DOI:
10.1039/D3AN00296A
(Paper)
Analyst, 2023,
148, 3036-3044
A dual paper-based nucleic acid extraction method from blood in under ten minutes for point-of-care diagnostics†
Received
23rd February 2023
, Accepted 28th May 2023
First published on 2nd June 2023
Abstract
Nucleic acid extraction (NAE) plays a crucial role for diagnostic testing procedures. For decades, dried blood spots (DBS) have been used for serology, drug monitoring, and molecular studies. However, extracting nucleic acids from DBS remains a significant challenge, especially when attempting to implement these applications to the point-of-care (POC). To address this issue, we have developed a paper-based NAE method using cellulose filter papers (DBSFP) that operates without the need for electricity (at room temperature). Our method allows for NAE in less than 7 min, and it involves grade 3 filter paper pre-treated with 8% (v/v) igepal surfactant, 1 min washing step with 1× PBS, and 5 min incubation at room temperature in 1× TE buffer. The performance of the methodology was assessed with loop-mediated isothermal amplification (LAMP), targeting the human reference gene beta-actin and the kelch 13 gene from P. falciparum. The developed method was evaluated against FTA cards and magnetic bead-based purification, using time-to-positive (min) for comparative analysis. Furthermore, we optimised our approach to take advantage of the dual functionality of the paper-based extraction, allowing for elution (eluted disk) as well as direct placement of the disk in the LAMP reaction (in situ disk). This flexibility extends to eukaryotic cells, bacterial cells, and viral particles. We successfully validated the method for RNA/DNA detection and demonstrated its compatibility with whole blood stored in anticoagulants. Additionally, we studied the compatibility of DBSFP with colorimetric and lateral flow detection, showcasing its potential for POC applications. Across various tested matrices, targets, and experimental conditions, our results were comparable to those obtained using gold standard methods, highlighting the versatility of our methodology. In summary, this manuscript presents a cost-effective solution for NAE from DBS, enabling molecular testing in virtually any POC setting. When combined with LAMP, our approach provides sample-to-result detection in under 35 minutes.
Introduction
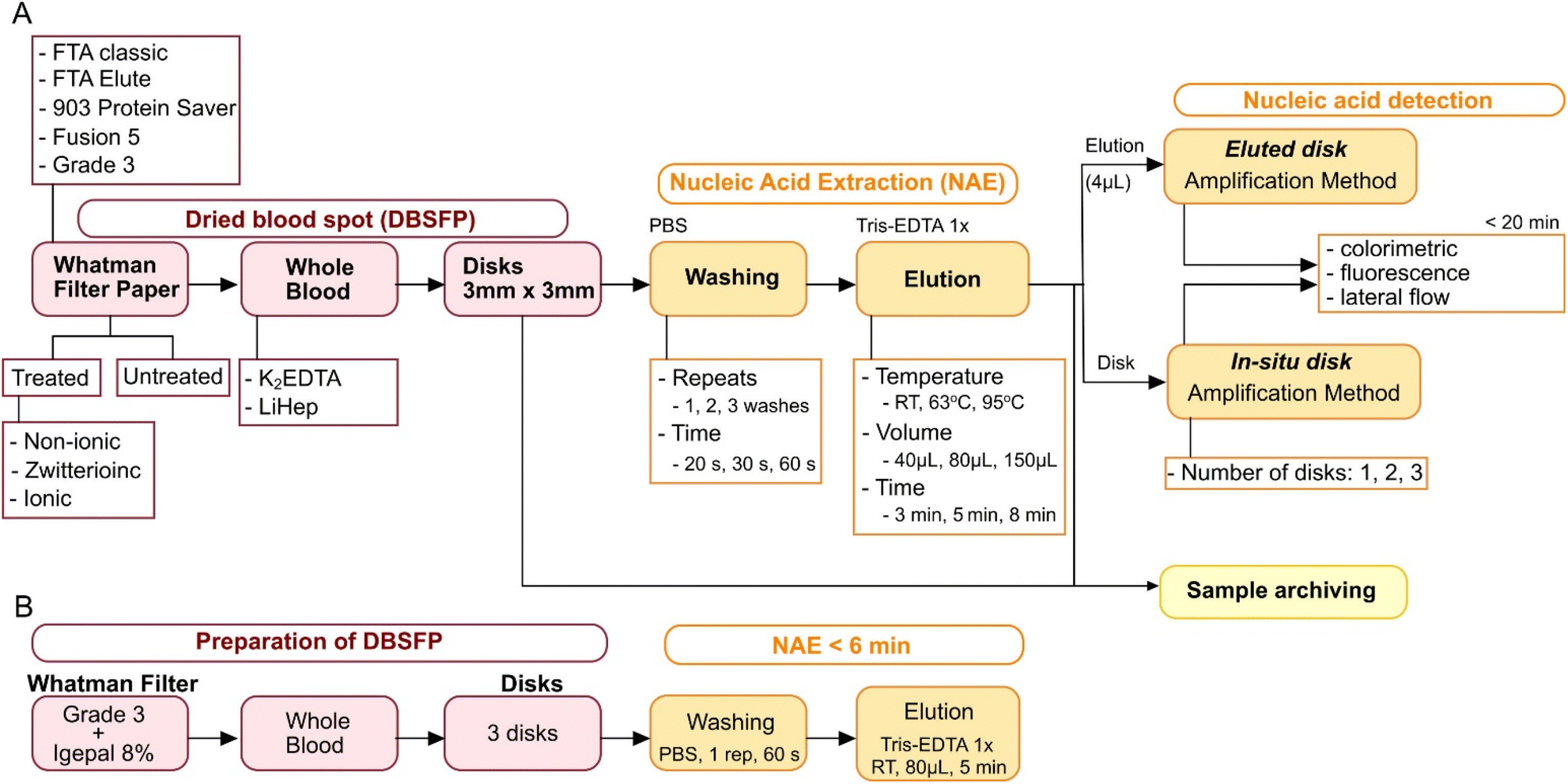
Dried blood spot (DBS) technology has been widely used for sample collection and diagnostic purposes.1–3 One of the most significant advantages of DBS is that whole blood can be safely stored and shipped at room temperature (RT) after simply spotting a few microliters onto cellulose filter paper (DBSFP).4–6 Several DBSFPs are commercially available for the collection of whole blood such as FTA Classic cards, Whatman 903 Protein Saver cards or most recently, FTA Elute cards (QIAGEN, Whatman, Cytiva); all of them allowing for cell lysis and the preservation of the genetic material for a long period of time without the need for cold chain storage. However, they are expensive (£3–5 per single card) and commonly treated with non-disclosed proprietary compounds (Table S1†). Alternatively, filter papers which have not been specifically designed for blood collection have been tested for nucleic acid extraction (NAE). Among others, Fusion 5, nitrocellulose, filter paper grade 1 or filter paper grade 3 have demonstrated their potential for nucleic acid preservation and extraction.5,7 Nevertheless, current strategies for NAE from DBSFP still rely on: (i) silica-based spin columns (e.g., QIAamp DNA Blood Mini kit, or Maxwell RSC DNA FFPE kit),8,9 (ii) magnetic beads,10 (iii) long incubations at high temperatures (e.g., 95 °C),11,12 or (iv) long protocols with hazardous chemicals.13,14 Although simplifications towards point-of-care (POC) applications have been developed, they have not been applied yet to whole blood samples15,16 or have only been directly used in combination with PCR;7,17 therefore limiting upstream and/or downstream analysis to laboratory-based settings. The high demand of rapid testing at the POC is creating a need for the development of NAE methods from DBSFPs that must be: (i) compatible with isothermal chemistries such as loop-mediated isothermal amplification (LAMP) to reduce the hardware complexity needed to perform nucleic acid amplification,18 (ii) independent of laboratory equipment to be accessible in limited-resource settings, (iii) affordable and compatible with broadly available molecular reagents to reduce the cost and increase scale-up capabilities, and (iv) stable at RT conditions to avoid the use of cold-chain storage.19 In this work, addressing aforementioned requirements, we report a fast (under 7 min) paper-based NAE method from blood which is electricity-free, performed at RT and relies on cellulose filter paper pre-treated with the surfactant igepal. The performance of the method was evaluated using LAMP and was further optimised to accommodate both elution (eluted disk) as well as direct placement of the disk in the LAMP reaction (in situ disk). The developed method allows cellular and cell-free RNA/DNA purification and was compatible with whole blood stored in anticoagulants. In addition, end-point colorimetric and lateral flow detection of amplified products was performed to show its applicability to the POC, showing a sample-to-result turnaround time under 35 min. The experimental design and optimised NAE protocol are depicted in Fig. 1.
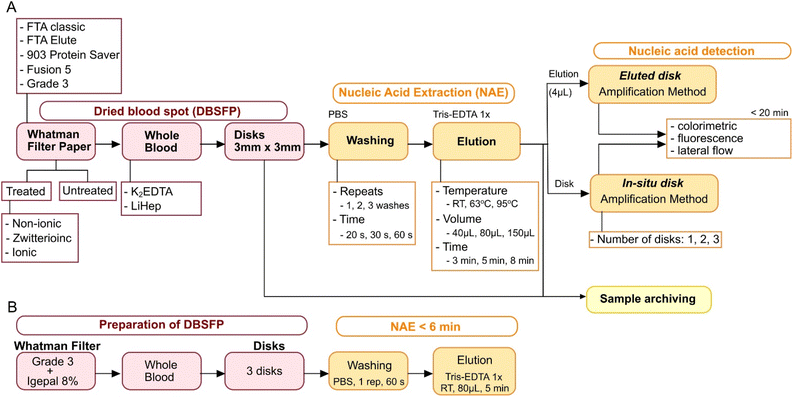 |
| | Fig. 1 Experimental design used in this study. (A) Workflow for optimisation of NAE from DBSFP. (B) Optimised DBSFP protocol. | |
Materials and methods
Samples and controls
Fresh and healthy whole blood stored in K2EDTA, or lithium heparin (LiHep) was purchased from Cambridge Bioscience. Blood was commercially available and collected under a comprehensive informed consent that permits the use of blood for research applications, therefore no further ethics were required. Then, it was dried onto filter papers by pipetting 2 μL each time to standardise the collection process and compare the filter papers without the influence of their physico-chemical properties at the time of generating the DBSFP. Human Genomic DNA (G1521, Promega) was used as positive control and synthetic DNA was used to validate the LAMP assay performance. A gBlock was purchased from Integrated DNA Technologies (IDT), resuspended in 1× TE buffer and quantified (copies per μL) using a Qubit fluorometer instrument; sequence is shown in Table S2.†
LAMP reaction conditions
(i) Fluorescent detection.
LAMP reactions were carried out at a final volume of 10 μL per reaction. Each mix contained the following: 1 μL of 10× custom isothermal buffer (pH 8.5–9), 0.5 μL of MgSO4 (100 mM stock), 0.56 μL of dNTPs (25 mM stock), 0.6 μL of BSA (20 mg mL−1 stock), 1 μL of 20× LAMP primer mix (F3/B3 5 μM, LF/LB 20 μM and FIP/BIP 40 μM), 0.25 μL of Syto9 dye (20 μM stock), 0.25 μL of NaOH (0.2 M stock), 0.04 μL of Bst 2.0 WarmStart DNA polymerase (120 kU mL−1 stock), 0.3 μL of WarmStart RTx Reverse Transcriptase (15 kU mL−1 stock), 4 μL sample, and enough nuclease-free water (Invitrogen) to bring the volume to 10 μL. In the case of the eluted disk method, 4 μL of elution from DBSFP and enough nuclease-free water (Invitrogen) were used to bring the final volume to 10 μL. For the in situ disk method, reactions were carried out at a final volume of 20 μL (2 × 10 μL reaction) adding enough nuclease-free water and immersing the disk(s) into the reaction; disk(s) still wet after the incubation during the elution step, without requiring any drying before placing it (them) into the reaction. Reactions were loaded into 96-well plates and were performed at 63 °C during 35 min using a LightCycler 96 Real-Time System (LC96) (Roche Diagnostics) or a CFX Real-Time PCR System (Bio-Rad). One melting cycle was performed at 0.1 °C s−1 from 63 °C up to 97 °C for validation of the specificity of the amplified products. Non-template control (NTC) was included.
(ii) Colorimetric detection.
LAMP reactions were performed at a final volume of 20 μL using the WarmStart Colorimetric LAMP 2× Master Mix (10 μL), 20× LAMP primer mix (2 μL), 4 μL of the elution and enough nuclease-free water to reach a final volume of 20 μL. Amplification reactions were carried out in a portable thermal block (N2400-4020 and N2400-4021, Starlab) during 35 min at 63 °C (checking at 20 min).
LAMP lateral flow detection
PCRD lateral flow tests (Abingdon Healthcare) were used following manufacturer's instructions for the detection of amplified products in lateral flow format. Briefly, 10 μL of 10× diluted amplified product was mixed with 140 μL of the provided buffer and the strip was inserted in the solution for 10 minutes. LAMP assays were modified to include labelled oligos, LF-DIG or LF-FAM and FIP-Biotin. The 10× LAMP primer mix consisted of F3/B3 2.5 μM, LB 10 μM, LF 3 μM, LF-DIG/FAM 7 μM, FIP 6 μM, FIP-Biotin 14 μM, and BIP 20 μM.
Analytical specificity of the LAMP-ACTB assay
Analytical specificity of the amplified products was evaluated by melting curve analysis. To further demonstrate the specificity among the proposed eluted disk and in situ disk methods, restriction digestion of the LAMP products was performed using the MseI enzyme (NEB, UK) which performs a single cut at T/TAA position. The restriction was performed for 15 min at 37 °C, using the rCutSmart buffer provided by the manufacturer. Enzyme was deactivated following manufacturer's protocol by incubating for 20 min at 65 °C. The digested products were analysed by electrophoresis on a 1.5% agarose gel stained with SYBR Safe DNA Gel Stain (InvitroGen). The experiment was run for 1 h at 145 V and the gel was visualized under UV light using a BioSpectrum Imaging System (Ultra-Violet Products Ltd). The Quick-Load 1 kb Plus DNA Ladder (NEB, UK) was used as reference.
Detergents and pre-treatment of filter papers
Detergents used in this study were purchased from Sigma-Aldrich and included: sodium dodecyl sulfate (L3771-25G), CHAPS (C9426-1G), Triton X-100 (T8787-100ML), Tween 20 (P9416-50ML) and igepal (I8896-50ML). Detergents were diluted in nuclease-free water at final concentrations of 2%, 4%, 6%, 8% and 10% w/v or v/v. The evaluated filter papers were spotted with 150 μL of each solution covering a circular surface area of 3 cm diameter, and were left to dry overnight at RT. Filter papers used in this work included: FTA Classic cards (Classic), Grade 3 Qualitative Filter Paper Standard Grade (Grade3), Fusion 5, 903 Protein Saver cards (Protein), and FTA Elute cards (FTA elute).
Experimental conditions to optimise the paper-based method for nucleic acid extraction
NAE was performed as follows: (i) whole blood was spotted on the pre-treated or untreated filter paper and was left to dry overnight (following standard practice20); (ii) 1, 2 or 3 disks with a diameter of 3 mm each were punched using the Harris Core puncher from the UniCore Punch kit 3.00 mm; disks were deposited into a 1.5 mL or 2.0 mL Eppendorf tube and the puncher was rinsed with 70% ethanol after every use; (iii) disk(s) washing was performed with 1× PBS by shaking, vortex or passively (not disturbed). The three washing types were assessed at varying conditions including number of washes (1, 2 or 3), time per wash (20 s, 30 s or 60 s), and volume of 1× PBS (400 μL, 600 μL, or 1500 μL); (iv) after discarding the PBS from the washing step, nucleic acid elution was performed in 1× TE buffer at different volumes (40 μL, 80 μL, or 150 μL), incubation times (3, 5 or 8 min), and at varying temperatures (RT, 63 °C, or 95 °C); (v) lastly, for the eluted disk method, 4 μL of the elution was added into the LAMP reaction (final volume of 10 μL), and for the in situ disk method, 1, 2 or 3 disks were added to a LAMP reaction with final volume of 20 μL to completely cover the disk in solution (disks still being wet when transferred). Higher volume of blood and therefore, disks with a higher diameter could be used. These modifications may require a proportional volume of 1× PBS for washing.
Magnetic bead-based nucleic acid extraction from whole blood
Magnetic bead-based extraction was performed using the Dynabeads DNA DIRECT Blood kit from Invitrogen (Life Technologies) following the manufacturer's instructions. Input sample volume was 100 μL.
Statistical analysis
Time-to-positive (TTP) data is presented as mean TTP ± standard deviation; p-values were calculated by Welch's unequal variance two sample t-test or one-way ANOVA with Fisher LSD means comparison in Origin 2019 (v9.6). A p-value equal to 0.05 was considered as the threshold for statistical significance.
Results and discussion
LAMP assay for detection of the house-keeping gene beta-actin
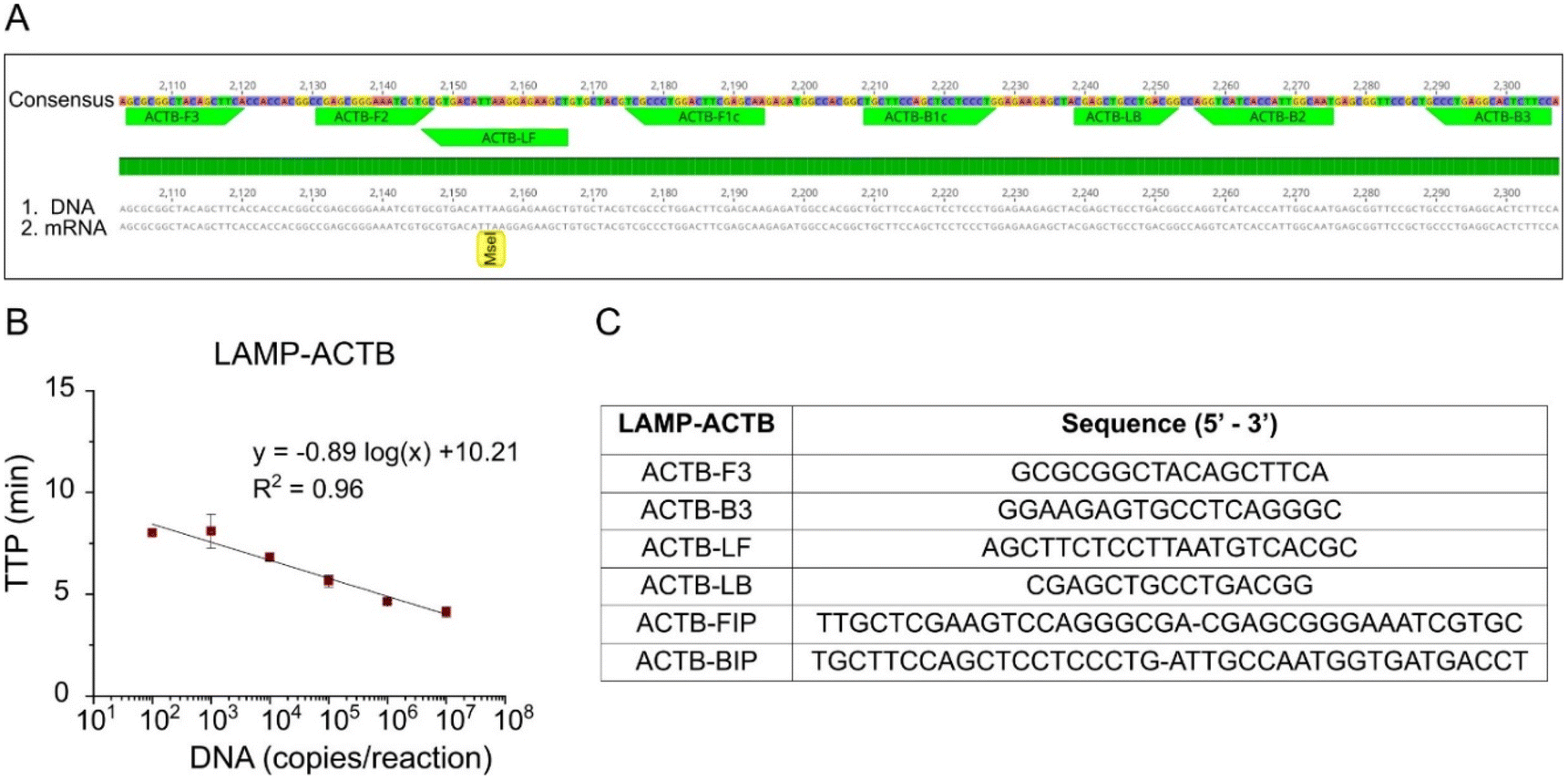
A novel LAMP assay, named LAMP-ACTB, was designed targeting the human house-keeping gene ACTB to detect both mRNA and DNA (Fig. 2A). Details of assay design in Table S3† and primer sequences shown in Fig. 2C. The ACTB gene is highly expressed, particularly in blood, and widely used as reference gene.21,22 Analytical sensitivity achieved a limit of detection of 100 copies per reaction within 10 min. The standard curve is shown in Fig. 2B, with a coefficient of determination R2 = 0.96.
 |
| | Fig. 2 LAMP assay for the detection of the human house-keeping gene beta-actin. (A) DNA and mRNA alignment showing the primer regions. (B) Standard curve with synthetic DNA dilutions ranging from 10 to 107 copies per reaction. (C) LAMP-ACTB primer sequences. NTCs were negative during the 35 cycles run, and non-specific products were not observed. | |
Evaluation of surfactants for enhanced lysis
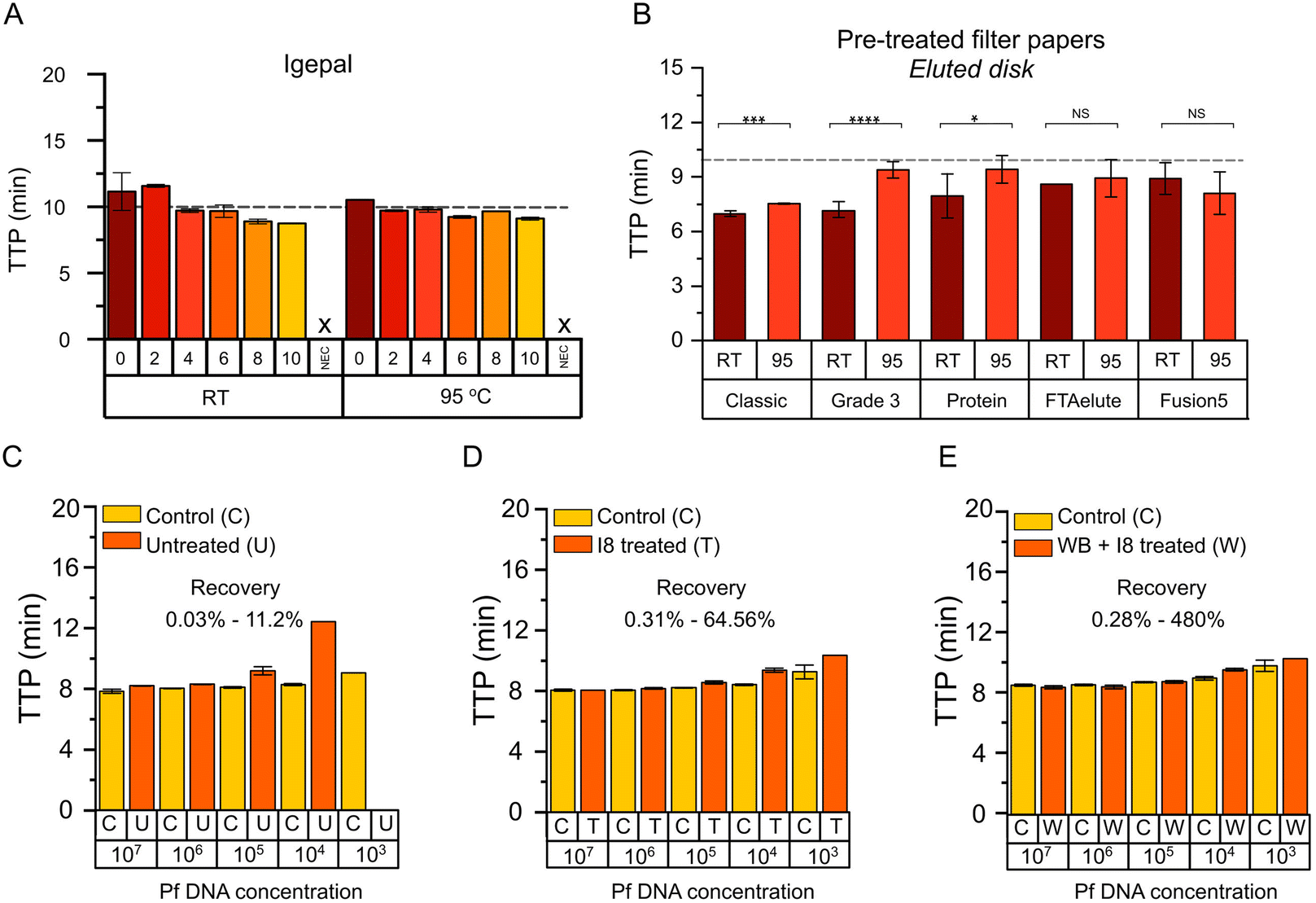
Surfactants are the main reagent that determine the lysis strength of a given lysis buffer.23 Therefore, surfactants with different physic–chemical properties were selected to evaluate their lysis effect on DBSFP. Their properties and applications are summarised in Table S4.† The surfactants were applied on FTA Classic cards at concentrations ranging from 0% to 10% v/v or w/v. Once dried, whole blood in K2EDTA was spotted. Nucleic acid extraction was performed using the eluted disk method including a washing step (3 washes of 20 s with 400 μL of 1× PBS) and incubation at RT, 63 °C or 95 °C during 8 min. Elutions were tested with the LAMP-ACTB assay targeting both DNA and RNA. Results obtained with the elutions from incubation at 63 °C where inconsistent and mostly negative (Fig. S1†). Nucleic acid amplification was comparable between incubation at RT and at 95 °C (p-value > 0.05), all with TTP values below 15 min. Among the different surfactants, igepal showed a linear trend as the concentration was increased (Fig. 3A and Fig. S2A†). To further evaluate the impact that detergent residues could have in the performance of LAMP, a titration of igepal directly in the reaction was carried out (Fig. S2B†). A high concentration of free igepal (>1.5%) in the reaction led to a decrease in the performance. Therefore, to minimise the risk of carry-over, igepal at 8% (p-value = 0.04, compared to 0%) was the condition selected for subsequent experiments instead of igepal at 10% (igepal at 8% and igepal at 10% were not statistically different). Results of all tested surfactants are shown in Fig. S3.†
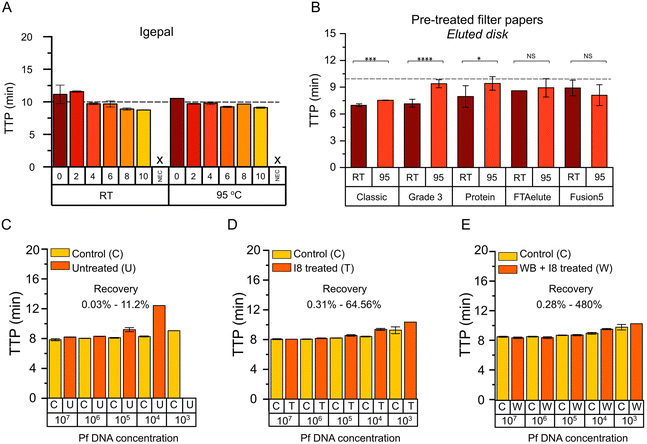 |
| | Fig. 3 Optimisation of NAE from DBSFP with eluted disk method. (A) Evaluation of igepal for DBSFP lysis with the eluted disk method. TTP values using the LAMP-ACTB assay plotted against the different detergent concentrations. The “0” denotes no detergent addition and NEC denotes negative extraction control which consisted of disks without dried blood. (B) TTP values from elutions after incubation at RT or 95 °C using different filter papers and the eluted disk method. (C) TTP values of recovery of spiked synthetic DNA of P. falciparum in untreated Grade3 filter. (D) TTP values of recovery of spiked synthetic DNA of P. falciparum in Grade3 filter treated with igepal 8% (I8). (E) TTP values of recovery of whole blood spiked with synthetic DNA of P. falciparum in Grade3 filter treated with I8. | |
Comparison of filter papers for nucleic acid extraction
Properties of the filter papers evaluated in this work are summarised in Table S1,† and photographs of the dried blood spots are shown in Fig. S4A and B.† Typically, FTA Classic cards are washed several times with proprietary FTA purification reagent and 1× TE buffer (∼10 min) and a drying step at 55 °C (∼15 min) is required prior to the use of the disks in molecular-based amplification; or in the case of FTA Elute cards, several washing steps with proprietary FTA purification reagent (∼10 min) and incubation at 95 °C (∼30 min) is required before the elution is used for downstream molecular applications.12,36 All the filter papers will be evaluated with the eluted disk method. Comparison between the recommended protocol for FTA Elute cards (30 min incubation at 95 °C) and the eluted disk method (8 min incubation at 95 °C) is shown in Fig. S4C† to demonstrate that the benefit of reducing the incubation time did not have a negative effect in the NAE. Untreated and pre-treated filter papers with igepal at 8% (I8) were evaluated. NAE was performed from whole blood in K2EDTA using the eluted disk method including a washing step (3 washes of 20 s with 400 μL of PBS 1×) and incubation at RT or 95 °C during 8 min. Elutions were tested with the LAMP-ACTB assay. NAE from untreated filter papers (Fig. S4D†) resulted in slower TTP values compared to pre-treated filter papers. Comparison between pre-treated and untreated filter papers eluted at RT or 95 °C are shown in Fig. S4E and F† respectively, where those pre-treated showed faster TTP values. Results obtained from pre-treated filter papers (Fig. 3B) showed faster TTP values after incubation at RT compared to 95 °C in the case of Classic (p-value < 0.001), Grade3 (p-value < 0.0001) and Protein filter papers (p-value < 0.05). In addition, Classic and Grade3 filter papers at RT were significantly faster compared to the rest (p-value < 0.05), with TTP values of 6.90 ± 0.16 min and 6.87 ± 0.43 min, respectively. Grade3 filter paper was selected for subsequent experiments, and FTA Classic cards as reference.
Optimisation of the paper-based nucleic acid extraction
Optimisation of nucleic acid extraction from DBSFP using whole blood in K2EDTA and the eluted disk method was carried out using pre-treated Classic and Grade3 filter papers to evaluate the effect of: (i) number of disks (D1, D2, D3); (ii) incubation time (3, 5, 8 min); (iii) volume of the elution (40, 80, 150 μL); and (iv) elution buffer (TE, water). Incubation temperatures at RT and 95 °C were tested across all the conditions, except for (iii) and (iv) which were only tested at RT. Across all the parameters assessed incubation during 5 min at RT in 80 μL of 1× TE, and the use of 3 disks were selected (Fig. S5A–C and F†). Incubation at 95 °C and the use of FTA Classic cards were performed as reference (Fig. S5D and E†), showing TTP values comparable to incubation at RT conditions and the use of Grade3 filter paper. Recovery of NAE with the eluted disk method was assessed by spiking synthetic DNA of P. falciparum in three scenarios: (i) deposition of synthetic DNA in untreated Grade3 filter, (ii) deposition of synthetic DNA in Grade3 filter treated with I8, (iii) deposition of whole blood spiked with synthetic DNA in Grade3 filter treated with I8. Results are shown in Fig. 3C–E where extracted DNA is compared to spiked DNA in the reactions. P. falciparum-specific LAMP assay24 is detailed in Table S5.† From the data points from the serial dilutions showing positive results, NAE recovery ranged between 0.03–11.2%, 0.31–64.56% and 0.28–480%, respectively in scenarios (i) to (iii) respectively.
Duality of the paper-based method
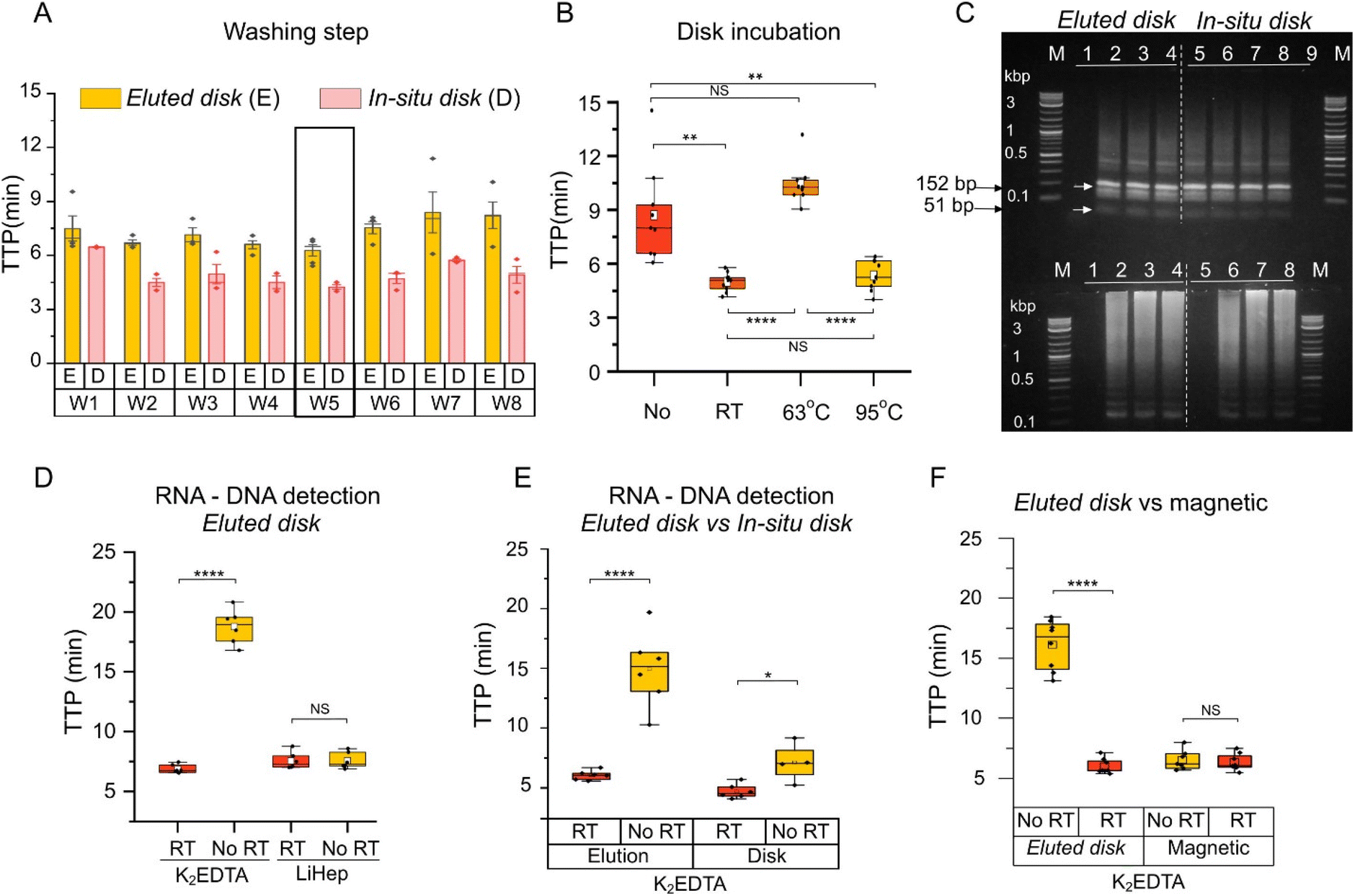
A second method was developed, named in situ disk method, which consisted of directly placing the eluted disks into the amplification reaction. Firstly, the in situ disk method was evaluated at different conditions including number of disks, incubation time, and elution volume using whole blood in K2EDTA. No significant differences were observed among them as shown in Fig. S5G–I.† Secondly, the washing step was evaluated at various conditions, including: (i) no wash (W1), (ii) the use of a vortex (W2 to W4), (iii) shaking (W5 and W6), (iv) and passive washing (W7 and W8). More details in Table S6.† Lower TTP values were obtained with W4 and W5 which consisted of 3 washes using vortex and 1 wash by shaking, respectively. Between them, W5 was selected for subsequent experiments (Fig. 4A). Thirdly, disks incubated at the three different temperatures (RT, 63 °C and 95 °C) were tested. Contrary to the eluted disk method, nucleic acid amplification was detected with the disks after incubation at the three conditions. TTP values were significantly lower at RT and 95 °C compared to 63 °C (Fig. 4B). Average TTP values were 5.00 ± 0.51 min, 10.45 ± 1.15 min and 5.27 ± 0.96 min for RT, 63 °C, and 95 °C, respectively. As reference, the absence of an incubation step was also evaluated. Results showed a higher variability, and TTP values were significantly higher (8.71 ± 2.65 min) than those obtained at RT or 95 °C. Also, the in situ disk method was performed with the different filter papers (Fig. S6†) and results agreed with those obtained in Fig. 3B. In order validate the specificity of the obtained amplification products, restriction analysis was performed demonstrating specific amplification with both, the eluted disk and in situ disk methods (Fig. 4C).
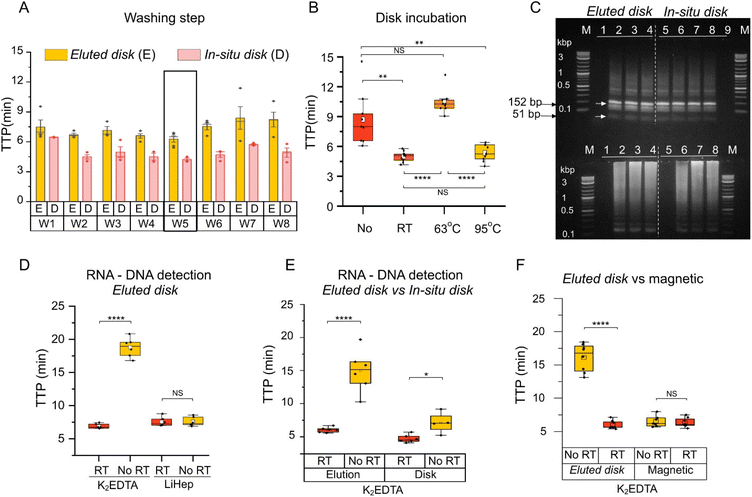 |
| | Fig. 4 Duality of the DBSFP. (A) Optimisation of the washing step in eluted disk and in situ disk methods. Further details in Table S6.† (B) Boxplot showing the effect of the incubation step and its temperature for the in situ disk method. “No” denotes the absence of incubation step after washing. Each dot represents a sample, n = 9; (C) restriction analysis and gel electrophoresis of amplified products with the RE MseI showing bands at 152 bp and 51 bp. Further details in Table S6.† (D) Boxplot showing RNA and DNA detection from whole blood stored in anticoagulants using the eluted disk method. (E) Boxplot showing RNA and DNA detection from whole blood in K2EDTA using the eluted disk and in situ disk method. (F) Boxplot showing RNA and DNA detection from whole blood in K2EDTA using the eluted disk and magnetic extraction with the Dynabeads kit. Each dot represents a sample, n = 6; horizontal lines in the boxes indicate medians; lower and upper edges of boxes indicate interquartile range and whiskers are <1 times the interquartile range. | |
Detection of RNA and DNA
Whole blood stored in anticoagulants were screened for RNA and DNA purification from DBSFP with the developed methods. Elutions (eluted disk method, Fig. 4D) from whole blood stored in K2EDTA spotted on Grade3 filter paper reported an average TTP value of 6.99 ± 0.45 min and 18.76 ± 1.34 min with and without the addition of the reverse transcriptase enzyme, respectively. Elutions (eluted disk method, Fig. 4D) from whole blood stored in LiHep reported TTP values of 7.62 ± 0.60 min and 7.50 ± 0.63 min with and without the reverse transcriptase with the Grade3 filter paper. NAE with the eluted disk method from blood stored in K2EDTA allowed RNA discrimination. However, this was not the case for whole blood stored in LiHep. To further investigate the role of the anticoagulants in the NAE recovery, an additional experiment was performed at different temperatures during the elution step (Fig. S1†). Contrary to K2EDTA, whole blood stored in LiHep showed comparable TTP values across all the tested temperatures (RT, 40, 63, 80 and 95 °C). LiHep may enhance the elution of DNA molecules such that the reverse transcriptase effect is negligible, which could be due to the negative charge of LiHep and its interaction with the DNA molecules in the matrix. Furthermore, this may explain a resilient behaviour to the different incubation temperatures during the elution step. Several studies have shown lower recovery of RNA from whole blood stored in LiHep compared to other anticoagulants such as K2EDTA.25–27 Nevertheless, further research is needed to fully understand the role of the anticoagulant in the preservation of RNA and DNA, since they could also be part of the pre-treatment of the DBSFP depending on the application and target.
The eluted disk and in situ disk methods were compared independently using whole blood stored in K2EDTA (Fig. 4E). Both methods showed a faster detection when the reverse transcriptase was incorporated in the reaction mix. A higher significant difference (p-value < 0.0001) with and without the reverse transcriptase was observed with the eluted disk method indicating that DNA molecules may be trapped in the cellulose and therefore it is harder to discriminate RNA from DNA using the in situ disk method.
The eluted disk method was compared against a commercially available magnetic bead-based NAE method (Dynabeads, Invitrogen). Average TTP values of 5.99 ± 0.62 min and 6.34 ± 0.68 were obtained with eluted disk and magnetic bead-based NAE methods (p-value > 0.05), respectively, in the presence of reverse transcriptase. Significant difference was observed with the eluted disk method with and without reverse transcriptase (5.99 ± 0.62 min and 16.12 ± 2.08, respectively). On the contrary, no significant difference was observed with the magnetic bead-based NAE method (6.34 ± 0.68 min and 6.52 ± 0.81, respectively) as shown in Fig. 4F.
Colorimetric and lateral flow detection
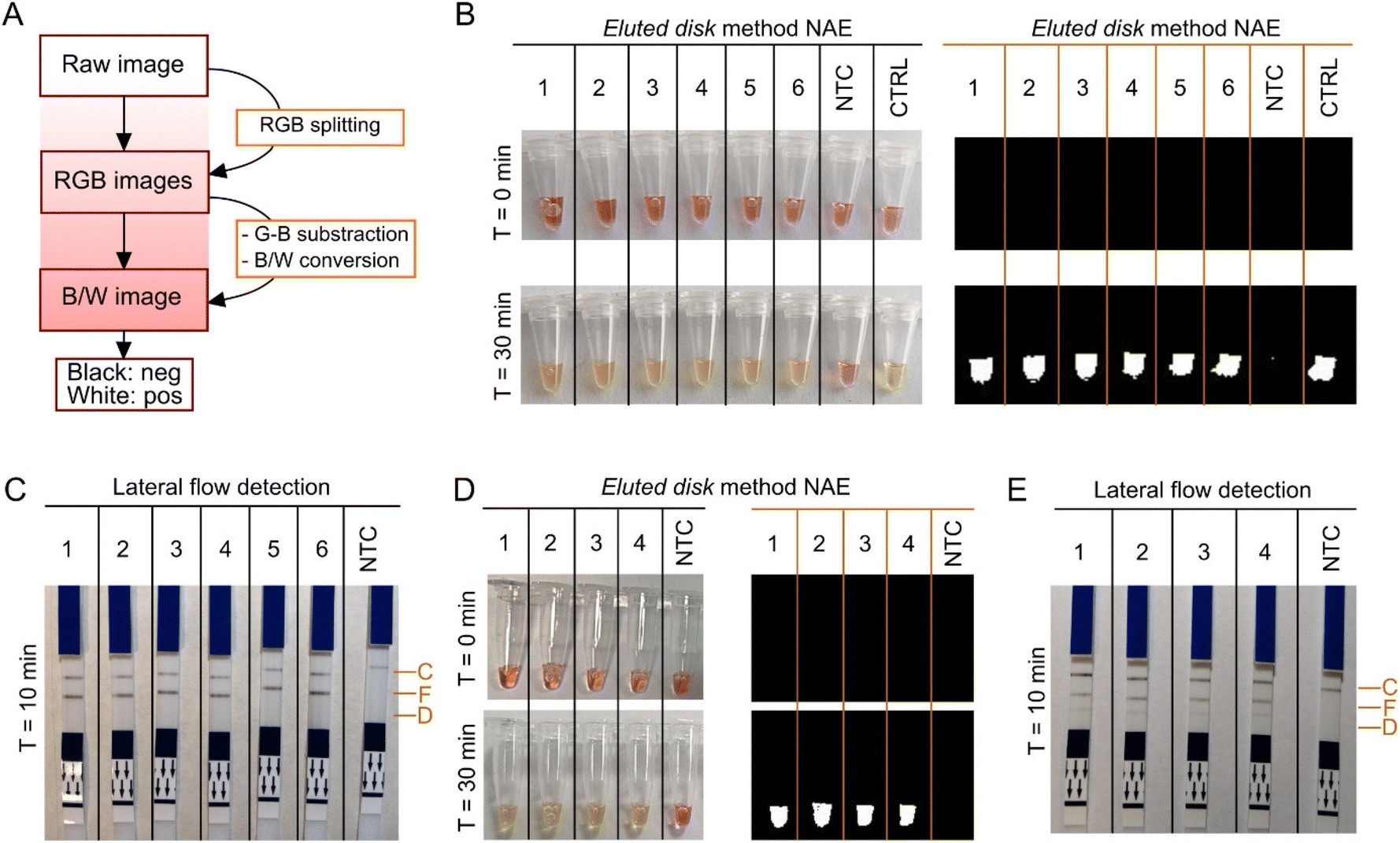
Real-time, colorimetric and lateral flow detection was performed with LAMP using extracted nucleic acids by the eluted disk method. For objective colorimetric readout, an algorithm was implemented in MATLAB based on the work reported by Rodriguez-Manzano et al.28 The script was adjusted for the discrimination between pink and yellow to obtain a binary output, where white corresponded to amplification and black to non-amplification. Workflow is shown in Fig. 5A and script in Fig. S7.†
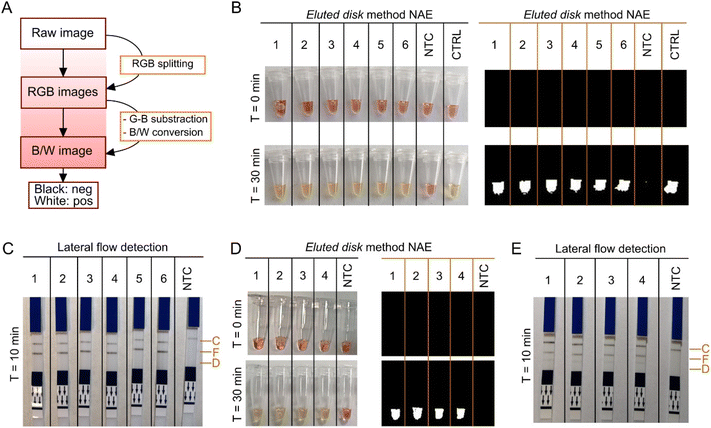 |
| | Fig. 5 Colorimetric and lateral flow detection. (A) Workflow for colorimetric image processing. Algorithm implemented in MATLAB from raw image to a binary output. (B) Colorimetric detection of ACTB from extracted nucleic acids with the eluted disk method, and post-processed images. Samples used included whole blood in K2EDTA (1–3) and whole blood in LiHep (4–6); non-template control (NTC); and positive control (CTRL), purified human genomic DNA (Promega). (C) Lateral flow detection of ACTB gene from extracted nucleic acids with the eluted disk method. (D) Colorimetric detection of P. falciparum DNA from extracted nucleic acids with the eluted disk method, and post-processed images. Samples included spiked synthetic DNA of P. falciparum in whole blood at final concentration per reaction as follows: 7.5 × 106, 7.5 × 105, 7.5 × 104, 7.5 × 103 copies per reaction, and NTC. (E) Lateral flow detection of P. falciparum DNA and ACTB from extracted nucleic acids with the eluted disk method. C – Control; F – FAM; D – DIG. | |
Results in Fig. 5B showed amplification of the ACTB gene from extracted nucleic acids using the eluted disk method as a colour change from pink to yellow, and as a binary output. Same samples were extracted with the magnetic bead-based method and colorimetric detection was performed; results are sown in Fig. S8.† Lateral flow detection of the extracted nucleic acids is shown in Fig. 5C, detected in the test line specific for FAM after 10 min incubation. Lastly, spiked whole blood stored in LiHep with synthetic DNA of P. falciparum at different concentrations (7.5 × 106, 7.5 × 105, 7.5 × 104, 7.5 × 103 copies per reaction) was tested with colorimetric and lateral flow detection. Colorimetric detection of the four concentrations is shown in Fig. 5D, and lateral flow detection in Fig. 5E. Bands specific for FAM (ACTB gene) and DIG (P. falciparum) can be observed. LAMP amplification using only the P. falciparum-specific assay is shown in Fig. S9†, where only bands specific for DIG are visible. Sample-to-result was obtained under 35 min with the colorimetric approach and under 45 min with the lateral flow approach.
Conclusions
Molecular diagnostics from DBS envisioned to be used at the POC are currently limited by the complex laboratory-based methods for NAE4,29 or their application to PCR which require high temperature incubation and thermal-cycling.7,17 To date, a simplified and rapid protocol for NAE and its subsequent downstream application for isothermal amplification has not been reported. In this work, we have developed a paper-based NAE method which consists of a pre-treatment of filter paper with a mild surfactant (e.g. igepal30–32), spotting of sample, a washing step (1 min) and incubation at RT conditions (5 min). We have shown high yield recovery of spiked synthetic DNA and RNA extraction without the need of equipment, long procedures or commercial kits.33–35 Leveraging on the duality of paper-based matrices, the elution obtained from the incubation step (eluted disk method) and the use of the disk itself (in situ disk method) were successfully applied in combination with LAMP for the lysis of eukaryotic cells, detection of the human reference gene ACTB and detection of cell-free synthetic DNA of P. falciparum (kelch 13 gene). This is the first study that has explored the duality of DBSFP. We have demonstrated the use of standard grade 3 filter paper (∼£0.10 per card) for accurate NAE from blood, and the potential use of more expensive (£3–5 per card) commercial filter papers (FTA Classic cards, FTA Elute cards, 903 Protein Saver and Fusion 5) with the developed eluted disk and in situ disk methods. The lower sample input for NAE with our methodology (2 μL × 3 disks) compared to standard methods such as magnetic bead-based extraction (100 μL) demonstrates that this method is less invasive but equivalent in performance. In future work, the applicability could be extended to the detection of bloodstream pathogens such as virus, bacteria or parasites,37–39 and also applications in genomics and transcriptomics involving the detection of endogenous genes with RNA-specific assays. Igepal has been previously described as a component in lysis buffers for bacterial, viarl or eukaryotic cells, therefore we anticipate that it could be compatible to lyse pathogenic material.31,40,41 The use of DBSFP without the need of prior cell or bacteria culture, will highly reduce the processing time of samples and allow the detection of pathogens which cannot be currently cultured. As proof-of-concept, we have successful preliminary data of NAE of RNA from SARS-CoV-2 viral particles and DNA from Escherichia coli using the eluted disk method and available LAMP assays42,43 (Fig. S10†). Given the reported applicability of FTA cards for sample preservation and NAE, we expect that the proposed methods could be compatible with other matrices such as buccal swabs, faecal swabs, urine or saliva.44 Since we have also demonstrated a sample-to-result proof-of-concept workflow through the combination of the eluted disk method with colorimetric LAMP and lateral flow detection, we envision that eluted disk and in situ disk could be used at the POC once embedded in a portable diagnostic format.45 Furthermore, we anticipate that once integrated they could be applied for sample archiving and improve the current pipeline for paper-based POC diagnostics.
Author contributions
Conceptualisation – KMC and JRM. Methodology, investigation, project administration, visualisation – KMC. Supervision – JRM and PG. Funding acquisition – KMC, JRM, PG, AC, JB. Writing original draft – KMC and JRM. Writing review and editing – all authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
EPSRC HiPEDS CDT (EP/L016796/1) and EPSRC Doctoral Prize Fellowship (EP/T51780X/1) to KMC; Wellcome Trust Investigator Award (100993/Z/13/Z) to JB; UKRI Global Challenge Research Fund (EP/T029005/1) to AC. JRM and PG are affiliated with the NIHR Health Protection Research Unit (HPRU) in Healthcare Associated Infections and Antimicrobial Resistance at Imperial College London in partnership with the UK Health Security Agency (previously PHE) in collaboration with, Imperial Healthcare Partners, the University of Cambridge and the University of Warwick. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health and Care Research, the Department of Health and Social Care, or the UK Health Security Agency. JRM, PG and KMC are affiliated with the Department of Health and Social Care, Centre for Antimicrobial Optimisation (CAMO) at Imperial College, London.
References
- J. D. Freeman, L. M. Rosman, J. D. Ratcliff, P. T. Strickland, D. R. Graham and E. K. Silbergeld, Clin. Chem., 2018, 64, 656–679 CrossRef CAS PubMed.
- R. Guthrie and A. Susi, Pediatrics, 1963, 32, 338–343 CrossRef CAS PubMed.
- S. P. Parker and W. D. Cubitt, J. Clin. Pathol., 1999, 52, 633–639 CrossRef CAS PubMed.
- J. R. Choi, R. Tang, S. Wang, W. A. B. Wan Abas, B. Pingguan-Murphy and F. Xu, Biosens. Bioelectron., 2015, 74, 427–439 CrossRef CAS PubMed.
- R. H. Tang, L. N. Liu, S. F. Zhang, X. C. He, X. J. Li, F. Xu, Y. H. Ni and F. Li, Microchim. Acta, 2019, 186, 521 CrossRef PubMed.
- S. Lehmann, C. Delaby, J. Vialaret, J. Ducos and C. Hirtz, Clin. Chem. Lab. Med., 2013, 51(10), 1897–1909 CrossRef CAS PubMed.
- W. Gan, B. Zhuang, P. Zhang, J. Han, C.-X. Li and P. Liu, Lab Chip, 2014, 14, 3719–3728 RSC.
- B. Aydin-Schmidt, W. Xu, I. J. González, S. D. Polley, D. Bell, D. Shakely, M. I. Msellem, A. Björkman and A. Mårtensson, PLoS One, 2014, 9, e103905 CrossRef PubMed.
- M. K. Hegazy, S. I. Awad, N. E. Saleh and M. M. Hegazy, Exp. Parasitol., 2020, 211, 107869 CrossRef CAS PubMed.
- J. J. C. de Vries, E. C. J. Claas, A. C. M. Kroes and A. C. T. M. Vossen, J. Clin. Virol., 2009, 46, S37–S42 CrossRef CAS PubMed.
- M. Hashimoto, M. Bando, J. Kido, K. Yokota, T. Mita, K. Kajimoto and M. Kataoka, Parasitol. Int., 2019, 73, 101941 CrossRef CAS PubMed.
- R. P. de Bie, C. E. Schmeink, J. M. J. E. Bakkers, P. J. F. Snijders, W. G. V. Quint, L. F. A. G. Massuger, R. L. M. Bekkers and W. J. G. Melchers, J. Mol. Diagn., 2011, 13, 371–376 CrossRef PubMed.
- A. N. Mohon, S. Getie, N. Jahan, M. S. Alam and D. R. Pillai, Malar. J., 2019, 18, 1–10 CrossRef CAS PubMed.
- E.-H. Choi, S. K. Lee, C. Ihm and Y.-H. Sohn, Osong Public Health Res. Perspect., 2014, 5, 351–357 CrossRef PubMed.
- G. Chondrogiannis, P. Réu, M. M. Hamedi, G. Chondrogiannis, P. Réu and M. M. Hamedi, Adv. Mater. Technol., 2023, 8, 2201004 CrossRef CAS.
- J. T. Connelly, J. P. Rolland and G. M. Whitesides, Anal. Chem., 2015, 87, 7595–7601 CrossRef CAS PubMed.
- O. W. Stringer, J. T. Bossé, S. Lacouture, M. Gottschalk, L. Fodor, Ø. Angen, E. Velazquez, P. Penny, L. Lei, P. R. Langford and Y. Li, Front. Vet. Sci., 2021, 8, 1–9 Search PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, E63 CrossRef CAS PubMed.
- A. J. Cunnington, PLOS Digital Health, 2022, 1, e0000064 CrossRef PubMed.
- N. Grüner, O. Stambouli and R. S. Ross, J. Visualized Exp., 2015, 2015, 1–9 Search PubMed.
- L. L. M. Poon, C. S. W. Leung, K. H. Chan, J. H. C. Lee, K. Y. Yuen, Y. Guan and J. S. M. Peiris, J. Clin. Microbiol., 2005, 43, 427–430 CrossRef CAS PubMed.
- M. N. Anahtar, G. E. G. McGrath, B. A. Rabe, N. A. Tanner, B. A. White, J. K. M. Lennerz, J. A. Branda, C. L. Cepko and E. S. Rosenberg, Open Forum Infect. Dis., 2021, 8(2), ofaa631 CrossRef PubMed.
- N. Ali, R. d. C. P. Rampazzo, A. D. T. Costa and M. A. Krieger, BioMed Res. Int., 2017, 2017, 9306564 Search PubMed.
- K. Malpartida-Cardenas, N. Miscourides, J. Rodriguez-Manzano, L. S. Yu, N. Moser, J. Baum and P. Georgiou, Biosens. Bioelectron., 2019, 145, 111678 CrossRef CAS PubMed.
- M. Ding, A. Bullotta, L. Caruso, P. Gupta, C. R. Rinaldo and Y. Chen, J. Virol. Methods, 2011, 176, 1–8 CrossRef CAS PubMed.
- P. Tiberio, L. De Cecco, M. Callari, E. Cavadini, M. G. Daidone, V. Appierto, D. M. Duelli, J. Palma, K. Beaman, M. L. Hastings, J. Kwak-Kim, S. Linnstaedt, J. C. Park and D.-J. Kim, J. Mol. Diagn., 2013, 15, 138–139 CrossRef PubMed.
- J. Kim, B. G. Park, D. H. Lim, W. S. Jang, J. Nam, D.-C. Mihn and C. S. Lim, PLoS One, 2021, 16, e0244753 CrossRef CAS PubMed.
- J. Rodriguez-Manzano, M. A. Karymov, S. Begolo, D. A. Selck, D. V. Zhukov, E. Jue and R. F. Ismagilov, ACS Nano, 2016, 10, 3102–3113 CrossRef CAS PubMed.
- P. Li, M. Li, D. Yue and H. Chen, J. Sep. Sci., 2022, 45(1), 172–184 CrossRef CAS PubMed.
- A. Castellanos-Gonzalez, T. R. Shelite, N. Lloyd, A. Sadiqova, R. Ping, N. Williams-Bouyer, P. C. Melby and B. L. Travi, Sci. Rep., 2021, 11, 14204 CrossRef CAS PubMed.
- A. Viet-Phuong Le, D. Huang, T. Blick, E. W. Thompson and A. Dobrovic, Sci. Rep., 2015, 5, 12859 CrossRef CAS PubMed.
- W. J. Xia and H. Onyuksel, Pharm. Res., 2000, 17, 612–618 CrossRef CAS PubMed.
- T. W. McDade, K. M. Ross, R. L. Fried, J. M. G. Arevalo, J. Ma, G. E. Miller and S. W. Cole, Biodemography Soc. Biol., 2016, 62, 182–197 CrossRef PubMed.
- M. J. Reust, M. H. Lee, J. Xiang, W. Zhang, D. Xu, T. Batson, T. Zhang, J. A. Downs and K. M. Dupnik, Am. J. Trop. Med. Hyg., 2018, 98, 1541–1546 CrossRef CAS PubMed.
- M. Monleau, C. Montavon, C. Laurent, M. Segondy, B. Montes, E. Delaporte, F. Boillot and M. Peeters, J. Clin. Microbiol., 2009, 47, 1107–1118 CrossRef CAS PubMed.
- D. Rajendram, R. Ayenza, F. M. Holder, B. Moran, T. Long and H. N. Shah, J. Microbiol. Methods, 2006, 67, 582–592 CrossRef CAS PubMed.
- G. Singh and R. Sehgal, Asian J. Transfus. Sci., 2010, 4, 73 CrossRef PubMed.
- S. A. Dunbar, C. Gardner and S. Das, Front. Cell. Infect. Microbiol., 2022, 12 DOI:10.3389/fcimb.2022.859935.
- D. J. Diekema, P.-R. Hsueh, R. E. Mendes, M. A. Pfaller, K. V. Rolston, H. S. Sader and R. N. Jones, Antimicrob. Agents Chemother., 2019, 63 DOI:10.1128/AAC.00355-19.
- F. Song, J. V. Kuehl, A. Chandran and A. P. Arkin, mSystems, 2021, 6 DOI:10.1128/MSYSTEMS.00224-21/SUPPL_FILE/REVIEWER-COMMENTS.PDF.
-
N. Craig, S. L. Fletcher, A. Daniels, C. Newman, M. O'Shea, A. Warr and C. Tait-Burkard, bioRxiv, 2021, preprint, DOI:10.1101/2021.11.30.470550.
- J. Rodriguez-Manzano, K. Malpartida-Cardenas, N. Moser, I. Pennisi, M. Cavuto, L. Miglietta, A. Moniri, R. Penn, G. Satta, P. Randell, F. Davies, F. Bolt, W. Barclay, A. Holmes and P. Georgiou, ACS Cent. Sci., 2021, 7(2), 307–317 CrossRef CAS PubMed.
- J. Hill, S. Beriwal, I. Chandra, V. K. Paul, A. Kapil, T. Singh, R. M. Wadowsky, V. Singh, A. Goyal, T. Jahnukainen, J. R. Johnson, P. I. Tarr and A. Vats, J. Clin. Microbiol., 2008, 46, 2800–2804 CrossRef CAS PubMed.
- D. Obino, M. Vassalli, A. Franceschi, A. Alessandrini, P. Facci and F. Viti, Sensors, 2021, 21, 3058 CrossRef CAS PubMed.
- K. Malpartida-Cardenas, N. Miscourides, J. Rodriguez-Manzano, L.-S. Yu, N. Moser, J. Baum and P. Georgiou, Biosens. Bioelectron., 2019, 145 DOI:10.1016/j.bios.2019.111678.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Jake
Baum
cd,
Aubrey
Cunnington
a,
Pantelis
Georgiou
b and
Jesus
Rodriguez-Manzano
*a
ab,
Jake
Baum
cd,
Aubrey
Cunnington
a,
Pantelis
Georgiou
b and
Jesus
Rodriguez-Manzano
*a