clin. Spectrosc., 2021, 3, 100011 CrossRef.
- Z. Farhane, F. Bonnier and H. J. Byrne, Anal. Bioanal. Chem., 2017, 409, 1333–1346 CrossRef CAS PubMed.
- L. E. Jamieson and H. J. Byrne, Vib. Spectrosc., 2017, 91, 16–30 CrossRef CAS.
- A. Derenne, R. Gasper and E. Goormaghtigh, Analyst, 2011, 136, 1134 RSC.
- J. L. Denbigh, D. Perez-Guaita, R. R. Vernooij, M. J. Tobin, K. R. Bambery, Y. Xu, A. D. Southam, F. L. Khanim, M. T. Drayson, N. P. Lockyer, R. Goodacre and B. R. Wood, Sci. Rep., 2017, 7, 2649 CrossRef PubMed.
- A. Mignolet, A. Derenne, M. Smolina, B. R. Wood and E. Goormaghtigh, Biochim. Biophys. Acta, Proteins Proteomics, 2016, 1864, 85–101 CrossRef CAS PubMed.
- J. A. Ferreira, A. Peixoto, M. Neves, C. Gaiteiro, C. A. Reis, Y. G. Assaraf and L. L. Santos, Drug Resist. Updates, 2016, 24, 34–54 CrossRef PubMed.
- E. Giorgini, S. Sabbatini, R. Rocchetti, V. Notarstefano, C. Rubini, C. Conti, G. Orilisi, E. Mitri, D. E. Bedolla and L. Vaccari, Analyst, 2018, 143, 3317–3326 RSC.
- L. Yuan, W.-M. Yu and C.-K. Qu, J. Biol. Chem., 2003, 278, 42812–42820 CrossRef CAS PubMed.
- H. Nawaz, F. Bonnier, P. Knief, O. Howe, F. M. Lyng, A. D. Meade and H. J. Byrne, Analyst, 2010, 135, 3070 RSC.
- V. Notarstefano, A. Belloni, S. Sabbatini, C. Pro, G. Orilisi, R. Monterubbianesi, V. Tosco, H. J. Byrne, L. Vaccari and E. Giorgini, Cells, 2021, 10, 2127 CrossRef CAS PubMed.
- V. Notarstefano, S. Sabbatini, C. Pro, A. Belloni, G. Orilisi, C. Rubini, H. J. Byrne, L. Vaccari and E. Giorgini, Analyst, 2020, 145, 8038–8049 RSC.
- A. de Juan and R. Tauler, Anal. Chim. Acta, 2021, 1145, 59–78 CrossRef CAS PubMed.
- M. Garrido, F. X. Rius and M. S. Larrechi, Anal. Bioanal. Chem., 2008, 390, 2059–2066 CrossRef CAS PubMed.
- D. Perez-Guaita, G. Quintas, Z. Farhane, R. Tauler and H. J. Byrne, Talanta, 2020, 208, 120386 CrossRef CAS PubMed.
- I. Matveeva, I. Bratchenko, Y. Khristoforova, L. Bratchenko, A. Moryatov, S. Kozlov, O. Kaganov and V. Zakharov, Sensors, 2022, 22, 9588 CrossRef CAS PubMed.
- H. Noothalapati, K. Iwasaki and T. Yamamoto, Anal. Sci., 2017, 33, 15–22 CrossRef CAS PubMed.
- H. J. Byrne, P. Knief, M. E. Keating and F. Bonnier, Chem. Soc. Rev., 2016, 45, 1865–1878 RSC.
- A. D. Meade, C. Clarke, F. Draux, G. D. Sockalingum, M. Manfait, F. M. Lyng and H. J. Byrne, Anal. Bioanal. Chem., 2010, 396, 1781–1791 CrossRef CAS PubMed.
- M. J. Baker, J. Trevisan, P. Bassan, R. Bhargava, H. J. Butler, K. M. Dorling, P. R. Fielden, S. W. Fogarty, N. J. Fullwood, K. A. Heys, C. Hughes, P. Lasch, P. L. Martin-Hirsch, B. Obinaju, G. D. Sockalingum, J. Sulé-Suso, R. J. Strong, M. J. Walsh, B. R. Wood, P. Gardner and F. L. Martin, Nat. Protoc., 2014, 9, 1771–1791 CrossRef CAS PubMed.
- E. Brauchle, S. Thude, S. Y. Brucker and K. Schenke-Layland, Sci. Rep., 2014, 4, 4698 CrossRef PubMed.
- J. Gala de Pablo, F. J. Armistead, S. A. Peyman, D. Bonthron, M. Lones, S. Smith and S. D. Evans, J. Raman Spectrosc., 2018, 49, 1323–1332 CrossRef CAS PubMed.
- M. Pudlas, S. Koch, C. Bolwien, S. Thude, N. Jenne, T. Hirth, H. Walles and K. Schenke-Layland, Tissue Eng., Part C, 2011, 17, 1027–1040 CrossRef PubMed.
- L. T. Kerr and B. M. Hennelly, Chemom. Intell. Lab. Syst., 2016, 158, 61–68 CrossRef CAS.
- A. de Juan, M. Maeder, M. Martínez and R. Tauler, Chemom. Intell. Lab. Syst., 2000, 54, 123–141 CrossRef CAS.
- J. Jaumot, R. Gargallo, A. de Juan and R. Tauler, Chemom. Intell. Lab. Syst., 2005, 76, 101–110 CrossRef CAS.
- A. de Juan, J. Jaumot and R. Tauler, Anal. Methods, 2014, 6, 4964–4976 RSC.
- F. Bonnier and H. J. Byrne, Analyst, 2012, 137, 322–332 RSC.
- OECD, OECD Series on Adverse Outcome Pathways, https://www.oecd-ilibrary.org/environment/deiodinase-2-inhibition-leading-to-increased-mortality-via-reduced-posterior-swim-bladder-inflation_dc406014-en, (accessed January 16, 2023).
- H. J. Byrne and M. A. Maher, Comput. Toxicol., 2019, 12, 100090 CrossRef.
- A. C. S. Talari, Z. Movasaghi, S. Rehman and I. ur Rehman, Appl. Spectrosc. Rev., 2015, 50, 46–111 CrossRef CAS.
- V. Notarstefano, M. Pisani, M. Bramucci, L. Quassinti, F. Maggi, L. Vaccari, M. Parlapiano, E. Giorgini and P. Astolfi, Spectrochim. Acta, Part A, 2022, 269, 120735 CrossRef CAS PubMed.
- H. Huang, H. Shi, S. Feng, W. Chen, Y. Yu, D. Lin and R. Chen, Anal. Methods, 2013, 5, 260–266 RSC.
- E. Efeoglu, A. Casey and H. J. Byrne, Analyst, 2017, 142, 3848–3856 RSC.
- G. Birarda, D. E. Bedolla, E. Mitri, S. Pacor, G. Grenci and L. Vaccari, Analyst, 2014, 139, 3097–3106 RSC.
| This journal is © The Royal Society of Chemistry 2023 |