
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ infrared imaging of the local orientation of cellulose fibrils in plant secondary cell walls†
Alexander
Veber
 ab,
Victor M. R.
Zancajo
a,
Ljiljana
Puskar
b,
Ulrich
Schade
b and
Janina
Kneipp
*a
ab,
Victor M. R.
Zancajo
a,
Ljiljana
Puskar
b,
Ulrich
Schade
b and
Janina
Kneipp
*a
aDepartment of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, 12489 Berlin, Germany. E-mail: janina.kneipp@chemie.hu-berlin.de
bInstitute for Electronic Structure Dynamics, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, Albert-Einstein-Straße 15, 12489 Berlin, Germany
First published on 20th July 2023
Abstract
The mechanical and chemical properties of plant cell walls greatly rely on the supramolecular assembly of cellulose fibrils. To study the local orientation of cellulose in secondary plant cell walls, diffraction limited infrared (IR) micro-spectroscopic mapping experiments were conducted at different orientation of transverse leaf section of the grass Sorghum bicolor with respect to the polarization direction of the IR radiation. Two-dimensional maps, based on polarization-sensitive absorption bands of cellulose were obtained for different polarization angles. They reveal a significant degree of anisotropy of the cellulose macromolecules as well as of other biopolymers in sclerenchyma and xylem regions of the cross section. Quantification of the signals assigned to polarization sensitive vibrational modes allowed to determine the preferential orientation of the sub-micron cellulose fibrils in single cell walls. A sample of crystalline nano-cellulose comprising both a single microcrystal as well as unordered layers of nanocrystals was used for validation of the approach. The results demonstrate that diffraction limited IR micro-spectroscopy can be used to study hierarchically structured materials with complex anisotropic behavior.
Introduction
Knowledge about the composition, structure, and biosynthesis of the cell walls of higher plants could offer solutions to a diverse range of problems of our time, from creating new types of composite materials1 over engineering healthier plants2 to better understanding of the climate change.3 Cell walls of plants possess unique mechanical and chemical properties, owing to a supramolecular, composite arrangement that is determined by the hierarchical assembly of cellulose fibrils and their interaction with hemicelluloses and pectin, as well as with lignin, peptides, and proteins.4–6 The structure and composition of the cell wall can be related to its physiological function, its dynamics during cell growth,7–10 and its mechanical stability. Effects on stability are caused directly by the molecular and supramolecular structure and interaction,11,12 but can also be the consequence of distinct biochemical remodeling based on signaling events13 or by facilitating biomineralization.14,15 Cellulose crystallinity greatly determines interaction with the other molecular constituents, especially in secondary cell walls.16,17 Information on the orientation of cellulose crystals in the plant material, also in situ in cell walls, can be retrieved by birefringence techniques,18 and to even greater detail by vibrational spectroscopies that can reveal anisotropy in supramolecular structure and orientation. Raman spectroscopy, when using signals of cellulose with a small depolarization ratio, reveals the orientation of purified cellulose fibers,19,20 of cellulose in wood,21,22 and in herbaceous plants such as equisetum23 and rice.24 The hierarchical organization of highly oriented cellulose crystals into microfibrils has also been addressed by probing the electronic anisotropy of cellulose fibrils using higher order polarizability in coherent anti-Stokes Raman scattering (CARS),25,26 as well as by vibrational sum frequency generation (SFG) spectroscopy27 and imaging by second harmonic generation (SHG) signals that ideally complement vibrational spectroscopic data.25,28The potential of infrared (IR) spectroscopy to indicate cellulose orientation was shown already in the first IR absorption experiments with flax fibers at defined polarization 65 years ago,29 and translates to IR measurements at higher lateral resolution when studying purified cellulose30,31 or the structure–function relationships of cellulose in plant tissues using FTIR microscopes.32–34 Compared to spontaneous Raman spectroscopy, IR absorption is found to be very sensitive and robust with respect to electronic resonances that often lead to selective signal enhancement, photobleaching, and/or excitation of autofluorescence35 or to thermal degradation of plant cell materials when excitation lasers in the visible region are used.36 The absence of selectivity caused by electronic resonances enables an unbiased probing of all IR active vibrations. Moreover, the origination of the IR absorption bands from dipole moment changes simplifies the analysis of the spectra and the quantification of the results, in particular in the case of anisotropy studies.37 These, among other advantages make FTIR microscopy very attractive for studies of cellulose orientation in situ in the cell walls of plants.
Plant tissues contain substructures and vessels of sizes that match the (diffraction-limited) spatial resolution of typical mid-IR microscopy experiments. When a high brightness broadband synchrotron is used as the IR light source, such diffraction limited IR microscopic mapping experiments can be done in reasonable periods of time38–40 and at highly defined polarization. Here, we use a synchrotron coupled FTIR microscope to investigate in situ cellulose and other biopolymers in plant cell walls of sorghum leaves by polarization-resolved chemical imaging.
The properties and functionality of individual cell walls are intimately related to both content and organization of different cell wall components, in particular orientation and packing of the cellulose microfibrils. We have previously studied the composition of secondary cell walls of different tissues of sorghum by IR and Raman micro-spectroscopies and one- and two-photon fluorescence, specifically with respect to lignin structure and composition,28,41 and analyzed cellulose microfibril orientation in these cells by SHG microscopy.28 As will be discussed later, in addition to the chemical composition and structure the polarization resolved IR imaging data reveal the organization of the cellulose in the cell walls in situ. Specifically, in the transverse sections studied here at diffraction limited spatial scale, the anisotropy can be traced down to individual cell walls in the sclerenchyma and xylem regions. The IR micro-spectroscopy has previously been used for chemical imaging of plant samples,40–42 however, the anisotropic measurements in plants are usually done either for longitudinal sections43,44 of the cells and/or averaging the information over rather large areas,33i.e. over many or several cells.
Materials and methods
Sample preparation
To study orientation of cellulose fibrils in plants secondary walls, Sorghum bicolor (L.) Moench plants (wild type; line BT × 623) were grown hydroponically for 9 weeks after germination using Hoagland solution. The leaves were dissected mechanically. After chemical fixation of the tissues with FAA a mixture of formaldehyde, ethanol, acetic acid, and water in a ratio of 10%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50%:5%:35% v/v, respectively, gradual dehydration in an ethanol series, and a following paraffin embedding, histological sections of 5 μm thickness were prepared with a microtome. The sections were mounted on 0.5 mm thick CaF2 windows and inspected using a Zeiss Axio Imager. A2m optical microscope (Carl Zeiss Microscopy, Germany) to determine the quality of the cut. A single section, which preserved the best morphology of the plant was selected for the investigation and all the experimental data related were collected from this plant section. The plant section was de-paraffinized with hexane just before IR analysis. A layer of crystalline nano-cellulose (NAVITAS, Slovenia) was drop casted on a 0.5 mm CaF2 window and was used as a reference sample for pure cellulose.
:50%:5%:35% v/v, respectively, gradual dehydration in an ethanol series, and a following paraffin embedding, histological sections of 5 μm thickness were prepared with a microtome. The sections were mounted on 0.5 mm thick CaF2 windows and inspected using a Zeiss Axio Imager. A2m optical microscope (Carl Zeiss Microscopy, Germany) to determine the quality of the cut. A single section, which preserved the best morphology of the plant was selected for the investigation and all the experimental data related were collected from this plant section. The plant section was de-paraffinized with hexane just before IR analysis. A layer of crystalline nano-cellulose (NAVITAS, Slovenia) was drop casted on a 0.5 mm CaF2 window and was used as a reference sample for pure cellulose.
FTIR microscopy experiments
The IR microscopy end-station at IRIS beamline, BESSY II Synchrotron, HZB Berlin was used for the experiments.45 The measurements were done in transmission mode with a confocal Continuμm FTIR microscope (Thermo Scientific Nicolet, Waltham, MA, USA) using a pair of 32× Schwarzschild objectives (N.A. 0.65). Synchrotron light was used for recording of the spectra with well-defined light polarization. Use of the in-ring plane component of the synchrotron IR radiation was ensured by placing and aligning a grid polarizer before the microscope optical entrance. Different areas of the sample were investigated (Fig. 1): (i) a wide overview area, covering different histological structures in the cross section of about 150 × 150 μm2 using an aperture size of 7 × 7 μm2, a 4 μm step size between sampled spots, and 32 interferograms co-added per spectrum; (ii) areas of more detailed measurements, using an aperture size of 5 × 5 μm2, sampling step size of 2 μm, and co-addition of 64 interferograms per spectrum in the sclerenchyma (Fig. 1, orange marking) and xylem (Fig. 1, blue marking) tissue regions of 20 × 20 μm2 and 20 × 40 μm2, respectively. The sample was rotated about the microscope optical axis, and the measurements of the same areas were done at 0°, 45°, 90°, and 135° positions. The initial angular position of the sample does not intentionally correspond to any features in the sample and was chosen arbitrary. | ||
| Fig. 1 Bright field microscope image of a transversal section of a sorghum leaf studied. The rectangles of different colors show the areas of FTIR mapping experiments that were conducted at 45° sample orientation. Red arrowheads point to the positions where single point spectra were obtained for a large set of different angles by rotation of the sample with respect to the orientation of the IR electric field (double-sided arrow in the bottom-right corner). Abbreviations: scler = sclerenchyma. | ||
Single point spectra from several positions of interest were collected in the sclerenchyma (S1–S4) and xylem (X1, X2, X3) tissue regions using a 5 × 5 μm2 aperture size and 128 co-added scans per spectrum. The locations of the single point measurement spots are shown in Fig. 1. The spectra were collected at different angular orientations of the sample: 0°, 15°, 30°, 45°, 60°, 75°, 90°, 105°, 120°, and 135°, with repeated measurements at some of the angles, yielding a total of 17 spectra for each point of interest.
The same experimental conditions were used to collect spectra of the crystalline nano-cellulose reference samples, from an area that appeared homogenous and unoriented at the microscopic scale, and from a highly oriented cellulose microcrystal of an approximate dimensions of 5 × 10 μm2, respectively. The reference spectra were collected at angles of 0°, 22.5°, 45°, 67.5°, 90°, 112.5°, and 135°.
Background spectra were taken at the same experimental conditions as were used for the corresponding samples and experiments from an empty area on the CaF2 substrate in the proximity to the sample.
Additional spectra were recorded with the light blocked at the sample position. IR light diffracted of the internal microscope aperture is observed by this configuration and this light directly reaches the detector. The diffracted light gives a significant contribution to the generally observed signal when using the aperture of 5 × 5 μm2 in the wavenumber range below 1100 cm−1 (see Fig. S1†).
In addition, reference spectra from the sclerenchyma and xylem regions as well as from nanocrystalline cellulose were collected with unpolarized light using the internal Globar IR-source and an aperture size of 20 × 20 μm2.
Atomic force microscopy
AFM imaging of individual cell-walls in the plant cross-section was done in non-contact mode with a neaSNOM AFM (attocube systems AG, Germany) using Arrow-NCPt (tip radius <25 nm) Pt/Ir-coated AFM probes (Nanoworld AG, Switzerland).Data analysis
The collected spectra were corrected for the background transmission spectra of a CaF2 window during the acquisition by OMNIC software (Thermo Scientific Nicolet, Waltham, MA, USA). The data collected using the aperture of 5 × 5 μm2 (both maps and single point data) were additionally corrected for the aperture light diffraction effect (described above) using the corresponding blocked light spectrum and a self-written script in SciLab open-source software package.Polyline baseline correction and extraction of the spectral region of interest was done using the Quasar46 software package before further quantification of the data.
Absorption of a polarized beam by a perfect polarizer obeys Malus’ law. Taking into account a finite anisotropy and absorption of the investigated sample, Malus’ law can be generalized and the absorption calculated in arbitrary units can be expressed as follows:
 | (1) |
It is important to note that A in eqn (1) is in absorption and not absorbance units. The absorption units are calculated from the transmission spectra as follows:
A = 1−T, where T ∈ [0;1] is the transmission.
Use of absorbance (Abs = −log(T)) rather than absorption would mathematically complicate the analysis of the data. Therefore, to determine the dichroic ratio and angle of preferential orientation, the collected transmission spectra were converted to relative absorption units. At the same time, the IR spectra presented in the work are in absorbance units, as specified in the individual axis titles.
The intensity of a specific peak was determined by integration of the relative absorption curve using a linear baseline with two anchor points at the ends of the integration range. The obtained dependencies of the integral intensities on the angular position were fitted using eqn (1).
Values of D and φ were determined during by the fitting of the experimental data. An adjusted R-squared (R2adj) value was used to assess the quality and the reliability of the fit.
The plotting was done in OriginPro (OriginLab, Northampton, MA, USA) software.
The Fiji47 open-source platform was used for the analysis of the microscope images, in particular to measure the local angular orientation of the cell walls and to determine their preferential directionality. Gwyddion48 open source software was used for the analysis and visualization of the AFM topography data.
Results and discussion
Overall plant tissue composition and cellulose content
IR mapping experiments were conducted on the transverse leaf tissue section of sorghum plant in transmission mode from the areas indicated in Fig. 1. The spectra obtained from the plant were compared with nanocrystalline cellulose as reference for pure cellulose. Fig. 2 shows example spectra extracted from the mapping experiments. The IR spectra of the plant vary in different tissue regions, clearly indicating differences in the chemical composition and contribution of different components to the overall absorption (Fig. 2). As expected and in agreement with our previous reports,41 the fingerprint region of the spectra of sorghum leaves shows numerous differences in compositional details of different types of carbohydrates and lignin. They differ significantly from spectra of pure cellulose (Fig. 2, upper panel, bottom trace), nevertheless cellulose significantly contributes to the spectra. The spectra shown in Fig. 2 are normalized to the band at 1160 cm−1, which is dominated by the cellulose contribution. Comparison of the area under the absorbance curves of the normalized spectra allows to estimate the cellulose contribution to the overall absorption in the region of 900–1800 cm−1, which is about 50 and 30% for xylem and sclerenchyma regions, respectively. | ||
| Fig. 2 Typical IR spectra of sclerenchyma (blue/top, upper panel) and xylem (red/middle, upper panel) regions of a sorghum leaf section as well as a spectrum of nanocrystalline cellulose (black/bottom, upper panel) and the corresponding difference spectra of the plant spectra and the reference nanocrystalline cellulose spectrum: Asclerenchyma–Ancc(blue/top, lower panel) and Axylem–Ancc (red/bottom, lower panel). The most probable assignment of the bands in the difference spectra is indicated in the peak position annotation (L, lignin, H, hemicellulose). The spectra were collected using unpolarized IR light. | ||
Contribution of the non-cellulosic components can be more clearly seen from the difference spectra between the plant and the reference nanocrystalline cellulose spectra (Fig. 2, lower panel). Bands that are not observed for nanocrystalline cellulose can be assigned to lignin,49–51 evidenced, e.g., by intense absorption bands assigned to vibrations of C–O groups at 990 cm−1, 1028 cm−1, and ∼1230 cm−1, of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups at 1140–1155 cm−1, 1230/1265 cm−1, and deformation modes of C–H groups at 1140–1155 cm−1, 1420 cm−1, or 1460 cm−1, as well as indicators of the aromatic character of the compound, such as the bands at 1510 cm−1, 1600 cm−1 and 1660 cm−1. Apart from lignin, the presence of hemicellulose is indicated by characteristic bands at 1020 cm−1, 1050 cm−1, 1110 cm−1, 1150 cm−1, 1240 cm−1, 1313 cm−1, 1380 cm−1, 1602 cm−1, and 1730 cm−1. Corresponding lignin and hemicellulose vibrational bands are marked in Fig. 2. The assignments of the cellulose, lignin and hemicellulose bands are also given in Table S1.† Additional bands can be assigned to pectin, polygalacturonic acid, and proteins.41,49
O groups at 1140–1155 cm−1, 1230/1265 cm−1, and deformation modes of C–H groups at 1140–1155 cm−1, 1420 cm−1, or 1460 cm−1, as well as indicators of the aromatic character of the compound, such as the bands at 1510 cm−1, 1600 cm−1 and 1660 cm−1. Apart from lignin, the presence of hemicellulose is indicated by characteristic bands at 1020 cm−1, 1050 cm−1, 1110 cm−1, 1150 cm−1, 1240 cm−1, 1313 cm−1, 1380 cm−1, 1602 cm−1, and 1730 cm−1. Corresponding lignin and hemicellulose vibrational bands are marked in Fig. 2. The assignments of the cellulose, lignin and hemicellulose bands are also given in Table S1.† Additional bands can be assigned to pectin, polygalacturonic acid, and proteins.41,49
It is important to note that in most polysaccharides, the skeletal vibrations involving the C–O–C glycosidic bonds appear in the wavenumber range from 1140–1160 cm−1.52 For the most part of the cross section shown in Fig. 1, the position of this peak is equal or slightly higher than 1160 cm−1, while lower values are observed mostly in the proximity of the larger vessels in the bundle sheath region (Fig. S2B†). At the same time, xylem and sclerenchyma tissue regions demonstrate the highest intensity of this band (Fig. 3A). As discussed previously,33 the peak position of this band is slightly higher for crystalline cellulose (∼1160 cm−1), in comparison to pectin (1142 cm−1), hemicellulose (1148 cm−1), and amorphous cellulose (1156 cm−1). This allows us to conclude that most of the crystalline cellulose of the investigated sorghum cross-section is present in the cell walls of xylem and sclerenchyma tissue. Considering that sorghum leaves contain up to 65% (w/w) of cellulose,53 an even higher concentration of cellulose can be expected in these tissues regions. Therefore, the characteristic sharp band at 1160 cm−1 in xylem and sclerenchyma regions must be dominated by the cellulose contribution.
 | ||
| Fig. 3 Absorption of the ∼1160 cm−1 band of cellulose in the IR spectra acquired for (A) wide, (B) sclerenchyma, and (C) xylem mapping regions of the plant at 0°, 45°, 90° and 135° anglular positions. IR light electric field is always oriented vertically (cf.Fig. 1), as it is indicated with double-sided arrows in the panels for 135° orientation. Some prominent lumina in the sclerenchyma region (B) are numbered from 1 to 5 to follow the orientation and changes upon rotation of the sample. Scale bars (A) 70 μm, (B) 10 μm, (C) 10 μm, valid for each respective mapping region (panel). The arrowhead in A points to the sclerenchyma region that is analyzed in Fig. 4. | ||
Directionality of cellulose revealed by infrared maps
The 1160 cm−1 band originating from the asymmetric C–O–C stretching vibration in the glycosidic linkage of cellulose29,54 is highly polarized along the fiber axis of cellulose. Therefore, this band was used to determine orientation of the cellulose microfibrils via anisotropy measurements, as it was previously done at the macroscopic scale.29,52,55 Chemical maps using the intensity of the C–O–C stretching vibration of the glycosidic bond of cellulose for different orientation of the sample with respect to the polarization direction of the IR light are shown in Fig. 3A–C. Due to the strong contribution of cellulose to the spectra of the xylem and sclerenchyma regions of the leaf sections, these two regions were chosen for a detailed investigation of the cellulose orientation (Fig. 3B and C). Chemical images obtained at four different angular orientations of the sample show significant variation of the 1160 cm−1 band intensity at spatial scales of >10 μm (Fig. 3A) but also for the smaller areas of ≤5 μm as it is seen in the more detailed maps of regions in sclerenchyma (Fig. 3B) and xylem (Fig. 3C).The higher absorption of those cell walls oriented vertically, along the IR radiation electric field, is clearly visible in the maps of the sclerenchyma region (Fig. 3B) that were obtained at higher spatial resolution than the overview maps (Fig. 3A). This result is in good agreement with a preferential macroscopic orientation of cellulose, and a microscopic structuring of individual cell walls in the sorghum leaf sections.28Fig. 3C shows detailed maps of the xylem region collected with a spot size of 5 μm and a step size of 2 μm. Here, individual cell walls could not be resolved. However, a decrease in absorption of the polarization sensitive cellulose mode when the sample is rotated by 90° (cf. first and third panel in Fig. 3C) is still obvious.
The typical thickness of the cell walls in the regions shown in Fig. 3B and C is 1.5 to 1.8 μm and 1.0 to 1.2 μm, respectively, and was determined from bright field microphotographs (Fig. S3†). The size of the lumina of individual cells in sclerenchyma varies between 5 μm and 12 μm (Fig. 3B, Fig. S3†) and in xylem between 2 μm and 5 μm (Fig. 3C, Fig. S3†). In the latter case the typical cell size is comparable to or less than the diffraction limited light spot size at 1160 cm−1 and therefore several cells contribute to the obtained spectra.
To better understand the origin of the anisotropy at a spatial scale that includes information form several cells (>10 μm) as shown in Fig. 3A, we analyzed the cell wall directionality in a binary map of the cell walls that we generated from the bright field image of the cross section (Fig. S3†). In the subsection of the sclerenchyma region (Fig. 3A, arrowhead), the binary map reveals that on average, there are more cell walls oriented at 0°, 40°, 80°, and 125°, whereas distribution minima are observed at 15°, 60°, 105°, 140° (Fig. 4). The distribution analysis does not consider orientation of the cellulose fibrils in the plane of the cell walls. Nevertheless, the presence of the preferential angles in the orientation of the individual cell walls will significantly influence the observed variation of the absorption obtained when probing several cells at once at the tissue level.
 | ||
| Fig. 4 The amount of cell walls in the sclerenchyma region for different angles with respect to the vertical direction that also corresponds to the orientation of the polarized IR light. The dashed lines indicate the 0°, 45°, 90° and 135° angular positions at which the mapping measurements in Fig. 3 were conducted. The binary skeleton of the sclerenchyma region at 0° position used for the analysis is shown in the figure inset. | ||
Orientation of cellulose in microscopic spots
As seen above and discussed in previous reports,56 the collection of spectra at a few different orientation angles is sufficient to determine the preferential orientation of the respective anisotropic molecule in samples with a high anisotropy ratio, in this case cellulose. The discussion of the mapping data above shows that the changes in the false color images indeed can be roughly connected to sample orientation. However, fitting of the data for single spatial points extracted from the map datasets using a sine function results in a typical error of the phase shift of ±25°. This is the case even for the rather well spatially resolved and clearly anisotropic sclerenchyma region (Fig. 3A and 4B). As already mentioned, the investigated cell walls have lengths close to the diffraction limited spatial resolution at ∼1160 cm−1, and also thicknesses comparable or smaller than the 2 μm step size used in the mapping experiments, and therefore performed raster scan experiments cannot guarantee the collection of the data from the same cell wall of interest.To increase reliability of the anisotropy analysis, spectra were collected at 10 different angular orientations of the sample, while the precise positioning was adjusted individually for each point of interest by using a bright field light microscope image of the sample.
The data collected at single spatial points for different sample orientation angles were used to estimate a dichroic ratio for cellulose based on the 1160 cm−1 vibrational band (Table 1). The strongest anisotropy is observed for the data obtained with a cellulose crystal of micron dimensions that was contained in a layer of crystalline nano-cellulose (CNC), prepared from a commercial sample as reference, exhibiting a dichroic ratio of ∼3.7 (Table 1). In contrast, spectra of the homogeneous CNC layer that was spread out on the IR transparent support change only slightly upon rotation with respect to the IR light, nevertheless a formal fit of the peak absorption results in a dichroic ratio of 1.33 (Table 1). The absorption spectra of the cellulose reference samples are shown in the ESI Fig. S4.†
| Reference | Sclerenchyma | Xylem | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MCa | CNCa | S1 | S2 | S3 | S4 | X1 | X2 | X3 | |
| a Abbreviations MC and CNC correspond to the reference data obtained for cellulose microcrystal and homogeneous crystalline nano-cellulose layer, respectively. b φ−90 OM value corresponds to the direction of the long axis of the crystal. | |||||||||
| D | 3.66 ± 0.29 | 1.33 ± 0.09 | 1.36 ± 0.08 | 1.81 ± 0.13 | 1.12 ± 0.03 | 1.1 ± 0.04 | 1.28 ± 0.04 | 1.35 ± 0.04 | 1.30 ± 0.03 |
| φ−90 IR | 112 ± 1.4 | 106 ± 8 | 105 ± 6 | 46 ± 3 | 25 ± 6 | 160 ± 10 | 30 ± 5 | 24 ± 5 | 47 ± 3 |
| R 2 adj | 0.99 | 0.72 | 0.65 | 0.84 | 0.63 | 0.20 | 0.77 | 0.89 | 0.90 |
| φ−90 OM | 111b | N.a. | 112 | 53 | 30 | 115 | 165 | 30 | 155 |
The anisotropy found in the two reference samples can be compared to that observed in the spectra of cell walls in sclerenchyma and xylem (Table 1). The highest anisotropy is observed for the measurement point S2 in the sclerenchyma region: for this point the dichroic ratio is significantly higher than that of the unoriented CNC reference and the phase shift value error is rather small (cf.Fig. 1, Table 1). The corresponding absorption spectra with the orientation dependence of the relative absorption at 1160 cm−1 for this measurement point of the cell wall are shown in Fig. 5A. The dichroic ratios obtained for other positions in the plant tissue do not differ significantly or are even lower than the value of the unordered sample of the CNC reference (Table 1). Nevertheless, the quality of the fits differs for the different tissue positions and samples. As an example, the dichroic ratio at position X2 is only slightly higher than that of the unoriented reference (Table 1), but the corresponding fit quality is high and results in smaller errors of all the determined parameters in comparison to the case of the nano-cellulose layer. The absorption spectra and the dependence of the absorption at 1160 cm−1 for position X2 in the xylem region are shown in Fig. 5B.
 | ||
| Fig. 5 IR absorption spectra with maximal and minimal intensity of the 1160 cm−1 band acquired at: (A) position S2 in sclerenchyma tissue and (B) position X2 in the xylem region of the leaf cross section. The insets show the corresponding dependencies of the relative absorption of the band as function of the orientation (experimental data and the fit). | ||
It is important to note that for both example positions, S2 and X2, the maximum absorption at 1160 cm−1 is observed when the polarization of the incident light is close to the corresponding cell wall direction (Fig. 5, insets). It is known that the cellulose fibrils lay in the plane of the cell wall,57 and their orientation has been experimentally studied by different techniques.58 The data shown here are from a cross section that is cut perpendicular to the direction of the cell growth. In this geometry, the projection of the cellulose chains onto the sample cut plane should be preferably oriented along the visible cell walls, which agrees with the observation reported here.
In the experimental geometry used here, the orientation of the microfibrils in the cell wall plane with respect to the long (growth) axis of the cells cannot be observed directly. Nevertheless, the microfibril orientation influences the determined dichroic ratio: a maximum anisotropy and isotropic behaviour will be observed when the microfibrils are oriented perpendicular to and along the cell wall growth axis, respectively.
In the case of the infrared spectroscopy linear dichroism measurement, the dichroism is related to the distribution of polymer chains in three dimensions with different orientation angle θ with respect to a reference axis by the following equation:59
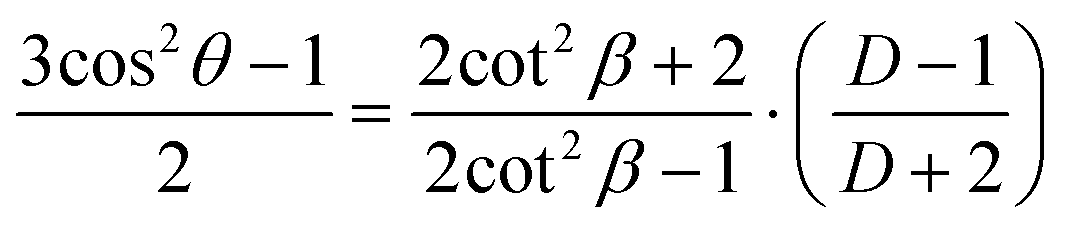 | (2) |
In case of an ideal sample with perfectly unidirectionally aligned polymer chains, the average over the angles in the equation simplifies as follows:
 | (3) |
Thus, the value of β can be determined from calibration experiments on highly oriented reference samples. To do so, the IR anisotropy measurements should be done in longitudinal geometry of the sample (the chain axis and IR light electric field vector lay in the same plane). In this configuration, angle θ in eqn (3) matches the experimental angle (e.g. polarizer angle) up to a constant and the left side of the equation can be determined experimentally. This has been previously done for the C–O–C bond vibration discussed here, and it has been shown that the transition dipole moment is oriented at about 30° with respect to the cellulose chain axis.33,43
An almost perfect alignment of cellulose microfibrils is observed in longitudinal sections of some plants, e.g. in flax43 and ramie,54 allowing to use only a single cos2θ term (eqn (3)), for determination of the microfibril orientation.
As first approximation one may assume a unidirectional orientation of the cellulose microfibrils in the investigated volume of the sorghum leaf cell walls. This allows one to convert the dichroic ratio values obtained in the experiment here (Table 1) to values for θ of 23°, and of 42° and 48° for the microcrystal in the cellulose reference, and for the positions S2 and X2, respectively. Here θ corresponds to the angle between the plane of the transversal section and the microfibril angle, i.e. θ = 0° and θ = 90° when the microfibrils are oriented perpendicular to and along the optical axis, respectively.
To understand the calculated θ values, it is important to consider the different possible terms that influence the observed result. In the section of the sorghum leaf sample, the observed dichroic ratio depends on the average orientation of the cellulose microfibrils and will influence the outcome by <cos2θ>, according to eqn (2).
The maximum dichroic ratio for the 1160 cm−1 vibration of a perfectly oriented cellulose sample according to the eqn (3) is equal to 6, which is higher than the value observed for the crystalline reference in our experiment. We attribute the lower value here to the non-ideal orientation of the crystal with respect to the plane of the IR substrate, as the crystalline reference used here was prepared by drop casting of a CNC aqueous suspension onto a substrate. Moreover, the presence of differently oriented smaller crystals in the area of the measurement cannot be completely excluded either.
In the case of the sampled spots in the plant cell walls, a spectrum always comprises information from two adjacent cells. Thus, the calculated microfibril orientational angles from the collected spectra comprises contributions of at least two microfibril orientations, that have a small likelihood to coincide.
A sub-micrometer resolution would be needed to resolve the contributions by the cell walls of the two neighbouring cells.21,60 In order to verify the obtained orientation angles of the cellulose fibrils we examined the cell wall corresponding to spot S2 (cf.Fig. 1) by means of AFM microscopy. The corresponding spot was found by means of the bright field optical microscope built in the AFM microscope used. The measurement was done close to the center of the cell wall. The topology and the mechanical phase profile of the central part of the cell wall are shown in Fig. 6. The structure of the individual microfibrils and fibril bunches that are forming the cell wall is well preserved during the sample preparation. The topology image allows to estimate the angle between the fibrils at the edge of the cell wall and the plane of the section. For the cell wall contained in spot S2, the values found for the left and the right side of the cell wall are 47 ± 3° and 32 ± 2°, respectively. Thus, its average can be estimated as 40 ± 3°, which is close to the value obtained from the dichroic measurement.
 | ||
| Fig. 6 AFM images of the cell wall located at S2 spot (cf.Fig. 1): (A) height profile; (B) mechanical phase profile. Scale bars: 500 nm. No data were collected for the dark red areas. | ||
In the xylem (Fig. 3C and Fig. 1, positions X1, X2, X3) and some points in sclerenchyma tissue regions (Fig. 1, positions S3, S4) where the size of the cells is small, the spectral data collected may include information originating from more than two cells, and may even include whole cell walls of different orientation. This results in lower dichroic ratios and the difference between the cellulose in plane orientation angle obtained from IR micro-spectroscopy data and the corresponding value expected from the cell walls orientation obtained from bright field optical microscopy (compare φ−90 IR with φ−90 OM values, Table 1).
Orientation of non-cellulosic components in the cell wall of sorghum
The anisotropy analysis based on the IR micro-spectroscopic data can also be used to understand the orientation of other molecules present in the plant cell walls,31,50 specifically those that are discussed to interact with the cellulose.17 From the experimental spectra it is possible to observe significant variation in intensity of the absorption bands at 1510 cm−1 and 1730 cm−1, which are quite well distinguished from the multiple peaks observed and are characteristic of CC vibrations of aromatic building blocks of lignin and a CO stretching mode assigned to the hemicellulose xylan, respectively. Fig. 5 shows the relative absorption data of these two bands at 1510 cm−1 (Fig. 7A and B) and 1730 cm−1 (Fig. 7C and D) for the positions S2 and X2 in sclerenchyma and xylem, respectively. Similar to the data corresponding to the cellulose signature (Insets in Fig. 5A and B), a higher degree of anisotropy is observed for the position in sclerenchyma compared to the values obtained for xylem (compare Fig. 7A with 7B and Fig. 7C with 7D). We ascribe this, as discussed above for the anisotropy of the cellulose signal, to the higher precision of the analysis of the individual cell walls that is possible in sclerenchyma tissue.
 | ||
| Fig. 7 Dependencies of the relative absorption of the bands at 1510 and 1730 cm−1 in sclerenchyma and xylem tissues versus angular position of the sample: (A) sclerenchyma, point S2, 1510 cm−1 band; (B) xylem, point X2, 1510 cm−1 band; (C) sclerenchyma, point S2, 1730 cm−1 band; (D) xylem, point X2, 1730 cm−1 band. The result values of the fit, namely D (dichroic ratio) and R2adj are given in each panel. Vertical dashed lines indicate an angle at which the IR-light polarization is parallel to the cell wall. | ||
The experimental data are rather well described by eqn (1) for the 1510 cm−1 vibrational band in both sclerenchyma and xylem regions, as evidenced by R2adj ≥ 0.75. The 1730 cm−1 band absorption also undergoes changes with the angle, however the fits are significantly less reliable (R2adj ∼ 0.6) than for the 1510 cm−1 band in both tissues (compare Fig. 7A with 7C and Fig. 7B with 7D).
Although it has been reported that the 1510 cm−1 and 1730 cm−1 vibrational modes demonstrate rather weak anisotropy,50 the data on the vibration at 1510 cm−1 enable us to infer that lignin (Fig. 7A and B) in sorghum leaves lays in the cell wall plane and most probably matches the orientation of the cellulose microfibrils, a finding that is important for understanding of lignocellulosic interaction. Previously the cellulose-lignin interaction and the mutual ordering of these macromolecules have been demonstrated using Raman spectroscopy in other herbaceous plants61,62 and IR spectroscopy conducted in longitudinal sections of cells in woods.32,63,64
The dipole moment of the band at 1730 cm−1 of a carbonyl stretching mode assigned mostly to xylan is oriented at about 54° relative to the polymer axis,31 making it difficult to determine its orientation, since the average β angle of 54.7° and a random orientation cannot be distinguished according to eqn (2). Nevertheless, the anisotropy of this IR absorption band in woods is high enough to be detected.31 Considering our results obtained here we assume that xylan is oriented perpendicularly to the cell walls of sorghum leaves. It should be noted though, that also other sugars, e.g., pectin65 could contribute to the band at 1730 cm−1, superimposing the absorption that can indicate xylan directions. This example shows that the unambiguous assignment of a specific band to a particular molecular compound is an important prerequisite for measurements of the anisotropy in the multicomponent mixture of the plant cell wall.
In the context of plant characterization, we expect that the acquisition of spectral information over several large sample areas simultaneously using focal plane array detectors66 will provide a more comprehensive picture with respect to the variation of cell wall macromolecular anisotropy in different plant tissues or stages of development.
Moreover in the recent past, a great leap in the near-field infrared spectroscopy techniques allowed to extend the benefits of the IR-spectroscopy to sub-diffractions scale down to 10–20 nm.67,68 Application of nanoscopic IR experiments to resolve polymer orientation by polarization sensitive characterization was successfully shown for isolated cellulose material,69 collagen polymers in tendon tissue,70 as well as in silk fibers.71 Also the methods to detect and quantify the anisotropy at the nm-scale are being actively studied.72,73
The FTIR experiments can be also done in native cells environment, e.g. in water. The liquid cuvettes or cells suitable for the appropriate in situ FTIR experiment have been successfully implemented for both diffraction limited and nano- FTIR spectroscopies.74,75
Nowadays broadband and tunable mid-IR lasers are starting to compete with the synchrotron based IR-sources,76 so that tabletop systems for diffraction limited and near-field sub-diffraction IR spectroscopic characterization of plant material hierarchical structure can become a standard tool soon.
Conclusions
Synchrotron FTIR micro-spectroscopy at defined polarization was used both for chemical imaging of cellulose and the investigation of its macromolecular organization in secondary cell walls of transversal section of Sorghum bicolor at the single cell level lateral resolution.The mapping experiments of different tissue types with varying characteristic cell sizes and the analysis of different vibrations in the cell wall spectra identified several factors being important to reveal structural anisotropies in the micro-spectroscopic data. These are (i) the typical size of the structures that contain the ordered macromolecules and their supramolecular organization, in our case plant cell walls and cells of different microscopic dimensions in relation to the diffraction limited lateral resolution of the IR microscope, (ii) the origin of the particular vibrational bands used for the analysis in the complex molecular mixture, and (iii) the orientation of the bonds relative to the polymer axis. The acquired data were used to determine a preferential direction of the submicron cellulose microfibrils.
It is shown that the selectivity of the method allows to simultaneously get information on several components present in the complex natural sample with diffraction limited spatial resolution. Relating biopolymer molecular structure and supramolecular structure and interaction in a micro-spectroscopic approach to biomolecular composition of a complex sample will help to better understand the deposition mechanisms of cellulose microfibrils in the context of other biomacromolecules, as well as the structural organization of different multicomponent samples of natural and artificial origin.
Author contributions
A.V. and J.K. conceived the research idea. A.V., V.M.R.Z., L.P., U.S. conducted the experiments. A.V. analyzed the data. A.V. and J.K. prepared the manuscript draft. All authors edited the manuscript draft. J.K., L.P., and U.S. provided resources, project administration, and funding acquisition. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Rivka Elbaum (Hebrew University of Jerusalem, Israel) for helpful discussions. We thank HZB-BESSY for the allocation of beam time at beamline IRIS. Funding by the German Federal Ministry for Education and Research (BMBF) project 05K19KH1 (SyMS) and EFRE 18/07 is gratefully acknowledged.References
- J. Xu, D. Sagnelli, M. Faisal, A. Perzon, V. Taresco, M. Mais, C. V. L. Giosafatto, K. H. Hebelstrup, P. Ulvskov, B. Jørgensen, L. Chen, S. M. Howdle and A. Blennow, Carbohydr. Polym., 2021, 253, 117277 CrossRef CAS PubMed.
- A. Molina, E. Miedes, L. Bacete, T. Rodríguez, H. Mélida, N. Denancé, A. Sánchez-Vallet, M.-P. Rivière, G. López, A. Freydier, X. Barlet, S. Pattathil, M. Hahn and D. Goffner, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2010243118 CrossRef CAS PubMed.
- L. de Vries, S. Guevara-Rozo, M. Cho, L.-Y. Liu, S. Renneckar and S. D. Mansfield, Biotechnol. Biofuels, 2021, 14, 167 CrossRef CAS PubMed.
- C. Somerville, S. Bauer, G. Brininstool, M. Facette, T. Hamann, J. Milne, E. Osborne, A. Paredez, S. Persson, T. Raab, S. Vorwerk and H. Youngs, Science, 2004, 306, 2206–2211 CrossRef CAS PubMed.
- D. J. Cosgrove, Curr. Opin. Plant Biol., 2014, 22, 122–131 CrossRef CAS PubMed.
- B. Zhang, Y. Gao, L. Zhang and Y. Zhou, J. Integr. Plant Biol., 2021, 63, 251–272 CrossRef PubMed.
- Y. Watanabe, M. J. Meents, L. M. McDonnell, S. Barkwill, A. Sampathkumar, H. N. Cartwright, T. Demura, D. W. Ehrhardt, A. L. Samuels and S. D. Mansfield, Science, 2015, 350, 198–203 CrossRef CAS PubMed.
- C. T. Anderson, A. Carroll, L. Akhmetova and C. Somerville, Plant Physiol., 2010, 152, 787–796 CrossRef CAS PubMed.
- B. Du, Q. Zhang, Q. Cao, Y. Xing, L. Qin and K. Fang, J. Plant Res., 2020, 133, 257–270 CrossRef CAS PubMed.
- T. Vellosillo, J. R. Dinneny, C. R. Somerville and D. W. Ehrhardt, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2021790118 CrossRef CAS PubMed.
- E. Toumpanaki, D. U. Shah and S. J. Eichhorn, Adv. Mater., 2021, 33, 2001613 CrossRef CAS PubMed.
- A. Khodayari, W. Thielemans, U. Hirn, A. W. Van Vuure and D. Seveno, Carbohydr. Polym., 2021, 270, 118364 CrossRef CAS PubMed.
- Q. Wu, Y. Li, M. Lyu, Y. Luo, H. Shi and S. Zhong, Sci. Adv., 2020, 6, eabc9294 CrossRef CAS PubMed.
- M. Soukup, V. M. R. Zancajo, J. Kneipp and R. Elbaum, J. Exp. Bot., 2020, 71, 6807–6817 CrossRef CAS PubMed.
- N. Zexer and R. Elbaum, J. Exp. Bot., 2020, 71, 6818–6829 CrossRef CAS PubMed.
- Y. Fridman, N. Holland, R. Elbaum and S. Savaldi-Goldstein, JoVE, 2016, 53707 Search PubMed.
- Y. Gao, A. S. Lipton, Y. Wittmer, D. T. Murray and J. C. Mortimer, Nat. Commun., 2020, 11, 6081 CrossRef CAS PubMed.
- Y. Abraham and R. Elbaum, New Phytol., 2013, 197, 1012–1019 CrossRef CAS PubMed.
- J. H. Wiley and R. H. Atalla, Carbohydr. Res., 1987, 160, 113–129 CrossRef CAS.
- L. G. Thygesen and N. Gierlinger, J. Struct. Biol., 2013, 182, 219–225 CrossRef CAS PubMed.
- U. P. Agarwal, Planta, 2006, 224, 1141–1153 CrossRef CAS PubMed.
- N. Gierlinger, S. Luss, C. Konig, J. Konnerth, M. Eder and P. Fratzl, J. Exp. Bot., 2010, 61, 587–595 CrossRef CAS PubMed.
- N. Gierlinger, L. Sapei and O. Paris, Planta, 2008, 227, 969–980 CrossRef CAS PubMed.
- L. Sun, S. Singh, M. Joo, M. Vega–Sanchez, P. Ronald, B. A. Simmons, P. Adams and M. Auer, Biotechnol. Bioeng., 2016, 113, 82–90 CrossRef CAS PubMed.
- M. Zimmerley, R. Younger, T. Valenton, D. C. Oertel, J. L. Ward and E. O. Potma, J. Phys. Chem. B, 2010, 114, 10200–10208 CrossRef CAS PubMed.
- C. Pohling, C. Brackmann, A. Duarte, T. Buckup, A. Enejder and M. Motzkus, J. Biophotonics, 2014, 7, 126–134 CrossRef CAS PubMed.
- C. M. Lee, K. Kafle, D. W. Belias, Y. B. Park, R. E. Glick, C. H. Haigler and S. H. Kim, Cellulose, 2015, 22, 971–989 CrossRef CAS.
- Z. Heiner, I. Zeise, R. Elbaum and J. Kneipp, J. Biophotonics, 2018, 11, e201700164 CrossRef PubMed.
- M. Tsuboi, J. Polym. Sci., 1957, 25, 159–171 CrossRef CAS.
- Y. Horikawa, T. Itoh and J. Sugiyama, Cellulose, 2006, 13, 309–316 CrossRef CAS.
- J. S. Stevanic and L. Salmén, Holzforschung, 2009, 63(5), 497–503 CrossRef CAS.
- A.-M. Olsson, I. Bjurhager, L. Gerber, B. Sundberg and L. Salmén, Planta, 2011, 233, 1277–1286 CrossRef CAS PubMed.
- K. Kafle, Y. B. Park, C. M. Lee, J. J. Stapleton, S. N. Kiemle, D. J. Cosgrove and S. H. Kim, Cellulose, 2017, 24, 3145–3154 CrossRef CAS.
- M. C. McCann, L. Chen, K. Roberts, E. K. Kemsley, C. Sene, N. C. Carpita, N. J. Stacey and R. H. Wilson, Physiol. Plant., 1997, 100, 729–738 CrossRef CAS.
- I. Zeise, Z. Heiner, S. Holz, M. Joester, C. Büttner and J. Kneipp, Plants, 2018, 7, 7 CrossRef PubMed.
- R. H. Atalla and U. P. Agarwal, J. Raman Spectrosc., 1986, 17, 229–231 CrossRef CAS.
- B. Jasse and J. L. Koenig, J. Macromol. Sci., Part C: Polym. Rev., 1979, 17, 61–135 CrossRef.
- D. L. Wetzel, J. A. Sweat and D. D. Panzer, AIP Conference Proceedings, 1998, 430, 354–357 CrossRef CAS.
- N. Jamin, P. Dumas, J. Moncuit, W.-H. Fridman, J.-L. Teillaud, G. L. Carr and G. P. Williams, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4837–4840 CrossRef CAS PubMed.
- P. Yu, J. J. McKinnon, C. R. Christensen, D. A. Christensen, N. S. Marinkovic and L. M. Miller, J. Agric. Food Chem., 2003, 51, 6062–6067 CrossRef CAS PubMed.
- V. M. R. Zancajo, S. Diehn, R. Elbaum and J. Kneipp, Analysis Sensing, 2022, 2, e202200006 CAS.
- J. Zhu, H. Wang, F. Guo, L. Salmén and Y. Yu, Carbohydr. Polym., 2021, 274, 118653 CrossRef CAS PubMed.
- A. Šturcová, I. His, T. J. Wess, G. Cameron and M. C. Jarvis, Biomacromolecules, 2003, 4, 1589–1595 CrossRef PubMed.
- J. Zhu, W. Ren, F. Guo, H. Wang and Y. Yu, Cellulose, 2022, 29, 3163–3176 CrossRef CAS.
- U. Schade, A. Röseler, E. H. Korte, F. Bartl, K. P. Hofmann, T. Noll and W. B. Peatman, Rev. Sci. Instrum., 2002, 73, 1568–1570 CrossRef CAS.
- M. Toplak, S. T. Read, C. Sandt and F. Borondics, Cells, 2021, 10, 2300 CrossRef PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- D. Nečas and P. Klapetek, Open Phys., 2012, 10, 181–188 Search PubMed.
- Y. Horikawa, S. Hirano, A. Mihashi, Y. Kobayashi, S. Zhai and J. Sugiyama, Appl. Biochem. Biotechnol., 2019, 188, 1066–1076 CrossRef CAS PubMed.
- R. H. Marchessault, Pure Appl. Chem., 1962, 5, 107–130 CAS.
- R. H. Marchessault and C. Y. Liang, J. Polym. Sci., 1962, 59, 357–378 CrossRef CAS.
- R. H. Wilson, A. C. Smith, M. Kačuráková, P. K. Saunders, N. Wellner and K. W. Waldron, Plant Physiol., 2000, 124, 397–406 CrossRef CAS PubMed.
- N. Reddy and Y. Yang, J. Agric. Food Chem., 2007, 55, 5569–5574 CrossRef CAS PubMed.
- C. Y. Liang and R. H. Marchessault, J. Polym. Sci., 1959, 39, 269–278 CrossRef CAS.
- S. E. Darmon and K. M. Rudall, Discuss. Faraday Soc., 1950, 9, 251 RSC.
- P. Koziol, D. Liberda, W. M. Kwiatek and T. P. Wrobel, Anal. Chem., 2020, 92, 13313–13318 CrossRef CAS PubMed.
- H. B. Peng and L. F. Jaffe, Planta, 1976, 133, 57–71 CrossRef CAS PubMed.
- L. Donaldson, IAWA J, 2008, 29, 345–386 Search PubMed.
- G. L. Wilkes, in Fortschritte der Hochpolymeren-Forschung, Springer Berlin Heidelberg, Berlin, Heidelberg, 1971, vol. 8, pp. 91–136 Search PubMed.
- T. Keplinger, J. Konnerth, V. Aguié-Béghin, M. Rüggeberg, N. Gierlinger and I. Burgert, Plant Methods, 2014, 10, 1 CrossRef PubMed.
- S. Richter, J. Müssig and N. Gierlinger, Planta, 2011, 233, 763–772 CrossRef CAS PubMed.
- Y. Cao, D. Shen, Y. Lu and Y. Huang, Ann. Bot., 2006, 97, 1091–1094 CrossRef CAS PubMed.
- J. Simonović, J. Stevanic, D. Djikanović, L. Salmén and K. Radotić, Cellulose, 2011, 18, 1433–1440 CrossRef.
- H. Peng, L. Salmén, J. S. Stevanic and J. Lu, Planta, 2019, 250, 163–171 CrossRef CAS PubMed.
- M. Kačuráková and R. H. Wilson, Carbohydr. Polym., 2001, 44, 291–303 CrossRef.
- M. J. Tobin, J. Vongsvivut, D. E. Martin, K. H. Sizeland, M. J. Hackett, R. Takechi, N. Fimorgnari, V. Lam, J. C. Mamo, E. A. Carter, B. Swarbrick, P. A. Lay, D. A. Christensen, D. Perez-Guaita, E. Lowery, P. Heraud, B. R. Wood, L. Puskar and K. R. Bambery, Infrared Phys. Technol., 2018, 94, 85–90 CrossRef.
- F. Huth, A. Govyadinov, S. Amarie, W. Nuansing, F. Keilmann and R. Hillenbrand, Nano Lett., 2012, 12, 3973–3978 CrossRef CAS PubMed.
- U. Schade, K. Holldack, P. Kuske, G. Wüstefeld and H.-W. Hübers, Appl. Phys. Lett., 2004, 84, 1422–1424 CrossRef CAS.
- B. Alonso-Lerma, L. Barandiaran, L. Ugarte, I. Larraza, A. Reifs, R. Olmos-Juste, N. Barruetabeña, I. Amenabar, R. Hillenbrand, A. Eceiza and R. Perez-Jimenez, Commun. Mater., 2020, 1, 1–10 CrossRef.
- G. Bakir, B. E. Girouard, R. Wiens, S. Mastel, E. Dillon, M. Kansiz and K. M. Gough, Molecules, 2020, 25, 4295 CrossRef CAS PubMed.
- M. Ryu, R. Honda, A. Reich, A. Cernescu, J.-L. Li, J. Hu, S. Juodkazis and J. Morikawa, Appl. Sci., 2019, 9, 3991 CrossRef CAS.
- K. Hinrichs and T. Shaykhutdinov, Appl. Spectrosc., 2018, 72, 817–832 CrossRef CAS PubMed.
- S. T. Chui, X. Chen, Z. Yao, H. A. Bechtel, M. C. Martin, G. L. Carr and M. Liu, J. Appl. Phys., 2021, 129, 083105 CrossRef CAS.
- M. Schmidt, N. Gierlinger, U. Schade, T. Rogge and M. Grunze, Biopolymers, 2006, 83, 546–555 CrossRef CAS PubMed.
- K. J. Kaltenecker, T. Gölz, E. Bau and F. Keilmann, Sci. Rep., 2021, 11, 21860 CrossRef CAS PubMed.
- C. R. Petersen, P. M. Moselund, L. Huot, L. Hooper and O. Bang, Infrared Phys. Technol., 2018, 91, 182–186 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an00897e |
| This journal is © The Royal Society of Chemistry 2023 |