
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reporter emission multiplexing in digital PCRs (REM-dPCRs): direct quantification of multiple target sequences per detection channel by population specific reporters†
Silvia
Calabrese‡
 a,
Anja M.
Markl‡
ab,
Maximilian
Neugebauer
ac,
Stefanie J.
Krauth
de,
Nadine
Borst
ac,
Felix
von Stetten
*ac and
Michael
Lehnert
a
a,
Anja M.
Markl‡
ab,
Maximilian
Neugebauer
ac,
Stefanie J.
Krauth
de,
Nadine
Borst
ac,
Felix
von Stetten
*ac and
Michael
Lehnert
a
aHahn-Schickard, 79110 Freiburg, Germany. E-mail: vstetten@imtek.de
bFaculty of Chemistry and Pharmacy, Albert-Ludwigs-Universität Freiburg, 79104 Freiburg, Germany
cLaboratory for MEMS Applications, IMTEK-Department of Microsystems Engineering, University of Freiburg, 79110 Freiburg, Germany
dSchool of Biodiversity, One Health, and Veterinary Medicine, University of Glasgow, Glasgow, UK
eSchool of Health and Wellbeing, General Practice and Primary Care, University of Glasgow, Glasgow, UK
First published on 13th September 2023
Abstract
Digital PCRs (dPCRs) are widely used methods for the detection and quantification of rare abundant sequences relevant to fields such as liquid biopsy or oncology. In order to increase the information content and save valuable sample materials, there is a significant need for digital multiplexing methods that are easy to establish, analyse, and interpret, and ideally allow the usage of existing lab equipment. Herein, we present a novel reporter emission multiplexing approach for the digital PCR method (REM-dPCR), which meets these requirements. It further increases the multiplexing capacity of commercial dPCR devices. For example, we present a stepwise increase in multiplexing degrees from a monochrome two-plex assay in one detection channel to a six-plex REM-dPCR assay in a three-color dPCR device for KRAS/BRAF single nucleotide polymorphism (SNP) target sequences. The guidelines for the REM-dPCR design are presented, and the process from duplex to six-plex assay establishment, taking into account the target sequence-dependent effects on assay performance, is discussed. Furthermore, the assay-specific, sensitive and precise quantification of different fractions of KRAS mutant and wild-type DNA sequences in different ratios is demonstrated. To increase the device capacitance and the degree of multiplexing, the REM-dPCR uses the advantage of n target-independent reporter molecules in combination with target sequence-specific mediator probes. Different reporter types are labelled with fluorophores of different signal intensities but not necessarily different emission spectra. This leads to the generation of n independent single-positive populations in the dataspace, created by k detection channels, whereby n > k and n ≥ 2. By usage of target-independent but population-specific reporter types, a fixed set of six optimized signalling molecules could be defined. This reporter set enables the robust generation and precise differentiation of multiple fluorescence signals in dPCRs and can be transferred to new target panels. The set which enables stable signal generation and differentiation in a specified device would allow easy transfer to new target panels.
Introduction
In recent years, digital PCRs (dPCRs) advanced to a widely used technology in molecular analysis and diagnostics, providing new approaches such as the detection of copy number variations,1 rare genetic variants, single nucleotide variations, detection of circulating tumor DNA or in infectiology.2,3–5 Especially in the field of liquid biopsy, dPCRs experience a strong increase due to their higher sensitivity than that of qPCRs,6 their specificity for mutation detection in a strong background of wild-type DNA and their robustness against inhibitory effects and interlaboratory transferability.7 In order to increase the information content and save valuable sample materials, multiplexing is the method of choice for the parallel detection of multiple DNA target sequences in one dPCR.In dPCRs, quantification solely depends on fluorescence signal counts (measurement of fluorescence intensity per compartment and detection channel). Hence, the fluorescence intensity is a free parameter that can be used to further differentiate signals within a detection channel. This is done by plotting fluorescence signal intensities and counts, whereby the number of dimensions in the dataspace increases with the number of used fluorescence detection channels. In distinct areas of those plots, fluorescence signals from reporter molecules such as hydrolysis probes group into clusters, also designated as populations. These populations can be assigned to different DNA target sequences.8
Current dPCR multiplexing methods can be roughly classified into colorimetric, amplitude, ratio-based and population pattern-based methods.9–11 Colorimetric multiplexing is based on the detection of one target sequence via one corresponding fluorescence signal per detection channel. Even though the degree of multiplexing is limited to the number of available fluorescence detection channels per device, colorimetric multiplexing enables precise, sensitive and robust detection of multiple target sequences in one PCR. In recent years, the number of available detection channels in commercial devices has increased from, on average, two to up to six detection channels. However, with the increase in fluorescence detection channels, there is an increase in spectral overlap. This may lead to signal spillover between detection channels, which in turn leads to reduced sensitivity and specificity of the assays.12,13 As a result, careful adjustment and optimization of assay design parameters such as the determination of adequate fluorophore and quencher pairs, oligonucleotides and cycling parameters are necessary. To counteract such spillover effects, compensation matrixes have to be applied, where single target reactions are run and measured in available detection channels. Once calibrated, the compensation matrix can be applied for this specific assay set-up only.5,14
To overcome the device limitations and increase the multiplexing degree, further amplitude and/or ratio-based multiplexing approaches were developed, where the additive character of fluorescence signals as well as different ratios of primer and probe concentrations were used to generate additional distinguishable populations of different intensities in the dataspace beyond the number of fluorescence channels.9 However, assay development requires complex and detailed optimization in order to achieve distinguishable populations, which makes development quite laborious and, therefore, not easily transferable to other target panels.9 Other approaches use either drop-off probes or classical hydrolysis probes to differentiate multiple targets per channel via combinatorial approaches. It was possible to distinguish up to 19 targets via a drop-off assay and 15 targets via a combinatorial approach.10,15 In the drop-off assay, target sequences were not detected particularly but multiple targets were grouped according to the applicable treatment scheme.10,11 Furthermore, hydrolysis probes were used for the drop-off assay development, which makes data analysis not intuitive and assay transfer to new target panels quite laborious. In conclusion, these different concepts show that the current bottle neck in dPCR multiplexing is to reach sufficient signal differentiation and simple data analysis. Even though the present technologies started to overcome this challenge, still a large degree of assay optimization is needed to create appropriate signal populations. This process has to be repeated for each new target sequence. As intensive signal optimization loops are often not feasible in daily clinical lab routine, rain may be tolerated to a certain amount and simpler single plex reactions are preferred over complex multiplex reactions, accepting loss of sample materials and/or information.
Therefore, there is a high need for a robust and intuitive method that extends colorimetric multiplexing, overcomes device limitations and is easy to adapt to new target panels. Schlenker et al.16 presented a photobleaching approach, in which there is an increase in the multiplexing degree by a factor of two per channel in a dPCR device. The dPCR-reporter molecules are labelled with either a photostable or a photosensitive fluorophore. Upon exposure to light, the photosensitive fluorophores lose their ability to be excited to a higher energetic state. Therefore, after the photobleaching process through an external light source, populations with photostable universal reporters can be detected at higher intensities and populations with photolabile universal reporters show lower signal intensities, doubling the number of targets to be detected per channel. The functionality of this monochrome multiplexing method could be shown for duplex reactions in three independent channels. Even though efficiently increasing the multiplexing degree, the process requires an additional light source and time for photobleaching, which poses a hurdle to direct implementation in workflows with standard laboratory devices. This photobleaching approach was furthermore based on Mediator Probe PCR (MP-PCR). This technology is an established PCR method in which target detection (by MPs) and signal generation (by fluorescence-labelled universal reporters) occur in two different steps.3,4,17 Optimized labelled reporter molecules can be used for different target panels, allowing easy assay design.18 Furthermore, this method has been demonstrated to be highly specific and sensitive for SNP detection and digital PCR applications, where cancer markers were detected in two- and four-plex reactions.17 The high specificity for SNP detection of this method is especially important for clinical applications such as cancer diagnosis and minimal residual disease monitoring, due to limited sample volume and the concentration of marker molecules.
In this work, we present a new method of reporter emission multiplexing in digital PCRs (REM-dPCRs), which allows the detection of multiple target sequences per fluorescence detection channel, and which can be implemented straight forward using standard laboratory instruments and workflows. We demonstrate specific detection of six target sequences in a three-color-dPCR device, for example, on a KRAS/BRAF target panel, which are relevant biomarkers in oncology. This was accomplished without the need for additional external devices, handling steps or combinatorial data interpretation. REM-dPCR is based on the reaction mechanism of MP-PCRs. By using fluorescence signal intensity variations of differently labelled molecules, population-specific reporters (PSR) are generated in dPCRs. Using PSRs, distinguishable populations could be generated per detection channel or within the respective multidimensional dataspace (Fig. 1). We demonstrate the REM-dPCR principle using MP and PSR molecules. As the PSRs are target independent, one set of reporter molecules could be defined for each dPCR device, which makes assay development and optimization more efficient. Starting from the establishment of REM-dPCR duplex assays in one detection channel, we provide guidance through the whole workflow for the creation of a six-plex assay in a standard three-color-dPCR device. Furthermore, we discuss the upcoming effects on the assay design caused by multiple SNP detection in the same gene region, using REM-dPCRs.
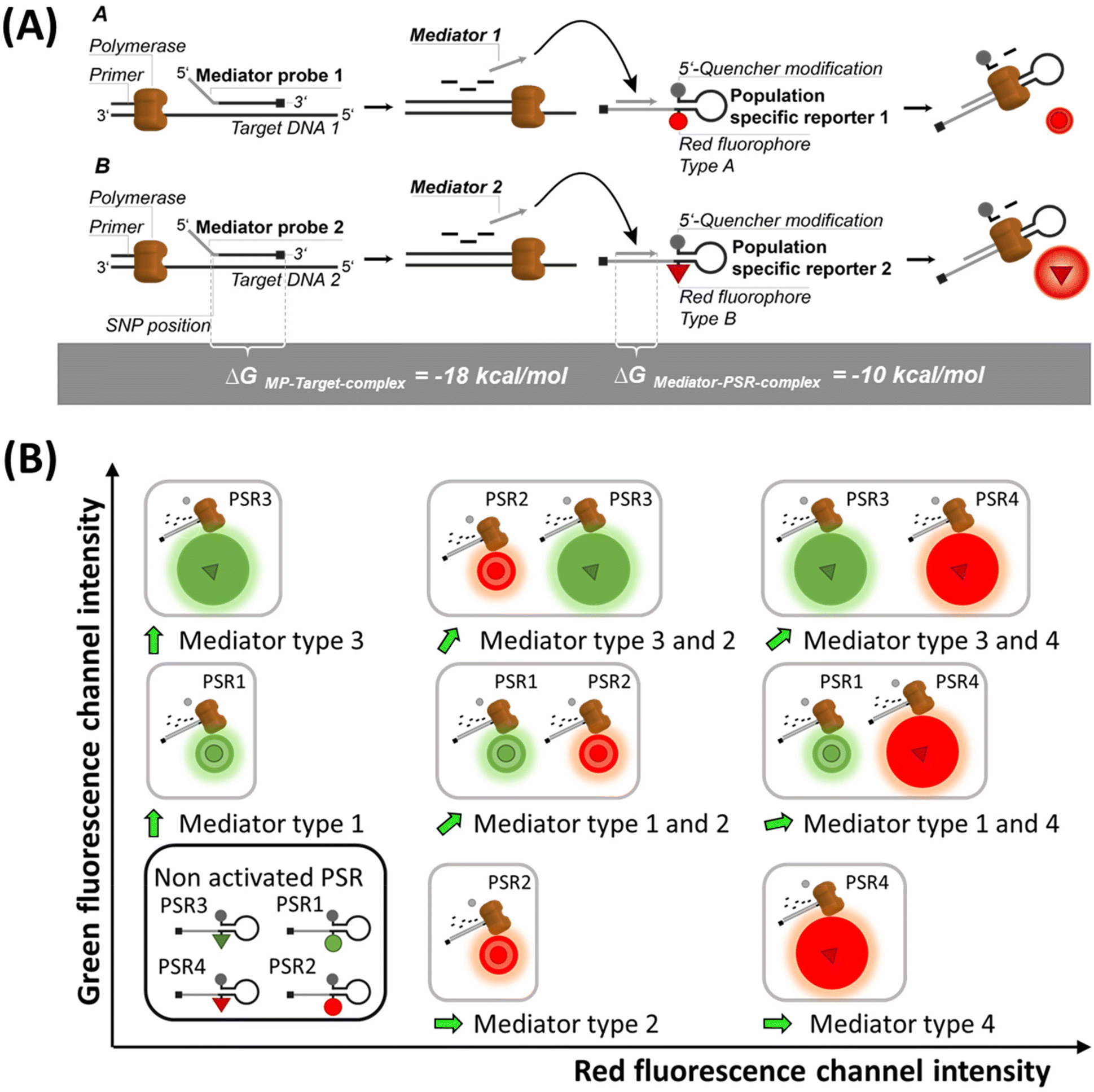 | ||
| Fig. 1 Schematic of the signal generation and population formation in the dataspace by REM-dPCRs. (A) Sequence-specific mediator probes (MPs) bind to their target sequence. Upon enzymatic cleavage, the released mediators bind to their target-independent population-specific reporter (PSR) types, which then generate a fluorescence signal. Therefore, generated signals from PSR1 and PSR2 can be detected in the same detection channel but can be differentiated by their signal intensities. (B) Schematic orientation of the signal populations in the dataspace of a four-plex REM-dPCR in two detection channels (green and red) using four different target-independent PSR types. In relation to each other, the target-independent PSR types form fixed populations in the dataspace of the respective detection channels. | ||
Experimental
Materials
Sequence information of all oligonucleotides and synthetic templates used is provided in ESI Table S1.† Customized primers, MPs and PSRs were synthesized and HPLC-purified at biomers.net. Oligonucleotides were dissolved in nuclease-free water to a stock concentration of 100 μM and stored at −20 °C. Synthetic templates (gBlocks) were purchased from Integrated DNA Technologies and resuspended in 1× TE-buffer (Sigma Aldrich) to a stock concentration of 10 ng μl−1 according to the manufacturer's instruction. Template concentrations were determined using a Qubit dsDNA high-sensitivity assay kit (Thermo Fisher Scientific Inc.). dPCRs were run in Sapphire chips using a Naica System (Prism3) from Stilla Technologies. Moreover, 5× PerfeCTa® MultiPlex qPCR ToughMix® was purchased from Quantabio and used at 1× final concentration. As background, fluorophore Alexa Fluor 488 Dye (Invitrogen by Thermo Fisher Scientific Inc.) was used at concentrations of 400 nM in two-plex experiments in green and red channels and 50 nM for further experiments, respectively.Assay design
PSRs were designed together with target-specific MPs according to Lehnert et al.19 using the software OligoPad version 0.3.9.4 (GNWI – Gesellschaft für naturwissenschaftliche Informatik GmbH) and VisualOMP version 7.8.42.0 (DNA Software Inc.).20 Sequence homologies to the human genome were analysed by nucleotide BLAST search on NCBI.21For the PSR and MP design, special attention was paid to the binding enthalpies and secondary structures. PSRs were designed as single-stranded oligonucleotides using a universal reporter basic structure with a hairpin at the 5′-end, followed by a mediator binding site. Overall, the stem loop structures had a length of 20 nt, whereby 6 bp with a ΔG value of around −4 kcal mol−1 formed the stem due to self-complementarity. Quenchers were linked to the 5′-end and fluorophores were coupled to the complementary base at the opposite position. To avoid G-quenching of green fluorophores, neighbouring nucleotides were selected from A/T bases. All PSRs were modified with a UR-block at the 3′-end. For MP design, it was of upmost importance to reach a binding ΔG value of approximately −18 kcal mol−1 at the probe hybridization side and a ΔG value of approximately −10 kcal mol−1 at the mediator hybridization side of the PSRs. Designed PSR types with respective quencher and fluorophore combinations are listed in Table 1.
| PSR type | Quencher | Fluoro-phore | Name | Target | |
|---|---|---|---|---|---|
| 02 | BMN-Q1 | DY-530 | KRAS | WT | III |
| 04 | BHQ-1 | BMN-536 | G12A | IV | |
| 05 | BHQ-2 | Cy5 | G12D | V | |
| 06 | BHQ-2 | Atto 647N | G12V | VI | |
| 01 | BHQ-2 | Cy5.5 | KRAS | G12V | VI |
| 06 | BHQ-2 | Atto680 | WT | III | |
| 07 | BMN-Q1 | Atto 488 | BRAF | WT | I |
| 08 | BHQ-1 | FAM | V600E | II | |
Multiplexing conditions
Initial dPCR parameters (C1, Table 2) were taken from Schlenker et al.16 As higher multiplexing degrees require a well-balanced PCR system, especially when multiple SNP sides were to be detected, PCR parameters were optimized with the increase in multiplexing degree.| Concentrations per target [nM] | ||||
|---|---|---|---|---|
| Condition | Primer–fwd | Primer–rev | MP | PSR |
| C1 | 500 | 500 | 1000 | 250 |
| C2 | 500 | 250 | 1200 | 600 |
| C3 | 500 | 250 | 1200 | 400 |
The annealing temperature was varied between 56 °C and 60 °C and set to 58 °C for conditions C2 and C3 (Table 3).
| Cycling protocols for: Naica Prism 3 | |||
|---|---|---|---|
| Condition | Temperature [°C] | Time [s] | Cycle |
| Partitioning | 40 | 720 | 1× |
| C1 | 95 | 300 | 1× |
| 95 | 15 | 45× | |
| 60 | 40 | ||
| C2/C3 | 95 | 300 | 1× |
| 95 | 15 | 45× | |
| 58 | 60 | ||
| Pressure equalization | 25 | 1980 | 1× |
Oligonucleotide titrations were performed for two-plex and four-plex reactions. Template concentrations were set to 3000 copies per reaction. For the duplex oligonucleotide titration, the following concentration ranges were tested: forward and reverse primers ranged from 500 to 1250 nM in 250 nM steps. The MPs ranged between 800 nM and 1400 nM in 200 nM steps and the PSR concentrations were tested at 200/240/400/600 nM. The resulting optimized two-plex condition C2 is listed in Table 2.
For the optimization of the four-plex, the following MP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PSR ratios were used: 3:1; 4:1, and 5:1, whereby the MP concentration was kept constant at 1200 nM.
:PSR ratios were used: 3:1; 4:1, and 5:1, whereby the MP concentration was kept constant at 1200 nM.
Forward-to-reverse primers ratios were 2:1, 1.5:1.5, and 1:2 μM. The resulting optimized four-plex condition C3 is listed in Table 2 and was applied also to the six-plex condition where oligonucleotide concentrations were set to 1.2 μM per MP, 0.4 μM per PSR, 2 μM forward primer KRAS, 1 μM reverse primer KRAS, 1 μM forward primer BRAF, and 0.5 μM reverse primer BRAF.
The quantification of KRAS G12 V mutant DNA together with KRAS wild-type DNA was done with a Naica Prism 6 (Stilla Technologies) using the cycling protocol given in Table 4:
The assay was performed in naica® multiplex PCR MIX buffer A in a 1× concentration with 4% of naica® multiplex PCR MIX buffer B. To avoid unspecific DNA adsorption, herring sperm DNA was added at a concentration of 10 μg ml−1. Primers were used at a final concentration of 2 μM (forward) and 1 μM (backward). The PSR concentrations were kept at 400 nM each. MPs were used at a concentration of 1.2 μM (KRAS wild type) and 1.0 μM (KRAS G12 V).
| Cycling protocols for: Naica Prism 6 | |||
|---|---|---|---|
| Condition | Temperature [°C] | Time [s] | Cycle |
| Partitioning | 40 | 720 | 1× |
| C2/C3 | 95 | 300 | 1× |
| 95 | 15 | 45× | |
| 58 | 60 | ||
| Pressure equalization | 25 | 1980 | 1× |
Data analysis
REM-dPCR data were evaluated using a commercial analysis software for dPCR (CrystalMiner Software version 3.1.6.3, Stilla® Technologies). Spillover compensation was performed with single target controls according to the recommendation of Stilla® Technologies: Stilla® Technologies user manual v2.2 “Crystal Miner for the Naica™ System”.14To qualitatively assess the improvement of clustering behaviour and compactness of positive populations, point pattern, analysis and variance-to-mean ratios were used in a one-dimensional approach. A scale was defined from the lowest to the highest fluorescence intensity, where each integer intensity was defined as one “square”. The value of each square depended on the droplet count of a defined fluorescence intensity. The mean square values, variances and sum of squared values were taken. Non-random or uniform distributions respectively were mirrored by high variance-to-mean ratio (VMR) values. Significance tests were based on the chi-square frequency distribution, where squared square values were integrated into the sum of squared differences divided by the mean value, whereby higher values indicated a stronger clustering.
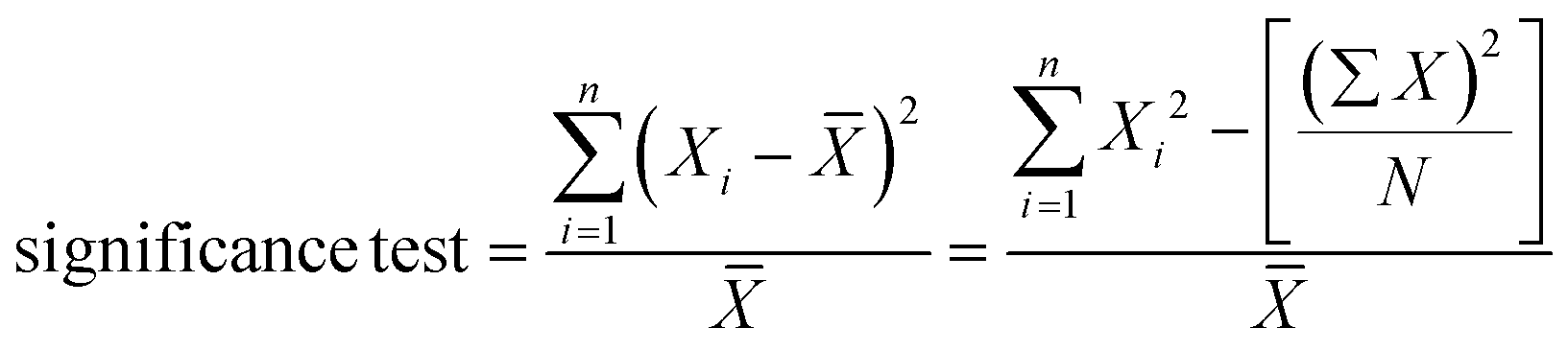
High-significance test values result from a non-random clustering. When the value was ten times higher than the number of analysed values, the probability of a random clustering was below 1%. Data were evaluated using Microsoft Excel. The VMR was applied in the comparison of reactions with the same PSR and target concentrations for optimization in one detection channel.
For the dilution row, the calculation of measured target molecule concentration (C) was performed using the following formula:
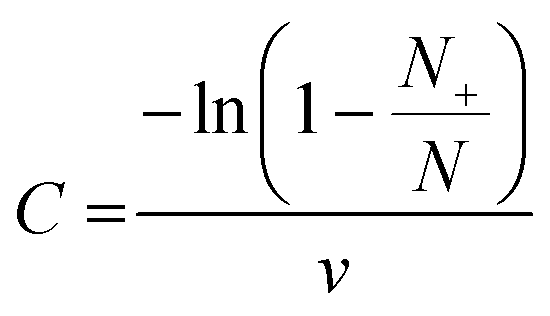
Results and discussion
Principle of REM-dPCRs
The REM-dPCR is a method that allows direct endpoint detection of n targets in k detection channels, where n > k and n ≥ 2. In contrast to other technologies, signals are not generated by target sequence-specific fluorogenic probes but by target sequence-independent fluorogenic reporters, so-called population-specific reporters (PSR), which can be clearly discriminated in the intensity domain and so population in data space. This adds additional degrees of freedom to the design process. Furthermore, it allows the use of reporter molecule structures with contact quenching for initial signal suppression, by which means a higher separation of positive and negative events can be achieved. Additionally, it allows a more controlled and robust signal generation because limitations due to target sequence effects can be ignored, making the reporter optimization process more effective.19 These PSR molecules are based on universal reporter structures and used in combination with MPs for target sequence detection.22The basic principle of REM-dPCRs is the detection of n target nucleic acid sequences by n target-specific MP types, which will activate n PSR types, whereby each PSR type is labelled with a unique fluorophore of defined optical properties (Fig. 1A). In the example of a duplex reaction, two fluorophores would be chosen, which emit light in the same wavelength range but differ in their intensities in the detection range. During PCR amplification, the bound label-free MPs are cleaved by the polymerase and release a mediator sequence. This mediator activates a PSR type, which will then generate a fluorescence signal. The sum of all droplets or compartments with a signal of a specific reporter type will form a population in a distinct area of the dataspace, according to the unique fluorescence label. Those PSR types labelled with fluorophores of a higher quantum yield will generate positive populations of higher fluorescence intensities (Fig. 1A bottom: high intensities are abstracted as bigger spheres), and those with lower quantum yields in the detection range will generate positive populations of lower fluorescence intensities (Fig. 1A top: low intensities are abstracted as smaller spheres). According to their position in the dataspace, formed populations can be assigned to a specific target DNA sequence to be detected inside the sample. It is worth to mention that there might be other ways to generate similar signal effects like usage of two different quencher types or two similar fluorophore labels at one PSR in a duplex REM-dPCR.
In a four-plex REM-dPCR assay, executed in two detection channels, the presence of multiple targets in a compartment will lead to the generation of additional populations. This means that by usage of four different PSR molecule types, up to sixteen different populations could theoretically be observed. However, as in digital assays, the distribution of target template follows Poisson statistics, and the generation of double and multiple positive populations depend on target template concentrations as well as on possible upcoming competing effects caused by the detection of gene loci sharing the same primer pairs.9 Therefore, only nine populations may have formed with the exemplary KRAS/BRAF target panel (Fig. 1B).
In comparison to the existing higher order multiplexing methods,9,23 REM-dPCR has the overall advantage that concentrations of target-independent reporter molecules can stay constant in different assays. This gives more freedom to the assay design, and fluorescence signal populations can be optimized with high efficiency. Regarding SNP detection, another advantage of combining MPs with target sequence-independent PSRs comes into play. During the elongation process, bound MPs are cleaved highly specifically by the polymerase. Thereby, the mediator section is cleaved off from the probe section. The cleavage side is located between the first and second nucleotides of the probe section. Only if cleavage was performed correctly, the released mediator can bind completely to the mediator binding side at its specific PSR type to initiate signal generation. If no correct cleavage occurred, the existing 3′-overhang will hinder the elongation process at the PSR and no signal will be generated. Therefore, in the case of SNP detection, the first nucleotide at the 5′-end of the target specific probe section of a MP has to be positioned over the SNP position in order to activate the specific PSR type.17,24
In this publication, a SNP detection REM-dPCR assay is presented, which demonstrates the functionality of a six-plex REM-dPCR and copes with upcoming population patterns of multiple positive populations caused by the same gene loci detection.
In the following paragraphs, the design guidelines for a six-plex REM-dPCR, starting from a two-plex to four-plex and six-plex reactions, are provided, targeting wild-type and mutant KRAS and BRAF sequences (Table 1; ESI Table S1†). Furthermore, the competing effect of shared primer pairs is visualized and its relevance for assay design discussed.
Two-plex REM-dPCRs
As the target panel for a proof of concept for REM-dPCRs, the genes BRAF (WT and V600E) and KRAS (WT, G12A, G12D, and G12 V) were selected (ESI Table S1†). With the aim to stepwise increase the multiplexing degree, first duplex reactions were defined targeting the genes BRAF WT and BRAF V600E in the blue, KRAS WT and KRAS G12A in the green and KRAS G12D and KRAS G12 V in the red detection channel of the PRISM3 in the Stilla Naica System (Table 1 and ESI Table S1†). Fluorophores were selected according to their optical properties (Table 1 and ESI Table S3†). In Fig. 2, the scatterplots of the three duplex reactions, with two different conditions C1 and C2 (Tables 2 and 3), are shown. Comparing the two conditions, one can see that the cycling condition had a substantial effect on signal generation and, therefore, also on the separation of positive populations and performance of a REM-dPCR assay. Under C1, all targets could be detected in the single target control, but a population differentiation in the duplex reaction was only possible in the red channel for the targets KRAS G12D and KRAS G12 V using the fluorophore–quencher pair combinations Atto647N/BHQ2 and Cy5/BHQ2. To enable population differentiation for the remaining four targets and reduce signal scattering and satellite populations, titrations of the PCR system as described above (Materials and methods, Multiplexing conditions) were performed with the least separable two-plex reaction targeting KRAS WT and KRAS G12A in the green detection channel (ESI Fig. S1–S4†). Scattering and therefore unspecific signals could be strongly reduced by decreasing the annealing temperature and prolongation of the elongation time (ESI Fig. S1†), resulting in the separation of two distinct positive populations. To further increase the compactness of the distinct population, an intense oligonucleotide titration was performed, varying the primer, MP and PSR concentrations and ratios (ESI Fig. S2–S4†). As here, the KRAS WT and G12A gene was targeted with the same primer pair it was assumed that PCR performance could be improved by varying the primer conditions. To support data evaluation, the compactness of positive populations, or rather the random distribution of positive data populations of beneficial titration conditions was analysed/evaluated by point-pattern analysis that takes into account the randomness of spatial distribution of data points (ESI Table S4†). As shown in the primer titration, the increased concentration of forward to reverse primers had indeed a major impact on population separation (ESI Fig. S2†). The VMR values increased with the increase in forward primer concentration from 12.9 at a forward-to-reverse primer ratio of 1/1 to up to 17.2 at a forward-to-reverse primer ratio of 2.5/1, indicating a stronger and more distinct clustering of positive signals (ESI Table S4†). Signal intensities could be further improved by an MP:PSR titration, whereby the increase in PSR concentration (ESI Fig. S3†) and a decrease in MP-to-PSR ratio improved signal generation and, therefore, also resulted in slightly improved population separation (ESI Fig. S3†). A combination and comparison of the most beneficial primer and MP-to-PSR ratios (ESI Fig. S4†) showed that the strongest clustering behaviour (VMR of 22.1, ESI Table S4†) and best population separation could be achieved at a 2/1 forward-to-reverse primer ratio and a 2/1 MP-to-PSR ratio, resulting in the definition of condition C2 (Table 2).
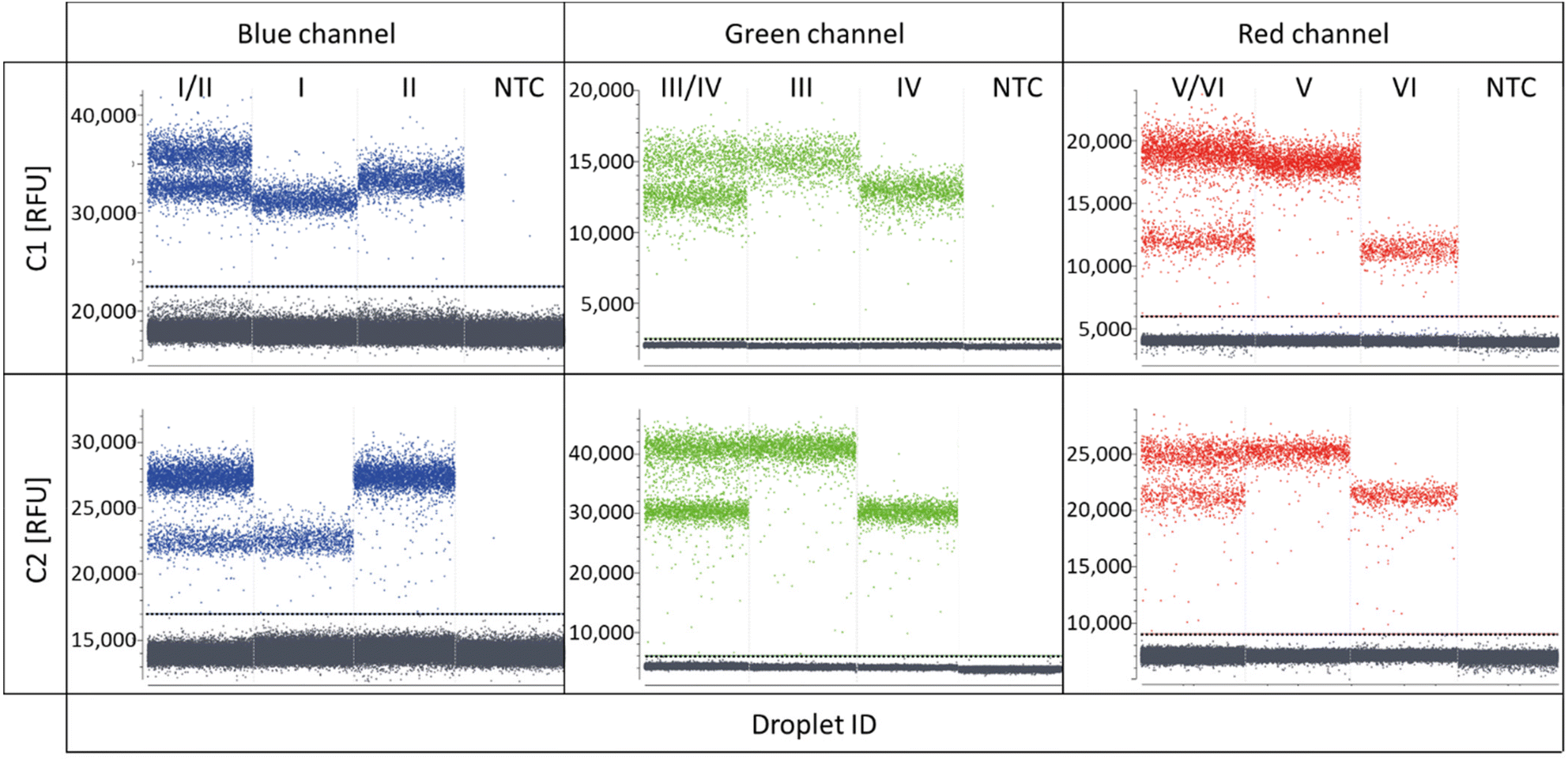 | ||
| Fig. 2 Scatterplots of two-plex REM-dPCRs comparing the two assay conditions C1 and C2. Shown are two-plex reactions and single target controls targeting BRAF WT (I) and BRAF V600E (II) in the blue channel, KRAS WT (III), KRAS G12A (IV), KRAS G12D (V) and KRAS G12 V (VI) in the green and red channels, respectively, as well as non-target controls in each channel (NTC). Plotted is the fluorescence intensity in relative fluorescence units (RFU) versus droplet ID. Thresholds are indicated by a horizontal line in the corresponding channel colour. | ||
The resulting condition C2 was applied to the two-plex assays run in the red and blue channels. A stronger clustering and, therefore, more distinct populations could be overserved for the targets BRAF WT and BRAF V600E (blue channel, VMR values increased from 28 to 61, ESI Table S4†), whereby in the red channel, the new assay parameters led to the reduced separation of the positive populations targeting KRAS G12D and KRAS G12 V (red channel, Fig. 2, condition C2), also mirrored by the reduced VMR value, which decreased from 28 in C1 down to 21 in C2 (ESI Table S4†). For the latter duplex reaction in the red channel, several reasons may have led to a decreased population separation. It may be that due to the increased PSR concentration, a non-proportional signal increase in the Atto647N-labelled PSR compared to the Cy5 labelled PSR occurred, which might be balanced by further optimization of assay parameters. Another reason may be a slightly different mediator performance. Due to increased MP and PSR (Atto647N/BHQ2) concentrations for the KRAS G12D target, signal saturation was reached at a later time point, meaning more PSR could be activated, resulting in an increase in the signal intensity. However, in order to identify the exact reason, more research efforts have to be devoted to the effects on sequence and fluorophore-dependent signal generation in future.
Concentrating on the KRAS duplex reactions, it is worth to mention that all four targets shared the same primer pair, which came into account once those duplex reactions were combined to a four-plex reaction, as presented in the next section.
Four-plex REM-dPCRs
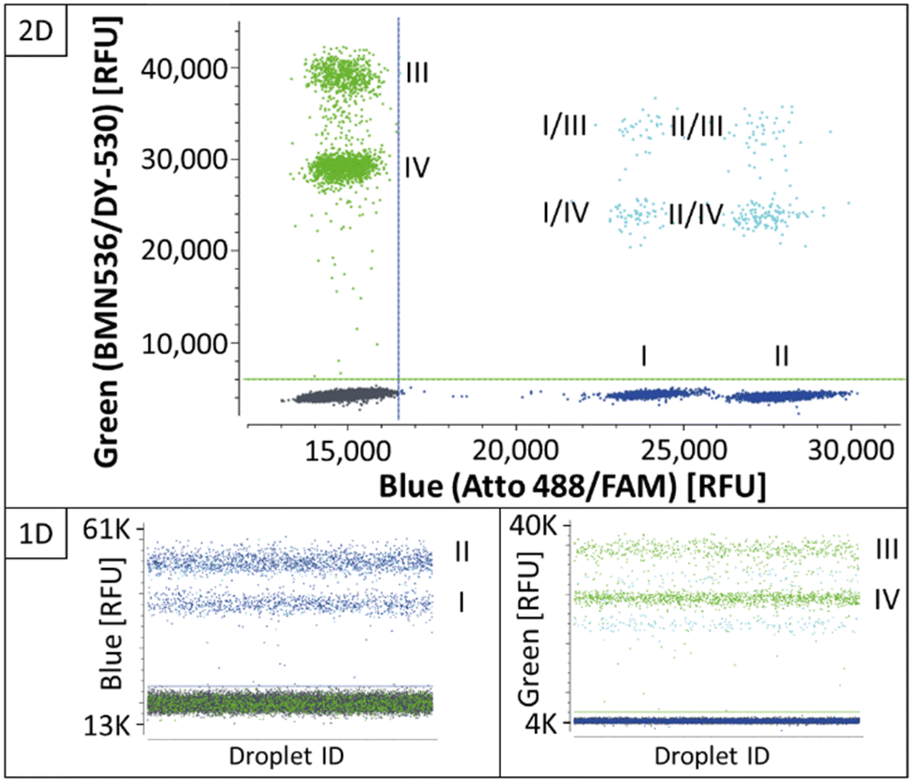 | ||
| Fig. 3 Four-plex REM-dPCR scatterplots targeting BRAF WT (I) and BRAF V600E (II) in the blue channel and KRAS WT (III) and KRAS G12A (IV) in the green channel, under C2 assay conditions. Double-positive populations are indicated in turquoise and negative population in dark grey. Thresholds were set as close as possible above the negative population and are indicated as lines in green and blue for the respective channel. The results are shown in 1D and 2D for the respective channels and channel combinations. | ||
As shown in the 2D plots in Fig. 3, single-positive populations formed distinct and differentiable population for the targets BRAF WT (I) and BRAF V600E (II) in the blue channel, and KRAS WT (III) and KRAS G12A (IV) in the green channel. Double-positive signals formed populations in the orthogonal space between the respective single-positive populations. Comparing the results in the 1D plots of Fig. 3 and C2 in Fig. 2, they show that the determined assay parameters of condition C2 enabled a direct transfer of assay parameters from two-plex to four-plex and a stable increase in the multiplexing degree.
:1, 4:1 and 5:1, and primers were set at 2:1; 1:1 and 1:2 forward-to-reverse ratios per MP:PSR condition. The titration showed that variation of the MP-to-PSR ratio had a major effect on positive population separation. While by increased PSR concentration, the separation was improved for targets KRAS WT (III) KRAS G12A (IV) in the green channel, the opposite effect could be observed for the targets KRAS G12D (V) and KRAS G12 V (VI) in the red channel, which corresponds to the observed effects in the two-plex reactions of C1 and C2 (Fig. 2). Including the effects of the primer ratios, the condition C3 is defined as the condition with best population separation for all four targets (Fig. 4), whereby the MP-to-PSR ratio was increased to 3:1 and a forward-to-reverse primer ratio was kept constant at 2:1 (Table 2; Fig. 4).
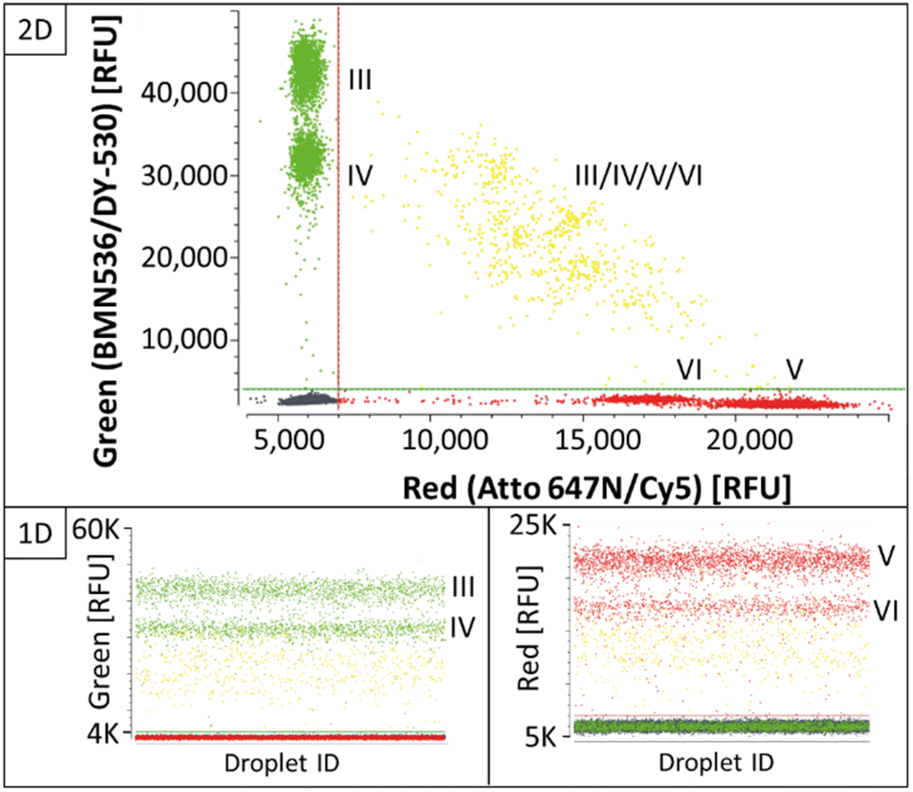 | ||
| Fig. 4 Four-plex REM-dPCR scatterplots with condition 3 assay parameters targeting KRAS WT (III) and KRAS G12A (IV) in the green channel and KRAS G12D (V) and KRAS G12 V (VI) in the red channel. Double-positive populations are indicated in yellow. Thresholds are indicated as green and red lines in the figure. The results are shown in 1D and 2D for the respective channels and channel combinations. | ||
Even though separation of single-positive populations in the KRAS four-plex assay could be improved from condition C2 (ESI Fig. S5†) to C3 (Fig. 4), the signals of double-positive populations did not form orthogonal clusters as observed for the BRAF/KRAS four-plex assay (Fig. 3). Instead, double-positive populations formed an arch-like cluster diagonal to the single-positive populations. Double-positive compartments showed quite diverging signal intensities, which lead to wide scattering of the double-positive populations (Fig. 4, highlighted in yellow). This is a known phenomenon already described as partition-specific competition effect.9 When multiple targets share the same primer pair, the outcompeting of one target amplicon over another causes a decrease in signal intensity and an arch-shaped clustering of the double-positive droplets can be observed. According to Whale et al.,9 the formation of a clear subpopulation may be possible if different starting conditions, such as multiple copies of one target per compartment/droplet, are provided, as the competing effects would provide an advantage for different target ratios.
This competing effect probably also accounts for the fact that double-positive signals of BRAF WT (I)/V600E (II) or KRAS WT (III)/G12A (IV) or KRAS G12D (V)/G12 V (VI) within the same fluorescence channel did not form a separate population with increased intensity on the same axis. Here, in this set-up, about 3000 copies were used per reaction for the illustration of the functionality of the assay. This means that depending on the generated number of droplets, here about 200 droplets were expected to be double positive. However, because of the decreased signal intensity due to the competing primer effect, the signals were rather incorporated in the single-positive populations of KRAS WT and KRAS G12D with higher signal intensities, respectively.
Six-plex REM-dPCRs
Finally, based on the results of the four-plex reactions, a six-plex REM-dPCR targeting BRAF WT (I; Atto488/BMNQ-1) and BRAF V600E (II; FAM/BHQ-1) in the blue channel, KRAS WT (III; DY-530/BMN-Q1) and KRAS G12A (IV; BMN536/BHQ-1) in the green channel and KRAS G12D (V; Cy5/BHQ-2) and KRAS G12 V (VI; Atto647N/BHQ-2) in the red channel was designed, applying determined assay condition C3 (Table 2). The assay was read out in all three detection channels of the dPCR device, and the results are analysed in 2D and 3D scatterplots, respectively. In Fig. 5, a 3D plot of the six-plex assay is shown where it becomes clear that a high information density could be achieved by REM-dPCRs as 16 populations can be clearly distinguished in one graph/reaction. Single-positive populations are oriented next to the main axes, and double-positive signals formed populations in the orthogonal position in between the respective single-positive populations for the targets BRAF WT and KRAS WT; BRAF WT and KRAS G12A; BRAF V600E and KRAS WT; and BRAF V600E and KRAS G12A (Fig. 5, highlighted in turquois), as well for BRAF WT and KRAS G12D; BRAF WT and KRAS G12 V; BRAF V600E and KRAS G12D and BRAF V600E and KRAS G12 V (Fig. 5, highlighted in purple). As already observed in the four-plex assay, double-positive populations for the targets KRAS WT, KRAS G12A, KRAS G12D and KRAS G12 V formed an arch-like population when multiple targets shared the same primer pair (Fig. 5, highlighted in yellow). The signals of multiple-positive droplets (containing more than two different target sequences) formed a more spacious population in the space between (Fig. 5, highlighted in light grey).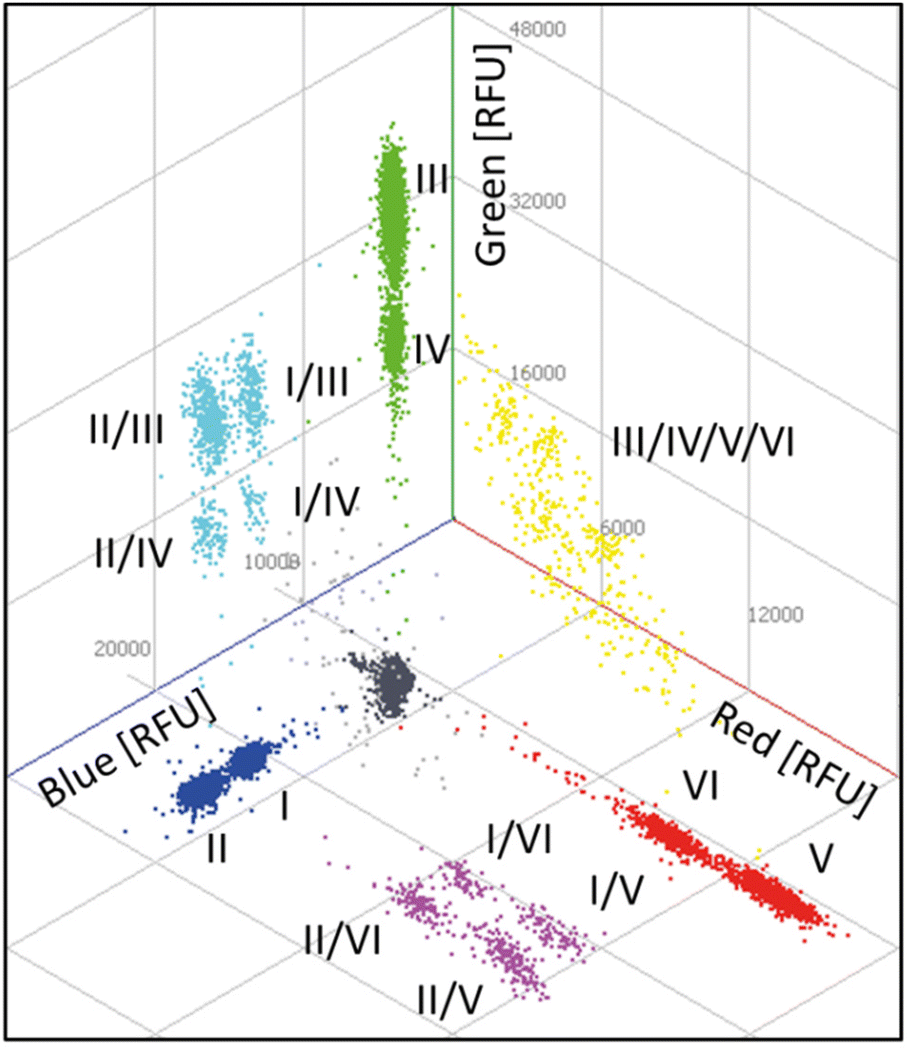 | ||
| Fig. 5 Three-dimensional scatterplot of a six-plex REM-dPCR. Six target genes were detected in three detection channels: BRAF WT (I; Atto488/BMNQ-1) and BRAF V600E (II; FAM/BHQ-1) in the blue channel, KRAS WT (III; DY-530/BMN-Q1) and KRAS G12A (IV; BMN536/BHQ-1) in the green channel and KRAS G12D (V; Cy5/BHQ-2) and KRAS G12 V (VI; Atto647N/BHQ-2) in the red channel. Respective double-positive populations are indicated in purple, turquoise and yellow. A multiple positive population is indicated in light grey, the negative population in dark grey. A .gif animation of this six-plex REM-dPCR can be seen in ESI Fig. S7.† | ||
As already shown by the increase from two-plex to four-plex reactions, the increase from the four-plex reaction to the six-plex reaction was according to the predetermined conditions, and the populations were allocated in the 3D space at expected positions, proving the stability of the REM-dPCR system. Positive populations formed by target-independent PSR labelled with fluorophores of higher quantum yield always formed clusters at higher intensities, while positive populations formed by target-independent PSR labelled with fluorophores of lower quantum yield always formed clusters of lower intensities. Therefore, one has to take into account that quantum yields are in dependency of buffer conditions and device-specific detection ranges.
According to Hugget et al.,5 rain are partitions of intermediate fluorescence caused by suboptimal PCR parameters, which can therefore be improved by optimizing the PCR system. During the process of REM-dPCR six-plex development, we could clearly show that by optimizing the PCR system, as well as the signalling system, the rain could be strongly reduced, resulting in clear populations that allowed even differentiation of two positive populations by monochrome multiplexing. Nevertheless, besides the optimization process, there will always remain a section of rain that may fall into a population of lower intensity (either a positive population or a negative population). In order to exclude false-negative signals, this rain has to be counted as positive for the overlying positive population.5 Here, in this set-up, the detection of KRAS WT was assigned to a target-independent reporter type, which, once activated, emits at a higher intensity (PSR02), and that of BRAF WT PSR07 emits at a lower intensity (Table 1). As WT sequences will always be present in high amounts, it would be advantageous for the future assay design to link WT sequence detection to lower positive populations to avoid the disappearance of rare mutant sequences in the rain of WT positive populations.
To demonstrate the functionality and stability of the six-plex REM-dPCR, an experimental set-up was chosen where BRAF WT and KRAS WT template sequences were added to the reaction, as well as one mutant sequence at a time. As shown in Fig. 6, the six-plex REM-dPCR assay formed clear clusters for present targets in expected positions of the dataspace, allowing clear identification of target sequences. Comparing Fig. 6A to B, it is well visible that the presence of KRAS 12A (IV) sequences leads to clear population formation below the KRAS WT (III) population, and double-positive populations (I/III and I/IV) were visualized in the orthogonal position to the BRAF WT (I) population. Here, in this case (Fig. 6B) also, around 28 double-positive signals were to be expected for KRAS WT and KRAS G12A targets. Such a double-positive population would have been expected to form a separate population of higher fluorescence intensity, distinctive from the KRAS WT and KRAS G12A populations. However, possibly due to the competing effect that causes a lowering of signal intensities in the respective compartments, the signals of double-positive droplets did not form a separate population above the KRAS WT population, and these signals probably got incorporated into the KRAS WT population. A distinct double-positive population may form, if different targets would be detected in the same wavelength range/detection channel, which has to be taken into consideration for future assay designs.
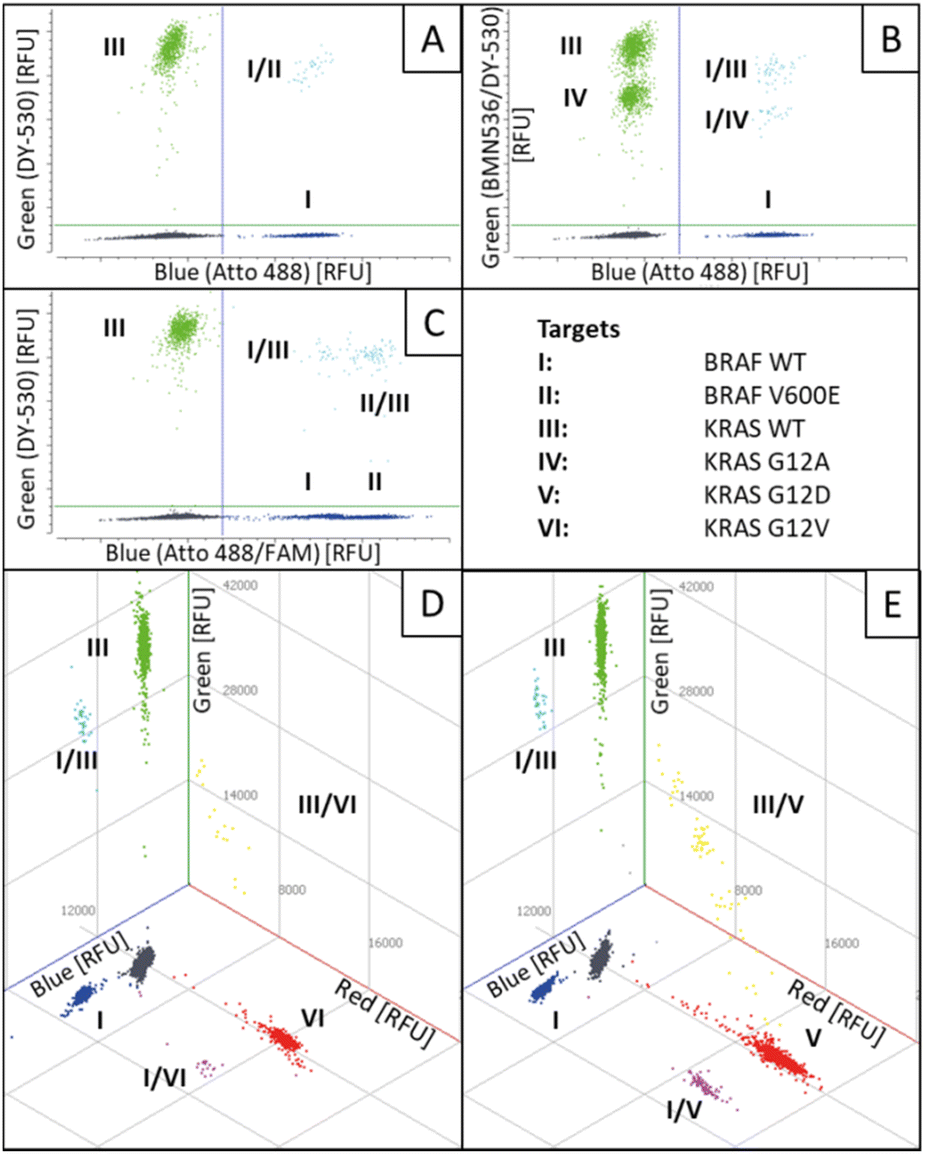 | ||
| Fig. 6 Scatterplot of six-plex REM-dPCRs with varying target concentrations. Two-dimensional (A)–(C) and three-dimensional (D)–(E) scatterplots are shown, in which both BRAF WT (I) and KRAS WT (III) targets were added to the reaction (A) as well as one additional target at a time: KRAS G12A (IV) (B); BRAF V600E (II) (C); KRAS G12 V (VI) (D) and KRAS G12D (V) (E). Double-positive partitions in the blue and green channels are indicated in turquoise, in blue and red channels in purple and in the green and red channels in yellow. Negative populations are indicated in dark grey. | ||
Nevertheless, this experimental set-up also showed that the functionality is not hampered by the presence or absence of target sequences. It is also worth mentioning that during the process of assay development, about 3000 copies per reaction and target were used, which mirrors an approximation of WT targets in real patient samples,17 but, for example, during oncology treatment and minimal residual disease monitoring, mutant sequences may appear in decreased copy numbers, which would also reduce the presence of double-positive populations, so that mainly single-positive populations should be observable.
For the validation of the transferability of the REM multiplexing concept to other detection channels/dPCR devices, a 2-plex assay was set up for the quantification of KRAS G12 V target DNA combined with KRAS WT DNA in the infra-red channel of a Prism6 (Stilla). For the KRAS G12 V target detection, the PSR was labeled with Cy5.5 – BHQ2 (upper population; Fig. 7) and for KRAS WT, the PSR was labelled ATTO680-BHQ2 (lower population; Fig. 7). MP sequences were adopted because both PSRs are activated by different mediator types then the PSRs used in previous experiments (see Table 1). As can be seen in Fig. 7, Fig. 8 and Table 5, specific, precise and sensitive quantification could be achieved in the same detection channel using REM. In addition, Fig. 7 shows that with the increase in template concentration and increased resolution of data points in the dataspace, subpopulations of the KRAS G12V-positive droplets can be observed. It may be that due to the high occurrences of the WT template DNA, a competition situation or FRET quenching may have occurred in droplets, in which multiple copies were present. However, comparable population formation was observed in colorimetric multiplex dPCRs.17
| KRAS G12V expected copies | 0 | 18 | 90 | 180 | 1800 | 9000 |
|---|---|---|---|---|---|---|
| Mean value measured copies (N = 3) | 1 | 21 | 128 | 286 | 2347 | 12435 |
| STD measured copies (N = 3) | 1 | 6 | 29 | 100 | 400 | 2174 |
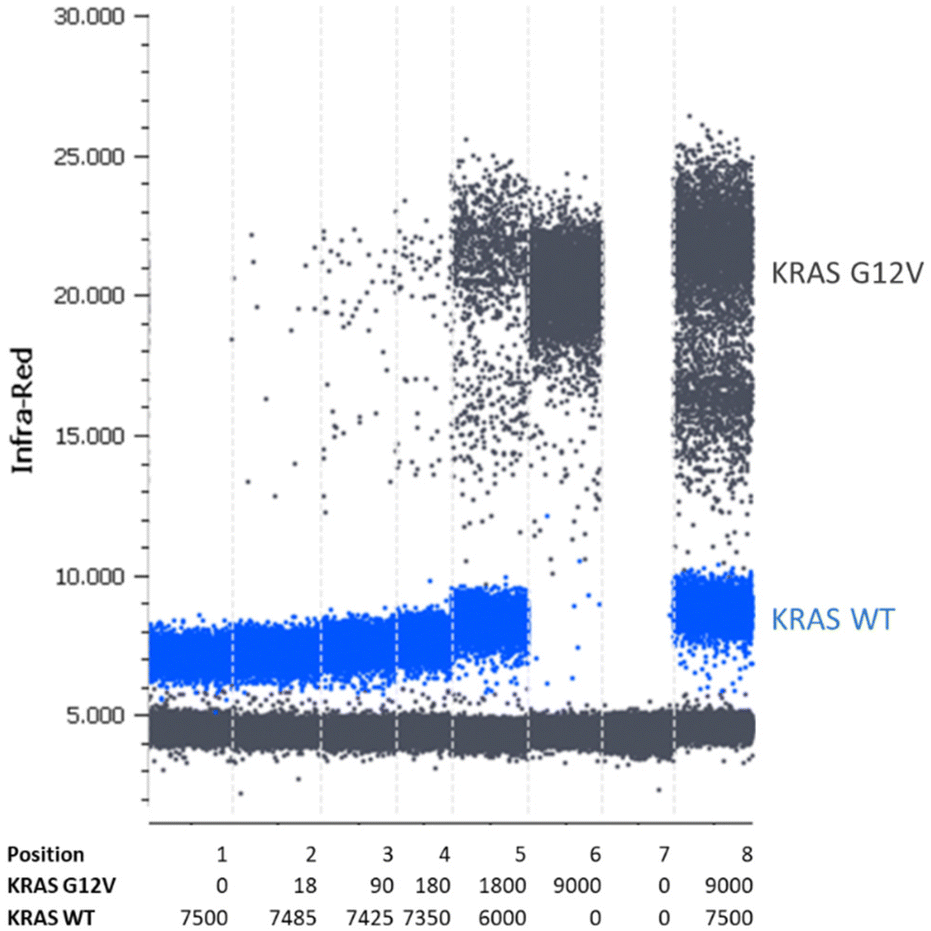 | ||
| Fig. 7 One-dimensional scatterplot of a duplex REM dilution row. The KRAS G12 V template was diluted in the KRAS WT template. Following conditions are shown: KRAS WT positive control (position 1); dilution of KRAS G12 V template in KRAS WT template (position 2–5); KRAS G12 V positive control (position 6); water control (position 7) and KRAS G12 V/WT double-positive control (position 8). Here, an exemplary scatterplot of one triplicate is shown. The table below indicates copy numbers per 25 μl reaction according to the manufacturer's information. | ||
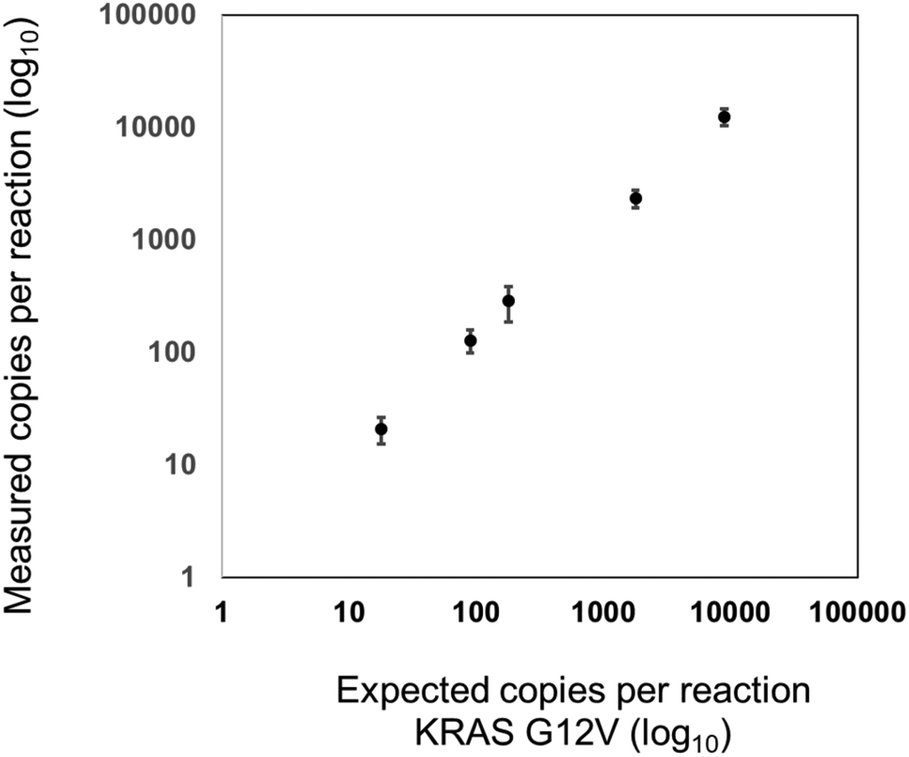 | ||
| Fig. 8 Correlation between the expected and measured KRAS G12 V copy numbers per reaction with a R2 value of 0.9998. KRAS G12 V templates were diluted in KRAS WT templates. Shown are the logarithmic mean values of calculated copy numbers per reaction (N = 3). The error bars represent the standard deviation of measured copy numbers. | ||
Conclusion
In this work, we presented a new technology that allows a direct increase in the multiplexing capacity of a dPCR device without the need for additional processing steps, pattern interpretation or external devices. We successfully demonstrated the establishment of a six-plex REM-dPCR assay, directly targeting two wild-type sequences and four SNP mutant sequences in three detection channels. We further demonstrated that once duplex reactions were established, higher multiplexing degrees can be reached easily by direct combination of the duplex and four-plex assays, respectively. Due to the target-independent reporter types, the presented six-plex REM-dPCR assay set may be transferred directly to new target sequences by simply adapting MPs to the new target sequences, when using the same dPCR fluorescence detection device. The use of the same preconditioned system makes this assay design more efficient than the other states of the art methods. Furthermore, we have shown that the transfer to other dPCR fluorescence detection devices/fluorescence detection channels can be achieved by exchanging the population-specific reporter molecules. Fluorophore–quencher modification of the adapted reporter molecules has to consider the wavelength of the fluorescence detection channel as well as their intensities, to achieve well-separated populations for different targets within the same detection channel. This transfer was validated by the quantification of KRAS G12 V mutant and wild-type DNA, without further optimization steps. One has to consider that the optical properties of the fluorophores of activated independent PSR types define the detectability of the signals in the dPCR device. Once another optical system is used, there may be a shift in the bandwidths of the detection channels, resulting in a shift in the signal intensities of the target-independent PSR types. Furthermore, one has to take into account that in case of an assay transfer to another dPCR system, buffer compositions may substantially affect the assay performance.For future establishment of even higher multiplexing degrees, an increase in detection channels may also raise the probability of signal spillover, and even more stringent fluorophore/quencher screening and selection as well as efficient spillover compensation have to be performed. With the increase in multiplexing levels, it might be necessary to perform more complex assay optimization. A workflow for the systematic multiplex MP PCR assembly and optimization in real-time PCRs has already been established.4 This workflow focusses on primer titration, which is independent of the fluorogenic properties of the selected reporter molecules. Therefore, it can be assumed that this approach will also be suitable for REM. For data analysis and data visualization, it was substantial during the development process to increase dataspace dimensions for correct data analysis, especially for the identification of multiple positive populations. However, current analysis software solutions may come to their limits once higher multiplexing degrees are to be achieved. Once more than three detection channels are used for the readout, populations could be mapped and theoretically separated in a dataspace of higher than three dimensions, which is not intuitively accessible. As could be seen already in Fig. 3, the increase in dataspace from 1D to 2D enabled improved population separation, whereas beforehand in 1D, double-positive populations were immersed in the single-positive population and could not be distinguished easily from single-positive populations. Furthermore, populations appear more distinguishable in higher dimensions than in lower dimensions, which is rather blurry. The additional degree of freedom lead to the inclusion of further fluorescence properties, which can and should be considered for data analysis. Therefore, additional detection channels will not only lead to higher multiplexing degrees, but also be important to separate the evolving fluorescence data clusters precisely. Advanced software solutions, as well as advanced statistical approaches, will be necessary to support improved data processing, analysis and assay establishment.
To facilitate population differentiation, future work could focus on an extended choice of fluorophores. This may allow the increase in the number of target-independent PSRs per channel from two to three, further increasing the multiplexing capacity per channel.
Author contributions
Conceptualization: M. L., S. C.; formal analysis: A. M. M., S. C., M. L., S. J. K.; investigation: A. M. M., M.N.; project administration: M. L., S. C.: funding acquisition: N. B., F. v. S., M. L.; methodology: A. M. M., S. C., M. L., S. J. K.; supervision: S. C., M. L.; visualization: A. M.M., S. C., M. L., M.N.; verification: A. M. M., S. C., M. L.; writing – original draft: S. C.; writing – review & editing: S. C., A. M. M., M. L., N. B., F. v. S., M. L., S. J. K, M.N. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare. The presented technology is part of an PCT patent application whereby several authors are listed as inventors.Acknowledgements
A selection of oligonucleotides were transferred from former already closed projects: IRMA-4-ALL (FKZ 01 EK1508B/INDIMED program) and ARTUR (grant number 13GW0198F) both funded by the Federal Ministry of Education and Research (BMBF), Germany; and the project PRIMO (AZ 3-4332.62-HSG/84) funded by the Ministry of Economic Affairs, Labor and Housing of the State of Baden-Württemberg, Germany, as well as the current project MelB (IGF-Vorhaben-Nr. 22486 N), founded by the AiF German Federation of Industrial Research Associations.References
- A D. Bell, C. L. Usher and S. A. McCarroll, Analyzing Copy Number Variation with Droplet Digital PCR, in Digital PCR: Methods and Protocols, ed. G. Karlin-Neumann and F. Bizouarn, Methods in Molecular Biology, Humana Press, New York, 2018, 1768, pp. 143–160 Search PubMed.
- (a) I. Palacín-Aliana, N. García-Romero, A. Asensi-Puig, J. Carrión-Navarro, V. González-Rumayor and Á. Ayuso-Sacido, Biomedicines, 2021, 9, 906 CrossRef PubMed; (b) U. P. Zmrzljak, R. Košir, Z. Krivokapić, D. Radojković and A. Nikolić, Genes, 2021, 12, 289 CrossRef CAS PubMed; (c) I. Merino, A. de la Fuente, M. Domínguez-Gil, J. M. Eiros, A. P. Tedim and J. F. Bermejo-Martín, Crit. Care, 2022, 26, 63 CrossRef PubMed.
- F. Schlenker, E. Kipf, N. Borst, N. Paust, R. Zengerle, F. von Stetten, P. Juelg and T. Hutzenlaub, Processes, 2021, 9, 97 CrossRef CAS.
- E. Kipf, F. Schlenker, N. Borst, M. Fillies, R. Kirschner-Schwabe, R. Zengerle, C. Eckert, F. von Stetten and M. Lehnert, J. Mol. Diagn., 2022, 24, 57–68 CrossRef CAS PubMed.
- J. F. Huggett, Clin. Chem., 2020, 66, 1012–1029 CrossRef PubMed.
- (a) A. S. Whale, J. F. Huggett, S. Cowen, V. Speirs, J. Shaw, S. Ellison, C. A. Foy and D. J. Scott, Nucleic Acids Res., 2012, 40, e82 CrossRef CAS PubMed; (b) P.-L. Quan, M. Sauzade and E. Brouzes, Sensors, 2018, 18, 1271 CrossRef PubMed; (c) R. Sanders, J. F. Huggett, C. A. Bushell, S. Cowen, D. J. Scott and C. A. Foy, Anal. Chem., 2011, 83, 6474–6484 CrossRef CAS PubMed.
- (a) A. S. Whale, A. S. Devonshire, G. Karlin-Neumann, J. Regan, L. Javier, S. Cowen, A. Fernandez-Gonzalez, G. M. Jones, N. Redshaw, J. Beck, A. W. Berger, V. Combaret, N. Dahl Kjersgaard, L. Davis, F. Fina, T. Forshew, R. Fredslund Andersen, S. Galbiati, Á. González Hernández, C. A. Haynes, F. Janku, R. Lacave, J. Lee, V. Mistry, A. Pender, A. Pradines, C. Proudhon, L. H. Saal, E. Stieglitz, B. Ulrich, C. A. Foy, H. Parkes, S. Tzonev and J. F. Huggett, Anal. Chem., 2017, 89, 1724–1733 CrossRef CAS PubMed; (b) L. B. Pinheiro, H. O'Brien, J. Druce, H. Do, P. Kay, M. Daniels, J. You, D. Burke, K. Griffiths and K. R. Emslie, Anal. Chem., 2017, 89, 11243–11251 CrossRef CAS PubMed.
- F. Bizouarn, Introduction to Digital PCR, in Quantitative Real-Time PCR, ed. R. Biassoni and A. Raso, Methods in Molecular Biology, Humana Press, New York, 2014, 1160, pp. 27–41 Search PubMed.
- A. S. Whale, J. F. Huggett and S. Tzonev, Biomol. Detect. Quantif., 2016, 10, 15–23 CrossRef CAS PubMed.
- J. Corné, F. Le Du, V. Quillien, F. Godey, L. Robert, H. Bourien, A. Brunot, L. Crouzet, C. Perrin, C. Lefeuvre-Plesse, V. Diéras and T. de la Motte Rouge, Sci. Rep., 2021, 11, 17316 CrossRef PubMed.
- J. Madic, C. Jovelet, J. Lopez, B. André, J. Fatien, I. Miran, A. Honoré, L. Mezquita, B. Besse, L. Lacroix and M. Droniou, Oncotarget, 2018, 9, 37393–37406 CrossRef PubMed.
- (a) C. B. Hughesman, X. J. D. Lu, K. Y. P. Liu, Y. Zhu, C. F. Poh and C. Haynes, PLoS One, 2016, 11, e0161274 CrossRef PubMed; (b) J. F. Huggett, S. Cowen and C. A. Foy, Clin. Chem., 2015, 61, 79–88 CrossRef CAS PubMed.
- R. Nguyen, S. Perfetto, Y. D. Mahnke, P. Chattopadhyay and M. Roederer, Cytometry, Part A, 2013, 83, 306–315 CrossRef PubMed.
- J. Madic, A. Zocevic, V. Senlis, E. Fradet, B. Andre, S. Muller, R. Dangla and M. E. Droniou, Biomol. Detect. Quantif., 2016, 10, 34–46 CrossRef CAS PubMed.
- I. Santos-Barriopedro, S. Ursuegui, E. Fradet and R. Dangla, Robust higher-order multiplexing in digital PCR by color-combination, bioRxiv, 2023, preprint, DOI:10.1101/2023.05.10.540190.
- F. Schlenker, E. Kipf, N. Borst, T. Hutzenlaub, R. Zengerle, F. von Stetten and P. Juelg, Anal. Chem., 2021, 93, 10538–10545 CrossRef CAS PubMed.
- F. Schlenker, E. Kipf, M. Deuter, I. Höffkes, M. Lehnert, R. Zengerle, F. von Stetten, F. Scherer, J. Wehrle, N. von Bubnoff, P. Juelg, T. Hutzenlaub and N. Borst, Cancers, 2021, 13, 5742 CrossRef CAS PubMed.
- S. Wadle, M. Lehnert, F. Schuler, R. Köppel, A. Serr, R. Zengerle and F. von Stetten, BioTechniques, 2016, 61, 123–128 CrossRef CAS PubMed.
- M. Lehnert, E. Kipf, F. Schlenker, N. Borst, R. Zengerle and F. von Stetten, Anal. Methods, 2018, 10, 3444–3454 RSC.
- J. SantaLucia, Physical Principles and Visual-OMP Software for Optimal PCR Design, in PCR Primer Design, ed. A. Yuryev, Methods in Molecular Biology, Humana Press, Totowa NJ, 2007, 402, pp. 3–33 Search PubMed.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef CAS PubMed.
- B. Faltin, S. Wadle, G. Roth, R. Zengerle and F. von Stetten, Clin. Chem., 2012, 58, 1546–1556 CrossRef CAS PubMed.
- B. Staff, Bioradiations, 2019, https://www.bioradiations.com/expanded-droplet-digital-pcr-multiplexing-capability-using-two-different-strategies/.
- V. Lyamichev, M. A. Brow and J. E. Dahlberg, Science, 1993, 260, 778–783 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an00191a |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |