
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative detection of RAS and KKS peptides in COVID-19 patient serum by stable isotope dimethyl labeling LC-MS†
Ben K.
Ahiadu
a,
Thomas
Ellis
a,
Adam
Graichen
a,
Richard B.
Kremer
b and
James F.
Rusling
 *acde
*acde
aDepartment of Chemistry, University of Connecticut, Storrs, Connecticut 06269, USA. E-mail: rusling.james@uconn.edu
bDepartment of Medicine, McGill University Health Centre, 1001 Decarie Blvd., Montreal, QC H4A, Canada
cDepartment of Surgery and Neag Cancer Center, UConn Health, Farmington, Connecticut 06232, USA
dSchool of Chemistry, National University of Ireland Galway, Galway, H91 TK33, Ireland
eInstitute of Materials Science, University of Connecticut, 97 N. Eagleville Road, Storrs, CT 06269, USA
First published on 10th October 2023
Abstract
Angiotensin and kinin metabolic pathways are reported to be altered by many diseases, including COVID-19. Monitoring levels of these peptide metabolites is important for understanding mechanisms of disease processes. In this paper, we report dimethyl labeling of amines in peptides by addition of formaldehyde to samples and deutero-formaldehyde to internal standards to generate nearly identical isotopic standards with 4 m/z units larger per amine group than the corresponding analyte. We apply this approach to rapid, multiplexed, absolute LC-MS/MS quantitation of renin angiotensin system (RAS) and kallikrein-kinin system (KKS) peptides in human blood serum. Limits of detection (LODs) were obtained in the low pg mL−1 range with 3 orders of magnitude dynamic ranges, appropriate for determinations of normal and elevated levels of the target peptides in blood serum and plasma. Accuracy is within ±15% at concentrations above the limit of quantitation, as validated by spike-recovery in serum samples. Applicability was demonstrated by measuring RAS and KKS peptides in serum from COVID-19 patients, but is extendable to any class of peptides or other small molecules bearing reactive –NH2 groups.
Introduction
Endogenous peptides regulate many human biological processes, including blood pressure, fluid, and electrolyte balance, and immune responses to foreign stimuli. Dysregulation of these peptides can cause undesirable biological effects, and these molecules can in principle be monitored to serve as biomarkers and reveal mechanisms underlying pathological conditions. While Immunoassays are valuable for detecting biomolecules, for small peptides they are often saddled with multiplexing, sensitivity, and selectivity issues1,2 although progress is being made in overcoming these limitations.3–5Liquid chromatography-mass spectrometry (LC-MS) offers high selectivity, easy multiplexing, and rapid assay development for multiplexed detection of peptides in biological samples.6 Quantitative LC-MS is often done using stable isotope dilution to account for variations in analyte recovery, ionization and/or detection. In the standard approach, a known amount of isotopically labeled standard is spiked into the sample at the earliest possible stage of preparation.7,8 While this approach offers high accuracy and selectivity in LC-MS quantitation of peptides, disadvantages include cost, time, and complexity of chemically synthesizing isotopically labeled standards for each analyte in a mixture.
Peptides can also be labeled using isobaric tags for relative and absolute quantitation (iTRAQ), isotope-coded affinity tags (iCAT), and tandem mass tags (TMT).9 Reductive amination has gained recent attention for fast, easy, low-cost isotopic labeling of peptides on amino groups enabling accurate, sensitive, selective, fast, multiplexed detection of proteins in comparative LC-MS proteomics.10 This method, often called stable isotope labeling (SIL), employs methylation of peptide amines using formaldehyde and subsequent reduction as first reported by Hsu et al.10 for comparative proteomic analyses of proteins in cell lysates.
We report here the application of SIL for absolute quantification of endogenous renin angiotensin system (RAS) and kallikrein-kinin system (KKS) peptide metabolites in serum from COVID-19 patients. Low-cost isotopic formaldehydes are used to differentially label multiple analyte peptides and standards that are mixed and analyzed in a single LC-MS/MS run. Deutero-formaldehyde and “light” formaldehyde react with primary and/or secondary amino groups in peptides to form Schiff's bases and are then reduced by sodium cyanoborohydride to add methyl groups on each reactive amino moiety (Scheme 1). Unmodified primary amino moieties on N-termini and lysine residues of peptides are dimethylated by this reaction, while N-terminal proline residues are mono-methylated. Formaldehyde addition followed by sodium cyanoborohydride reduction converts free amino groups on analytes to dimethyl amines to produce a mass shift of 28 Da. Deutero-formaldehyde is added to standards followed by reduction to introduce a 32 Da mass shift. Thus, high-resolution accurate mass (HRAM) spectrometers enable selective detection of the deuterated standards and undeuterated sample species that differ in m/z but have very similar LC retention times.11 Since first reported in 2003,10 stable isotope labeling by reductive amination has been further optimized12 and widely used for relative quantitation of peptides from protein digestion in proteomics.13,14 Triple-quadrupole mass spectrometry in selected reaction monitoring (SRM) mode (or multiple reaction monitoring, MRM) is often used for LC-MS/MS quantification of peptides.15 However, identifying and constructing transitions for MRM can be labor-intensive. Parallel reaction monitoring (PRM) with high resolution accurate MS such as quadrupole-time of flight (Q-TOF) or quadrupole-Orbitrap spectrometers16 has primarily been utilized for targeted protein quantification via fragments of chosen surrogate peptide precursor ions. MRM and PRM have comparable sensitivity, precision, and linear dynamic range,17,18 but PRM has better selectivity, and enables faster method development.
 | ||
| Scheme 1 Dimethyl labeling of amines in peptides. Light formaldehyde labeling is used for analytes; Mid or deutero-formaldehyde labeling is used for internal standards. | ||
In this paper, we describe stable isotope dimethyl-amination for quantitative PRM LC-MS determination of endogenous renin angiotensin system (RAS) and kallikrein-kinin system (KKS) peptide metabolites whose levels are reportedly altered in patients with COVID-19 and other diseases.19–22
RAS and KKS metabolites play regulatory roles in humans (Scheme 2). RAS is a major regulator of blood pressure, fluid, and electrolyte balance23,24 while KKS controls vascular permeability, vasodilation, release of inflammatory cytokines during tissue injury, and cell proliferation.19,25 As discussed above, we utilize in situ SIL of amines with formaldehyde and internal standards with deutero-formaldehyde with cyanoborohydride reduction to generate isotopic standards differing in mass by +4 Da per reacted primary amine compared to sample peptides (Scheme 1). We demonstrate herein a wide linear dynamic range and limits of detection (LOD) in the low pg mL−1 range for this new SIL peptide assay that was validated according to US Food and Drug Administration guidelines.26 Applicability was demonstrated by measuring RAS and KKS metabolites in serum of COVID-19 patients.
 | ||
| Scheme 2 Kinin (left) and angiotensin peptide metabolic pathways that utilize enzymes angiotensin converting enzyme (ACE) and ACE2. | ||
Experimental
Peptides and reagents
See ESI† for details about source of chemicals and reagents. Sample preparation and analysis procedures are further elaborated in the ESI.†Stock solutions
To inhibit adsorption of peptides on reaction and storage vials, and enhance solubility, 1 mg mL−1 stock solutions of individual peptides were made in 25% acetonitrile containing 1% formic acid and stored at −80 °C until use. Appropriate volumes of each peptide stock solutions were mixed so that each analyte was 5 μg mL−1 in 0.1 M sodium acetate buffer, pH 5.5. Calibration standards were made from this working solution in pH 5.5 buffer containing 10% acetonitrile and stored in 90 μL aliquots at −20 °C for subsequent labeling. Similarly, quality control (QC) samples were made from a separate 5 μg mL−1 combined stock solution and stored at −20 °C until use. External standards and QC samples were spiked with 10 μL of 0.2% human serum to mimic the sample matrix. Pipette tips were pre-rinsed 3× with respective solutions to saturate binding sites before solutions were transferred.Internal standards
A deutero-formaldehyde-labeled version of each analyte served as internal standard for quantifying formaldehyde-labeled endogenous metabolites. Internal standards were generated by reacting 8 μL of 4% (v/v) deutero-formaldehyde with 100 μL of 5 ng mL−1 peptide standard for 3 min followed by reduction with 8 μL of 0.6 M sodium cyanoborohydride for 1.5 h. LC-MS analyses confirmed that reactions (Scheme 1) are complete (see Results).Methylated internal standards were spiked into calibration standards, quality control (QC), and real samples at 300 pg mL−1 final concentration. To derivatize the analyte peptides, 16 μL of 4% normal formaldehyde (v/v) was added, vortexed, and allowed to stand for 3 min. The mixture was then incubated with shaking for 1.5 h at ambient temperature after adding 16 μL 0.6 M sodium cyanoborohydride (not deuterated) as reducing agent. The reaction was quenched with 16 μL of 1% ammonium hydroxide (v/v), then acidified and further quenched with 90 μL 0.1% formic acid. Reaction products were then isolated with solid-phase extraction (SPE).
Safety note
Because of toxicities of formaldehyde, sodium cyanoborohydride, and hydrogen cyanide released as a result of quenching and acidification of the samples, all reaction and quenching steps were performed under a fume hood.Analysis of serum
Serum from COVID-19 patients (n = 80) were collected at McGill University Health Centre Research Institute (MUHC-RI) with approval (#2021-6081) from the center's Ethics Board. Serum samples of 30 μL were analyzed in duplicate. Samples were subjected to dimethyl labeling with formaldehyde as described above, and the deuterium-labeled internal standard was spiked into the serum at 300 pg mL−1 final concentration.Solid phase extraction (SPE)
Salts and other interfering substances were removed from standards and serum samples using SPE. The SPE cartridges used were in a 96-well filter plate format (Oasis® HLB μElution Plate 30 μm, 186001828BA, Waters). Wells of the 96-well plate were conditioned using 200 μL methanol (2×) followed by equilibration using 200 μL water (2×). Samples acidified to pH 2.8 were then loaded into the wells and allowed to drain at 1 mL min−1 under vacuum, followed by washing with 0.1% formic acid in water (12×) and a final wash with 5% acetonitrile in water containing 0.1% formic acid. Analytes were then eluted from the wells using 150 μL each of 30% acetonitrile, and 60% acetonitrile containing 0.1% formic acid. This effectively eluted hydrophilic and hydrophobic analytes. Eluates were then dried in a vacuum centrifuge at 60 °C and reconstituted in 50 μL of 10% acetonitrile containing 0.1% formic acid.Instrumentation and software
Liquid chromatography was done using a Thermo Fisher Scientific UPLC with temperature control at 45 °C. 25 μL of reconstituted sample or standard was injected and separated on a Phenomenex® Kinetex® 1.7 μm C18 100 Å LC column, 50 mm × 2.1 mm, fitted with a Phenomenex SecurityGuard Ultra C18 (AJ0-8782) precolumn. The autosampler was operated at 4 °C. Water containing 0.1% formic acid was used as mobile phase A, while mobile phase B contained acetonitrile with 0.1% formic acid. A 7 min binary gradient elution was used at a flow rate of 300 μL min−1, starting with a 6 min column equilibration step at 10% B prior to the gradient. Mobile phase B was then increased linearly to 24% until 7.3 min, to 95% until 7.4 min and kept at 95% to remove strongly retained impurities for 1.9 min before decreasing to 10% after 20 s.The LC was coupled to an Orbitrap Exploris 480 mass spectrometer (Thermo) via a heated electrospray ionization (HESI) source (KQ Integrated Solutions, Inc.). The ionization source was operated in the positive ion mode at spray voltage of 4000 V, with sheath gas at 60 arbitrary units and auxiliary and sweep gases at 15 and 2 arbitrary units, respectively. Ion transfer tube temperature was 300 °C, while the vaporizer was at 400 °C. Thermo XCalibur software was used to control the system, while an open-source software Skyline (version 22.2.0.351) was used to analyze raw data. To verify proper peak detection and integration, every peak integrated was inspected manually.
Parallel reaction monitoring (PRM) was utilized for quantification, and a full scan was done to evaluate the efficacy of dimethyl labeling and to select the most intense, interference-free precursors for each peptide. The precursors and collision energies optimized for specific peptides are listed in ESI Table S1.† To ensure that all metabolites reacted fully, a full scan was done to confirm that the highest concentration standard in the calibration curves did not contain peaks for unreacted peptides.
A full scan (MS) was initially run on light and medium-labeled standards to identify the most intense precursor peaks and charge states. Precursors were subjected to fragmentation, and all fragment ions were monitored and recorded. Chromatogram traces of the fragment ions were then used to optimize the LC separation and ion source conditions. The MS/MS chromatograms were imported into Skyline to identify the retention time of each peptide. The retention times were then exported back into the XCalibur method editor to create a retention time-scheduled parallel reaction monitoring (PRM) method (ESI Table S1†). Next, collision energies were optimized for each peptide by comparing the energy distribution and fragment ion intensities of each peptide at each tested normalized collision energy. A sum of at least three most intense co-eluting fragment peaks having zero background interference were used for quantifying each peptide. A peptide was deemed detected when (i) its light-labeled version co-elutes with the respective labeled internal standard; and (ii) the mass error of the representative fragments does not exceed 6 ppm.
Method validation
Validation was according to US Food and Drug Administration guidelines26 in terms of linearity, accuracy, precision, recovery, sensitivity, carryover, and analyte stability.Carryover
This is a measure of the extent to which analytes from previous injections remain in the LC-MS (especially on the chromatographic column) and get detected in subsequent runs. Carryover was assessed by measuring analytes in a solvent blank after measuring the standard with highest concentration. A solvent blank was injected after the highest concentration standard was run alternately five times and the average peak areas of the analytes were computed.Stability test
Stability of the samples was assessed at two handling conditions namely, the autosampler temperature and storage at −20 °C. First, analytes were spiked into a 100% pooled human serum, labeled, and analyzed immediately. Then, they were stored in the autosampler at 4 °C for three days. The samples were re-analyzed on the third day, and the peak areas on the first and third days were compared to determine the extent of analyte stability. Also, the stability of endogenous metabolites was assessed by spiking the metabolites into pooled human serum and storage at −20 °C for 16 days. Signals were compared to spiked samples that were freshly prepared and analyzed without storage.Results
The SIL LC-MS peptide analysis method utilizing PRM was validated and applied to quantitatively determine key RAS and KKS metabolites. COVID-19 patient serum samples, calibration and internal standards were isotopically labeled using the dimethylation protocol (Scheme 1 and Fig. S1†). Calibration standards and COVID-19 samples were labeled using light-formaldehyde while internal standards were labeled with deutero-formaldehyde. Optimized reaction conditions ensured that both formaldehyde- and D2-formaldehyde-labeled isoforms of each peptide were successfully generated. No peaks were found in full scan MS-detected chromatograms of any of the labeled peptides that would indicate side products or unreacted peptides when the reducing agent was added immediately after 3 min reaction with formaldehyde, and reduction was done for 1.5 h (Fig. 1 and ESI Fig. S2, S3†). Labeling efficiency of 98 ± 2% was estimated by comparing full scan chromatograms and mass spectra of samples before and after labeling, as discussed below. Fig. 1 shows a single full MS scan chromatographic peak for each of 3 labeled peptides. A full scan chromatogram of the unlabeled complete peptide mixture showed seven major peaks (Fig. S2A†), representing the unlabeled peptides while a similar scan of the labeled sample also shows seven major peaks for formaldehyde-labeled peptides with similar retention times (Fig. S2B†). No other peaks were observed apart from the labeled peptide peaks. Fig. S3† shows similar full scan LCs of those labeled peptides not shown in Fig. 1. Fig. S4–S10† show mass spectra of each of the major peaks in the chromatogram with m/z ratios for the z = 1, 2 and 3 (in some cases) ions. m/z differences between D2-formaldsehyde- and formaldehyde-labeled peptides are consistent with the differences in mass of the labels and indicate again that all starting peptides have been quantitatively labeled.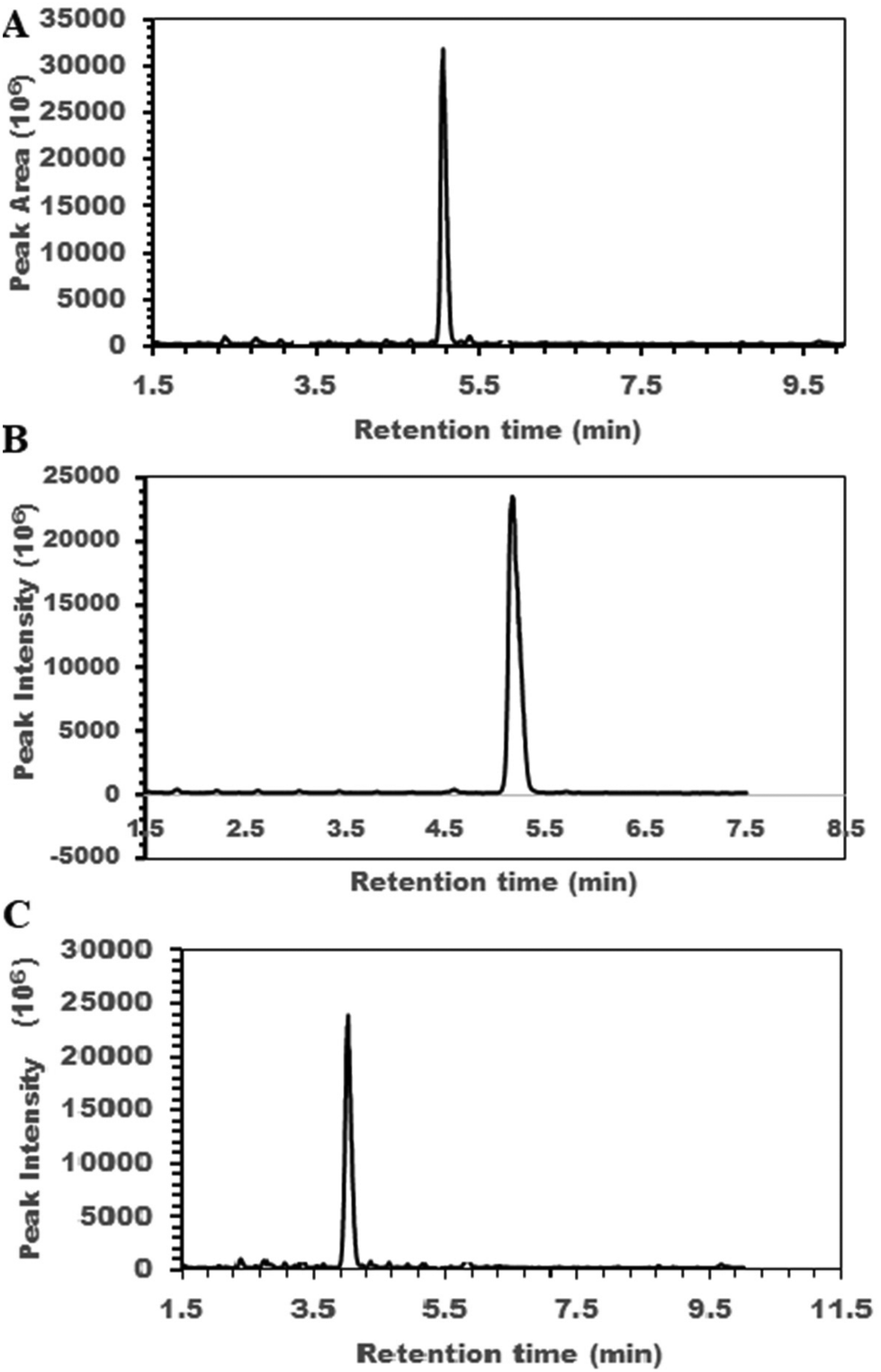 | ||
| Fig. 1 LC chromatograms of 3 of the labeled peptides obtained using full MS1 scan, consistent with successful quantitative labeling of peptides with no side products. Chromatograms of (A) formaldehyde-reacted Brad; (B) formaldehyde-reacted des-R9-Brad; and (C) formaldehyde-reacted Brad 1–7. Also see ESI Fig. S2–S10.† | ||
Optimized LC-MS provided sufficiently resolved chromatograms for all target metabolites (Fig. 2). Both light and medium labeled versions of each peptide had the same chromatographic retention times (inset in Fig. 2), a crucial requirement for precise and accurate quantitation.
 | ||
| Fig. 2 Chromatographic elution profiles of peptide analytes. Both formaldehyde and D2-formaldehyde labeled versions of each analyte have nearly identical retention times (inset is an example of formaldehyde and D2-formaldehyde labeled kinins with approximately the same retention times) on the C18 column. Chromatograms were obtained using a full MS1 scan. | ||
Linearity, accuracy, precision (intra- and inter-day) and sensitivity
Calibration curves were constructed for each labeled metabolite (Fig. 3 and Fig. S11†) in 0.02% human serum to assess linearity. Best fits were estimated using linear regression with standard concentrations from low pg mL−1 to 3000 pg mL−1 for all analytes except Ang 1 whose upper range extends to 5000 pg mL−1. Each analyte gave a linear plot with correlation coefficients r > 0.99. Lower limits of detection (LOD) ranging from 1.7 pg mL−1 to 67 pg mL−1 for all the different peptides (Table 1) were obtained.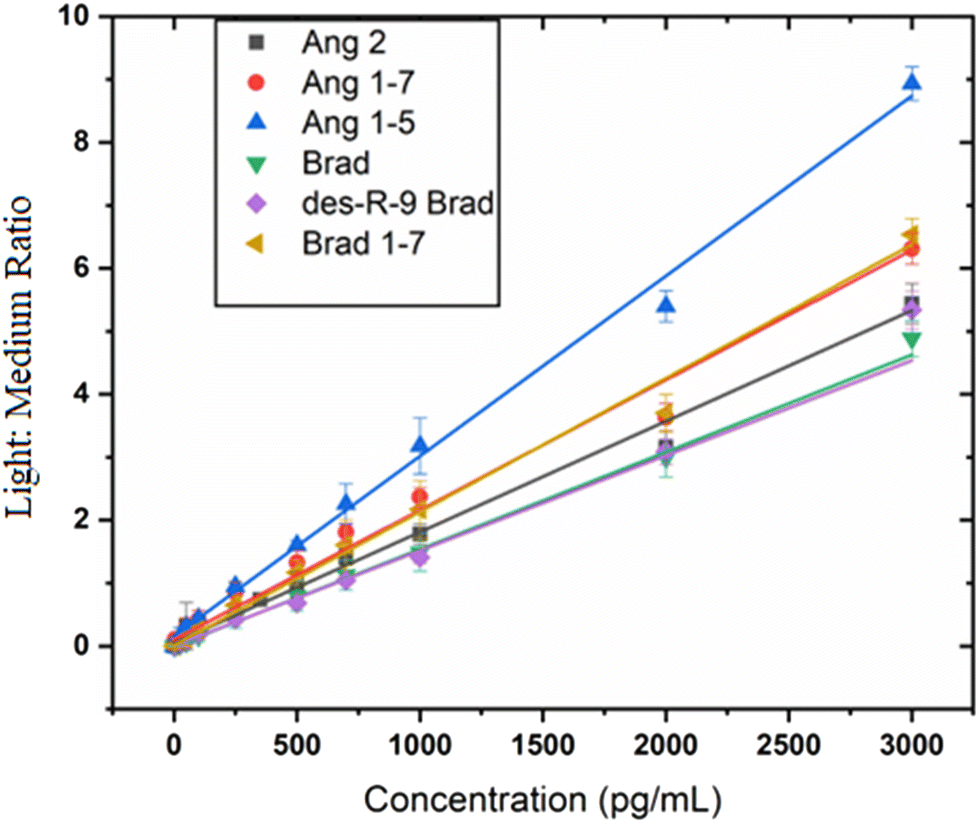 | ||
| Fig. 3 Calibration curves using chromatographic peak area ratio of sample/deuterated standard showing linear response for each analyte. Analyte detection and quantitation was done using parallel reaction monitoring (PRM). | ||
| Peptide | Peptide sequence | r-Value | LOQ, pg mL−1 |
|---|---|---|---|
| Ang 1 | DRVYIHPFHL | 0.9954 | 200.0 |
| Ang 2 | DRVYIHPF | 0.9977 | 233.0 |
| Ang 1–7 | DRVYIHP | 0.9981 | 44.0 |
| Ang 1–5 | DRVYI | 0.9977 | 57.0 |
| Brad | RPPGFSPFR | 0.9959 | 40.0 |
| Des-R9-Brad | RPPGFSPF | 0.9976 | 5.3 |
| Brad 1–7 | RPPGFSP | 0.9992 | 5.7 |
Spike-recovery studies
Intra- and inter-day accuracy and precision were estimated using pooled human serum spiked with three or four different concentrations of analyte, (depending on LOD of analyte) in triplicate. The intra-day accuracy and precision were within ±20% as stipulated for concentrations at the lower limit of quantitation (LLOQs), and ±15% for quality control samples (QCs) at all other concentrations. Accuracy and precision were ≤±15%, in compliance with the US FDA guidelines.26 Individual accuracy and precision values are shown in ESI Table S2.† Solvent blanks run after the most concentrated external standard showed no signals for the analytes, indicating no carryover of analytes to successive runs.Stability studies indicated that short-term storage of the native peptide samples at −20 °C and labeled peptides at autosampler temperature of 4 °C is appropriate. The stability in the autosampler was tested on labeled samples after extraction, and showed no effects of running long autosampler queues on the stability of the labeled analytes. Signal intensities of freshly prepared samples and those stored at −20 °C for 16 days were almost identical (ESI Fig. S12†). Also, analyte signals remained stable (ESI Fig. S13 and S14†) during storage at the autosampler temperature of 4 °C for at least three days.
COVID-19 samples
Serum samples from COVID-19 patients were analyzed, and averages for the analyte peptides were compared to averages obtained for 3 different batches of pooled human serum from healthy donors as surrogate controls. Age and sex matching of samples from surrogate healthy donors with those from COVID patients was not possible as the healthy samples were obtained from commercial sources. Results expressed in box and whisker plots indicate that RAS and KKS peptides in the 80 COVID-19 patients vary widely in concentration (Fig. 4). Indicated by low p-values except for Ang 1–7 (Table 2), average peptide concentrations in healthy controls differ significantly from values obtained in COVID-19 patients. All RAS peptides were downregulated in COVID-19 relative to the surrogate controls while the KKS counterparts were upregulated on average. | ||
| Fig. 4 Box and Whisker plots showing the distributions of mean peptide concentrations in non-COVID pooled human sera (blue) and serum from 80 COVID-19 patients (dark red). A. Normal and COVID-19 serum levels of KKS peptides. B. Normal and COVID-19 serum levels of RAS peptides. | ||
| Analyte | Average ± SD in healthy subjects (ng mL−1) | Average ± SD in COVID patients (ng mL−1) | Fold change (%) | p-Value |
|---|---|---|---|---|
| Ang 1 | 8.8 (±1.7) | 4.50 (±2.5) | −95.6 | 0.04 |
| Ang 2 | 1.33 (±0.3) | 0.38 (±0.2) | −71.7 | 0.03 |
| Ang 1–7 | 0.47 (±0.13) | 0.24 (±0.16) | −96.7 | 0.08 |
| Ang 1–5 | 0.68 (±0.12) | 0.20 (±0.28) | −71.2 | 0.01 |
| Brad | 0.20 (±0.001) | 1.73 (±1.3) | 756.4 | <0.001 |
| Brad 1–8 | 3.51 (±0.5) | 5.4 (±1.9) | 53.8 | 0.004 |
| Brad 1–7 | 0.07 (±0.02) | 0.28 (±0.1) | 308.9 | <0.001 |
Discussion
SIL was successfully applied to LC-MS analyses of key RAS and KKS metabolites in COVID-19 patient serum (Fig. 4). LODs were in the 1.6–13.2 pg mL−1 range for all but Ang1 and Ang2 which were 60 and 67 pg mL−1 (Table 1), respectively, sufficiently low to detect these peptides in normal patient blood serum. Accuracy and precision are better than ±15%, at peptide levels above the limits of quantitation. Wide dynamic ranges obtained (Fig. 3) are also important for detecting varied levels of RAS and KKS metabolites in other biofluids.27,28 The method utilizes low-cost commercially available reagents to rapidly generate isotopic labels in serum samples and internal standards in situ, at a reagent cost of ∼$1 per sample. This is the first assay to our knowledge designed for multiplexed endogenous peptide determinations by SIL-LC-MS in human biofluids. The method utilizes relatively small sample volumes and was successfully applied to an initial investigation of these two sister peptide metabolite systems in patients with COVID-19 infections (Fig. 4).Side-reactions in formaldehyde-based SIL, with products described as N-methyl-4-imidazolidinone moieties, have been recently reported for reductive dimethylation of peptides using formaldehyde and sodium cyanoborohydride.29 They form by slow intramolecular nucleophilic addition, and can influence accuracy and precision of subsequent quantitative LC-MS. We optimized SIL reaction conditions to enable complete dimethylation of each free amine in the peptides with no detectable side-products, intermediates, or unlabeled metabolites (Fig. 1, Fig. S2–S10†). Reducing agent added after the formaldehyde and briefly mixed with the sample blocks side-reaction pathways and enables rapid reduction of the Schiff's base. Excess amounts of reagents coupled with amine-free buffer ensure a complete, efficient reaction. We observed as reported previously29 that reductive amination is pH sensitive and occurs at lower pHs without side-product formation. We thus utilized sodium acetate buffer pH 5.5 to provide optimum pH.
SIL forms of each peptide analyte and standard have nearly identical retention times, ensuring accurate and precise quantitation. This is attributable to three factors: (i) the small size of the SIL agent relative to the peptides, (ii) the relatively small number of deuterium atoms in each standard label, and (iii) the nearness of the deuterium isotopes to a hydrophilic site in the peptide.30 Methyl groups bearing two or no deuterium atoms are too small to significantly influence the retention time of peptides with at least 5 amino acids. In addition, the peptides are derivatized at only one polar site which naturally has little hydrophobic interaction with the reversed-phase stationary phase. The fewer the labeling sites, the smaller the isotope effect on LC retention time.
Linear calibration curves were obtained with correlation coefficients greater than >0.99 for each analyte (Fig. 3). Internal standards were spiked into samples prior to labeling of the sample to minimize variations between samples, and loss of peptides. Once labeled with deutero-formaldehyde, the internal standard was no longer reactive towards formaldehyde. Therefore, internal standard was spiked into the samples prior to their derivatization with formaldehyde. Preliminary results underscored the importance of this, as calibrations curves generated using internal standards spiked at latter stages of sample preparation were not linear.
The sensitivities of the new SIL method are comparable to sensitivities of a recently reported method to determine kinin peptides27 but are far better than sensitivities reported for other studies.28,31 Spike-recovery results were within the acceptable 85%–115% range except for Ang 2 at low level of 50 pg mL−1 and Brad at 800 pg mL−1. Carry-over in the LC was minimal, and analytes were stable for short-term storage at −20 °C and in the autosampler at 4 °C.
We found a decrease in average levels of all RAS peptides in serum from COVID-19 patients compared to levels in pooled human sera from healthy donors, while all KKS peptides in COVID-19 patients were upregulated from these controls. Thus, dysregulation of RAS and KKS peptides may play a role in the pathogenesis of COVID-19. Other studies have investigated RAS and KKS peptides separately.32–34 Our findings agreed with earlier downregulations reported for RAS peptides32 in COVID-19, though our values are higher than previously reported. Similar observations were made by Martins et al. but in their system, Ang 1 concentrations remained unchanged while Ang 1–7 levels were elevated in plasma samples from critically ill COVID-19 patients.35 The differences in analyte concentrations reported by different research groups may be due to such factors as the geographical origin of the samples and the specific strain of the SARS-CoV-2 virus, among others. However, serum levels of some RAS peptides were also reported to be unchanged in COVID patients33,34 and differ from other reports that suggested an upregulation of some of the RAS peptides and downregulation of others.36,37
Our results show an increase in averages of all KKS peptides in COVID-19 patients relative to controls, which had not previously been reported for COVID-19. Bradykinin is known to induce vasodilation and vascular permeability.39 Its increase in COVID-19 may be responsible for the difficulty in gas exchange experienced by critically ill patients as more fluids leak into their lungs than needed. The current results on the KKS agree with the observations made by Garvin et al.38 about the upregulation of KKS components necessary to produce Bradykinin. Their study on fluids and cells from the lungs of COVID-19 patients observed an increase in kininogen and kallikreins that are required to produce Brad. Additionally, Garvin et al. reported that angiotensin converting enzyme (ACE) levels were lower in bronchoalveolar fluids (BALF) from COVID-19 patients. This decrease in ACE further explains our observed accumulation of Brad in serum samples from COVID-19 patients. It is also expected that the upsurge in Brad during SARS-CoV-2 infection would lead to the activation of the KKS receptors, especially the BDKRB2. Stimulation of this receptor leads to inflammation,39 which further induces the release of des-R9-Brad and its receptor, BDKRB1.40 The increased release of these two KKS receptors in the BALF of COVID-19 patients has been reported.38 Furthermore, Garvin et al. reported that angiotensin converting enzyme 2 (ACE2) was over expressed in COVID, which may explain the elevated concentrations of Brad 1–7 we observed in the serum samples from COVID-19 patients.
It was recently reported that des-R9-Brad and Brad 1–7 levels in plasma and bronchoalveolar lavage fluids of COVID-19 patients were significantly higher, but with lower Brad levels compared to baseline concentrations.41–43 This slight difference in Brad regulation in the current and earlier studies is likely to be caused by the difference in the biofluids analyzed. While our preliminary analysis suggests that dysregulation of both RAS and KSS may be involved in pathogenesis of COVID-19, further data analyses related to disease severity, and studies of additional patient cohorts are needed before a definitive conclusion can be reached.
In summary, our results demonstrate an efficient SIL LC-MS method for quantitative, multiplexed analysis of RAS and KSS peptides at levels applicable to human serum. This cost-efficient strategy can be utilized for absolute quantitation without the common problems previously reported for dimethyl labeling and without laborious synthesis and purification of isotopic standards. This quantitative SIL method detects peptides in the low pg mL−1 to high pg mL−1 ranges with excellent precision and accuracy. Its use can contribute to valuable future insights into metabolic alterations of peptide families that may occur in patients with different diseases, providing improved understanding of pathogenesis and biomarker guides for targeted therapies.
Author contributions
The project plan was conceived by Richard B. Kremer and James F. Rusling while Ben K. Ahiadu and Thomas Ellis developed the LC-MS/MS method with technical guidance and contributions from Adam Graichen. COVID data were collected and analyzed by Ben Ahiadu under the supervision of James F. Rusling. The manuscript was written by Ben Ahiadu and James Rusling while Richard B. Kremer contributed to the revisions of the text and supplied the COVID-19 patient samples.Ethical statement
All experiments were performed in accordance with Canadian Guidelines stated in “regulatory framework in health research at the McGill University Health Center” and approved by the ethics committee of the “McGill University Health Center”. Informed consents were obtained from human participants of this study.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful for funding from and the University of Connecticut Research Excellence Program and the Paul Krenicki Professorship. We thank Dr Catalin Mihalcioiu from the McGill UHC for providing access to his biorepository.References
- R. D. Grange, J. P. Thompson and D. G. Lambert, Br. J. Anaesth., 2014, 112(2), 213–216 CrossRef CAS PubMed.
- G. W. Boyd and W. S. Peart, in Handbook of Experimental Pharmacology, ed. I. H. Page and F. M. Bumpus, Springer, Berlin, Heidelberg, 1974, p. 37, DOI:10.1007/978-3-642-65600-2_11.
- X. Wang, L. Cohen, J. Wang and D. R. Walt, J. Am. Chem. Soc., 2018, 140(51), 18132–18139 CrossRef CAS PubMed.
- Y. Li, G. Zhang, X. Mao, S. Yang, K. De Ruyck and Y. Wu, TrAC, Trends Anal. Chem., 2018, 103, 198–208 CrossRef CAS.
- P. Chen, M. T. Chung, W. McHugh, R. Nidetz, Y. Li, J. Fu, T. T. Cornell, T. P. Shanley and K. Kurabayashi, ACS Nano, 2015, 9(4), 4173–4181 CrossRef CAS PubMed.
- S. Zhang (Weihua) and W. Jian, Rev. Anal. Chem., 2014, 33(1), 31–47 Search PubMed.
- C. Dass, G. H. Fridland, P. W. Tinsley, J. T. Killmar and D. M. Desiderio, Int. J. Pept. Protein Res., 1989, 34(2), 81–87 CrossRef CAS PubMed.
- J. R. Barr, V. L. Maggio, D. G. Patterson, G. R. Cooper, L. O. Henderson, W. E. Turner, S. J. Smith, W. H. Hannon, L. L. Needham and E. J. Sampson, Clin. Chem., 1996, 42(10), 1676–1682 CrossRef CAS.
- E. Ciccimaro and I. A. Blair, Bioanalysis, 2010, 2(2), 311–341 CrossRef CAS PubMed.
- J.-L. Hsu, S.-Y. Huang, N.-H. Chow and S.-H. C. Chen, Anal. Chem., 2003, 75(24), 6843–6852 CrossRef CAS PubMed.
- A. C. Tolonen and W. Haas, JoVE, 2014, 89, 51416 Search PubMed.
- P. J. Boersema, R. Raijmakers, S. Lemeer, S. Mohammed and A. J. R. Heck, Nat. Protoc., 2009, 4(4), 484–494 CrossRef CAS PubMed.
- A. C. Tolonen, W. Haas, A. C. Chilaka, J. Aach, S. P. Gygi and G. M. Church, Mol. Syst. Biol., 2011, 7(1), 461 CrossRef PubMed.
- P. J. Boersema, T. T. Aye, T. A. B. van Veen, A. J. R. Heck and S. Mohammed, Proteomics, 2008, 8(22), 4624–4632 CrossRef CAS PubMed.
- F. Pailleux and F. Beaudry, Biomed. Chromatogr., 2012, 26(8), 881–891 CrossRef CAS PubMed.
- A. C. Peterson, J. D. Russell, D. J. Bailey, M. S. Westphall and J. J. Coon, Mol. Cell. Proteomics, 2012, 11(11), 1475–1488 CrossRef PubMed.
- N. Rauniyar, Int. J. Mol. Sci., 2015, 16(12), 28566–28581 CrossRef CAS PubMed.
- G. E. Ronsein, N. Pamir, P. D. von Haller, D. S. Kim, M. N. Oda, G. P. Jarvik, T. Vaisar and J. W. Heinecke, J. Proteomics, 2015, 113, 388–399 CrossRef CAS PubMed.
- E. Kashuba, J. Bailey, D. Allsup and L. Cawkwell, Biomarkers, 2013, 18(4), 279–296 Search PubMed.
- F. S. de Miranda, J. P. T. Guimarães, K. R. Menikdiwela, B. Mabry, R. Dhakal, R. l. Rahman, H. Moussa and N. Moustaid-Moussa, Mol. Cell. Endocrinol., 2021, 528, 111245 CrossRef CAS PubMed.
- H. Jia, Shock, 2016, 46(3), 239–248 CrossRef CAS PubMed.
- K. Deepak, P. K. Roy, P. Kola, B. Mukherjee and M. Mandal, Biochim. Biophys. Acta, Rev. Cancer, 2022, 1877(6), 188807 CrossRef CAS PubMed.
- M. Pahlavani, N. S. Kalupahana, L. Ramalingam and N. Moustaid-Moussa, in Comprehensive Physiology, ed. R. Terjung, Wiley, 2017, pp. 1137–1150 Search PubMed.
- Physiology, Renin Angiotensin System - StatPearls - NCBI Bookshelf.Html. https://www.ncbi.nlm.nih.gov/pubmed/29261862, last accessed 9 June 2023.
- S. L. Cooper, E. Boyle, S. R. Jefferson, C. R. A. Heslop, P. Mohan, G. G. J. Mohanraj, H. A. Sidow, R. C. P. Tan, S. J. Hill and J. Woolard, Int. J. Mol. Sci., 2021, 22(15), 8255 CrossRef CAS PubMed.
- U.S. Food and Drug Administration, Bioanalytical Method Validation. Guidance for Industry, 2018, https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf Search PubMed.
- T. Gangnus and B. B. Burckhardt, Sci. Rep., 2021, 11(1), 3061 CrossRef CAS PubMed.
- I. van den Broek, R. W. Sparidans, J. H. M. Schellens and J. H. Beijnen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878(5–6), 590–602 CrossRef CAS PubMed.
- R. Liao, Y. Gao, M. Chen, L. Li and X. Hu, Anal. Chem., 2018, 90(22), 13533–13540 CrossRef CAS PubMed.
- R. Zhang, C. S. Sioma, R. A. Thompson, L. Xiong and F. E. Regnier, Anal. Chem., 2002, 74(15), 3662–3669 CrossRef CAS PubMed.
- E. Baralla, M. Nieddu, G. Boatto, M. V. Varoni, D. Palomba, M. P. Demontis, V. Pasciu and V. Anania, J. Pharm. Biomed. Anal., 2011, 54(3), 557–561 CrossRef CAS PubMed.
- A. Kutz, A. Conen, C. Gregoriano, S. Haubitz, D. Koch, O. Domenig, L. Bernasconi, B. Mueller and P. Schuetz, Eur. J. Endocrinol., 2021, 184(4), 543–552 CAS.
- M. Rieder, L. Wirth, L. Pollmeier, M. Jeserich, I. Goller, N. Baldus, B. Schmid, H.-J. Busch, M. Hofmann, W. Kern, C. Bode, D. Duerschmied and A. Lother, Am. J. Hypertens., 2021, 34(3), 278–281 CrossRef CAS PubMed.
- U. Kintscher, A. Slagman, O. Domenig, R. Röhle, F. Konietschke, M. Poglitsch and M. Möckel, Hypertension, 2020, 76(5), e34–e36 CrossRef CAS PubMed.
- A. L. V. Martins, F. A. da Silva, L. Bolais-Ramos, G. C. de Oliveira, R. C. Ribeiro, D. A. A. Pereira, F. Annoni, M. M. L. Diniz, T. G. F. Silva, B. Zivianni, A. C. Cardoso, J. C. Martins, D. Motta-Santos, M. J. Campagnole-Santos, F. S. Taccone, T. Verano-Braga and R. A. S. Santos, ERJ Open Res., 2021, 7, 00114–2021 Search PubMed.
- D. C. Files, K. W. Gibbs, C. L. Schaich, S. P. Collins, T. M. Gwathmey, J. D. Casey, W. H. Self and M. C. Chappell, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2021, 321(1), L213–L218 CrossRef CAS PubMed.
- D. van Lier, M. Kox, K. Santos, H. van der Hoeven, J. Pillay and P. Pickkers, ERJ Open Res., 2021, 7(1), 00848–02020 Search PubMed.
- M. R. Garvin, C. Alvarez, J. I. Miller, E. T. Prates, A. M. Walker, B. K. Amos, A. E. Mast, A. Justice, B. Aronow and D. A. Jacobson, eLife, 2020, 9, e59177 CrossRef CAS PubMed.
- D. A. B. Rex, N. Vaid, K. Deepak, S. Dagamajalu and T. S. K. Prasad, Mol. Biol. Rep., 2022, 49(10), 9915–9927 CrossRef CAS PubMed.
- E. Kashuba, J. Bailey, D. Allsup and L. Cawkwell, Biomarkers, 2013, 18(4), 279–296 Search PubMed.
- G. M. d. M. Mendes, I. J. B. Do Nascimento, P. H. S. Marazzi-Diniz, I. B. Da Silveira, M. F. Itaborahy, L. E. Viana, F. A. Silva, M. F. Santana, R. A. A. Pinto, B. G. Dutra, M. V. G. Lacerda, S. A. Araujo, D. Wanderley, P. V. T. Vidigal, P. H. Diniz, T. Verano-Braga, R. A. S. Santos and M. F. Leite, Front. Physiol., 2022, 13, 1080837 Search PubMed.
- C. P. Martens, P. Van Mol, J. Wauters, E. Wauters, T. Gangnus, B. Noppen, H. Callewaert, J. H. M. Feyen, L. Liesenborghs, E. Heylen, S. Jansen, L. C. V. Pereira, S. Kraisin, I. Guler, M. M. Engelen, A. Ockerman, A. Van Herck, R. Vos, C. Vandenbriele, P. Meersseman, G. Hermans, A. Wilmer, K. Martinod, B. B. Burckhardt, M. Vanhove, M. Jacquemin, P. Verhamme, J. Neyts and T. Vanassche, eBioMedicine, 2022, 83, 104195 Search PubMed.
- E. Alfaro, E. Díaz-García, S. García-Tovar, E. Zamarrón, A. Mangas, R. Galera, K. Nanwani-Nanwani, R. Pérez-de-Diego, E. López-Collazo, F. García-Río and C. Cubillos-Zapata, Front. Immunol., 2022, 13, 909342 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an00943b |
| This journal is © The Royal Society of Chemistry 2023 |